
Abstract
Background
Hypermutated colorectal cancer (CRC) reportedly accounts for 15%–17% of all cases of CRC. However, the proportion and number of patients with hypermutated CRC cannot be unappreciated. Additionally, therapy options for these patients differ from those for CRC patients, with a greater potential benefit from immunotherapy.
Materials and methods
We sequenced the tumor mucosa of CRC patients with >24 months of follow-up data at our center and identified mutation profiles of hypermutated CRC as a training data set (Zhejiang University [ZJU]); we then collected patients from The Cancer Genome Atlas (TCGA) as a validation data set. Recurrently mutated genes were combined to calculate a compound score via Cox proportional hazards model. Patients with higher-than-median scores were segregated as the high-risk group. Outcomes were analyzed by Kaplan–Meier and Cox regression analyses using Python (3.6.0) and R (3.4.0).
Results
We constructed a 4-gene signature (ACVR2A, APC, DOCK2, and POLE), with training in 45 hypermutated patients at ZJU and validation in 24 hypermutated patients from TCGA. Patients in the high-risk group showed poor survival (adjusted HR =9.85, 95% CI: 2.07–46.81, P=0.004). Further subgroup analysis was performed for stage II and III colon cancer (HR =10.91, 95% CI: 1.36–87.5, P=0.005) and high microsatellite instability (MSI-H) CRC (HR =12.57, 95% CI: 1.57–100.69, P=0.002) subgroups, which verified that our signature is universal. We then compared our prognostic signature with other risk factors (including MSI status, POLE driver mutation, BRAF-p.V600E, tumor mutational burden, and TNM staging). The results proved that our 4-gene signature is better than the other risk factor for prognosis in hypermutated CRC.
Conclusion
Our 4-gene signature is a good predictor of survival for hypermutated CRC, and this signature is powerful in stage II and III colon cancer and MSI-H CRC. Future prospective studies are needed to confirm the power of the 4-gene signature in patients receiving immunotherapy.
Introduction
Colorectal cancer (CRC) is one of the most commonly diagnosed cancers worldwide. Indeed, most recent data show that CRC is the second leading cause of cancer death, with over 500,000 deaths annually.Citation1
Hypermutation is characterized by mismatch repair (MMR) deficiency or POLE/ POLD1 driver mutations, indicating a high mutational rate. Accumulating evidence confirms that hypermutation occurs in many cancer types, such as melanoma,Citation2 lung cancer,Citation3 and bladder cancer.Citation4
Acquisition of genomic instability is a crucial feature of CRC development, and the microsatellite instability (MSI) pathway, which in involved in 15%Citation5,Citation6 of CRC pathogeneses, is known to arise from inactivation of one of four MMR genes: MSH2, MLH1, MSH6, and PMS2.Citation7–Citation10 An aberrant MMR process leads to additive mutations throughout the genome and ultimately a “hypermutator” phenotype.Citation10,Citation11 The results of a recent large-scale sequencing analysis also suggest that hypermutation in CRC should not be underestimated.Citation12 Moreover, hypermutation is associated with a predicted high neo-epitope load, which can be exploited for immunotherapy in selected patients who experience conventional therapy failure.Citation13
Because hypermutated CRC differs from CRC with regard to tumorigenesis, prognosis, and treatment, it is unreasonable to use the same prognostic biomarkers as employed for patients without hypermutations. Here, we propose a prognostic prediction signature for patients with hypermutated CRC. This signature can determine the hypermutated status of tumors and distinguish between patients who have a high or low risk of disease-related death and predict those who are likely to benefit from immunotherapy.
Materials and methods
Sample collection and DNA extraction
Fresh tissues were flash frozen in liquid nitrogen after surgery and stored at −80°C until genomic DNA isolation. Only samples from patients with a pathologic diagnosis of CRC were evaluated, but samples were excluded if they contained <40% tumor cells. Tumor-and matched normal mucosa-derived DNA was purified using the QIAamp DNA mini kit (Qiagen NV, Venlo, The Netherlands).
All patients signed a consent form when the frozen tissue was obtained. This consent form authorized us to conduct scientific research and publish the results anonymously with the approval of the Institutional Review Board (IRB). All protocols and procedures of this study were approved under The Second Affiliated Hospital, School of Medicine of Zhejiang University IRB protocol 2013-042. This study was conducted in accordance with the Declaration of Helsinki.
DNA sequencing and MSI status detection
Panel sequencing was performed using a custom-designed panel utilizing Agilent SureSelect capture kit technology (Agilent Technologies, Santa Clara, CA, USA), targeting the exonic region of 524 genes (), with HiSeq 2000 (Illumina, San Diego, CA, USA). Sequence reads were aligned to the human genome (GRCh37/hg19), and unique pairs were used for variant calling. Candidate variants and indels were detected using GATK. Somatic mutations and indels were then identified using MuTect and Strelka, respectively. These variants were annotated with ANNOVAR (Openbioinformatics, USA).Citation14 MSI status was detected using mSINGS.Citation15
Detection of hypermutated samples and candidate gene screening
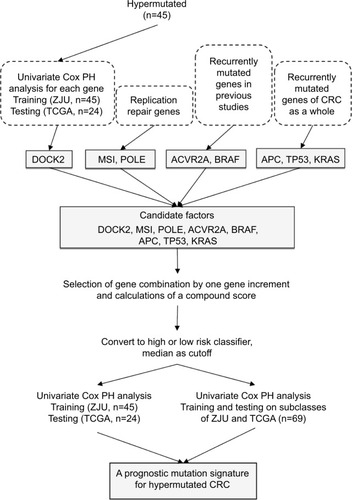
The threshold of hypermutation was higher than 10 Mut/ Mb, which was determined according to a recently published large-scale analysis.Citation12 As there are hundreds of mutated genes in a hypermutated tumor, it is impractical to use a mutation frequency ≥5% as a filtering criterion, which will include most of the genes sequenced. Therefore, we employed multiple strategies to screen for candidate prognostic genes in hypermutated CRC. First, we employed a univariate Cox proportional hazard (PH) model to evaluate the association between gene mutations and overall survival (OS) for each gene. Dedicator of cytokinesis 2 (DOCK2) was the only gene significantly associated with OS. Second, POLE driver mutation and MSI statuses were selected as candidates because replication repair mutations and MSI are observed in most types of hypermutated cancer. We identified POLE driver mutations based on the results of Campbell et al.Citation12 However, POLD1 was not included because no driver mutations were found in the present study. Third, activin A receptor type 2A (ACVR2A) and BRAF were included according to previous studies. APC, TP53, and KRAS, which have an overall higher mutation frequency in CRC, were also included. Thus, we obtained eight candidate factors (DOCK2, ACVR2A, BRAF, APC, TP53, KRAS, POLE, and MSI status) to construct a prognostic model. We summarize the selection process in .
Figure 1 Flowchart of prognostic signatures generation and validation procedures.
Abbreviations: TCGA, The Cancer Genome Atlas; ZJU, Zhejiang University; PH, proportional hazard; CRC, colorectal cancer.
Compound score calculation and high-risk patient detection
The final follow-up date was October 1, 2016 for patients from The Second Affiliated Hospital, School of Medicine of Zhejiang University (the ZJU cohort) and August 20, 2015 for patients from The Cancer Genome Atlas (TCGA) cohort. Only patients with >24 months of follow-up survival data were used for the prognosis-related analysis. OS was measured in months from the date of surgery to the date that the patient died. Stage was assessed according to the seventh version of the American Joint Commission on Cancer guidelines.
The compound score was calculated based on the sum of candidate gene mutations for each patient using a Cox PH model. Patients with higher-than-median scores were segregated as the high-risk group; others were defined as the low-risk group. The Cox PH model was employed to evaluate the association between the mutation signature and the clinical endpoint. To rule out over-fitting of the model, MSI data, somatic mutation data, and clinicopathological information for the TCGA cohort were obtained from the TCGA project data portal (http://www.cbioportal.org) on July 3, 2017.Citation16 These patients were used as an independent cohort for verification of the significance between the panel and OS.
Statistical analyses
Kaplan–Meier survival curve analysis with the log-rank test was applied to estimate the mutation signature in relation to OS. Fisher’s exact test, Student’s t-test, and the Mann–Whitney U-test were used to determine differences in clinicopathological variables between subgroups (high-risk vs low-risk/mutant vs wild-type). Multivariate Cox regression was performed to determine the contribution of the mutation signature to survival, adjusting for age, sex, and stage; the Wald test was employed in this analysis. Clinical subgroups of training and testing cohorts were also used to verify the prognostic prediction of the panel. Harrell’s concordance index (C-index) was used to quantify predictive accuracies. In addition, multiple permutation tests were performed on both cohorts. To obtain a sufficient sample size for clinical subclass analysis, we pooled the two cohorts and utilized 5-fold cross-validation. All statistical analyses were two-sided. A P-value of <0.05 was considered statistically significant. All analyses were performed using Python 3.6.0 (https://www.python.org/) and R 3.4.0 (https://www.r-project.org/).
Results
Patients’ basic characteristics
We sequenced 338 samples in our center, from which we identified 45 (13.30%) hypermutated patients as the training data set to construct the prognostic mutation signature. The proportion of hypermutation obtained in this study coincides with that of a previous report. Samples that met the criteria (63 patients) of hypermutation were selected from the TCGA project data portal for an independent testing data set. The demographics of the two initial cohorts, with a total of 108 patients, are shown in . Due to the lack of information on specific staging, location, and survival data in the TCGA data set, 24 patients from TGCA were ultimately selected as the testing group for prognosis analysis. The baseline characteristics of the two groups are presented in .
Table 1 Characteristics of included patients
Identified potential genes for constructing the signature
According to the three procedures described above, DOCK2 was found to be the only gene significantly associated with OS in both the ZJU and TCGA cohorts (HR =1.73, P=0.007 and HR =2.08, P=0.001, respectively). Combining the remaining selected genes, DOCK2, ACVR2A, BRAF, APC, TP53, KRAS, and POLE as well as MSI status were chosen as candidate factors for constructing the prognostic signature.
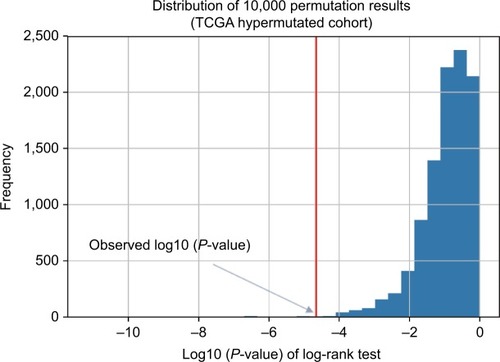
To determine the minimum number of genes able to discriminate outcomes, we began training in the ZJU cohort and stopped increasing the size of the signature when we obtained the maximal C-index. We next constructed a risk classifier based on four genes (DOCK2, ACVR2A, APC, and POLE), with good concordance (C-index =0.748). We further used multiple permutation testing to confirm the robustness of this signature, which indicated that our signature was not generated by coincidence ().
Figure 2 Distribution of 10,000 permutation results.
Abbreviation: TCGA, The Cancer Genome Atlas.
Construction of a 4-gene signature for high-risk hypermutated CRC patients
We divided patients into two groups by calculating the compound score for four genes. The compound score was generated from the ZJU cohort and further calculated for each patient in the TCGA cohort. Patients with higher-than-median scores were segregated as the high-risk group, and others were defined as the low-risk group. Patients with a high-risk classifier in the ZJU (HR =10.19, 95% CI: 1.25– 83.23, P=0.007) and TCGA (HR =8.62, 95% CI: 1.87–39.67, P=0.001) cohorts had significantly worse survival.
Due to the limited number of patients exhibiting hypermutation (n=45 in ZJU and n=24 in TCGA), we pooled the two cohorts to display the results and perform further subgroup analysis. A high-risk classifier was significantly associated with poor survival (HR =8.83, 95% CI: 2.00–39.04, P=0.001; ).
Figure 3 The pool data demonstrating effectiveness of the 4-gene signature.
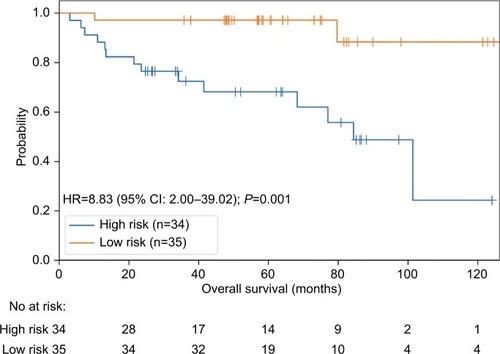
We also employed a multivariate Cox regression analysis including our 4-gene risk classifier, age, sex, and stage to reveal that the 4-gene risk classifier can serve as an independent determinant of OS in patients with hypermutated CRC (adjusted HR =9.85, 95% CI: 2.07–46.81, P=0.004) (). There was no statistically significant difference in the proportion of patients receiving adjuvant chemotherapy between the high-risk and low-risk groups (P=0.457, ).
Table 2 Multivariate Cox regression analysis of 4-gene signature
The 4-gene signature is powerful in specific subgroups
We first employed the 4-gene signature for subgroup analysis of stage II and III colon cancer patients, accounting for 74% (78/105) of all included patients. Twenty-two patients were in the high-risk group and twenty-eight in the low-risk group according to the compound score. Because the high-risk patients shared a higher death risk, the results suggested that our 4-gene signature is a powerful prognostic tool for this subgroup (HR =10.91, 95% CI: 1.36–87.50, P=0.005; ).
Figure 4 Analysis of different groups of patients.
Abbreviation: MSI, microsatellite instability.
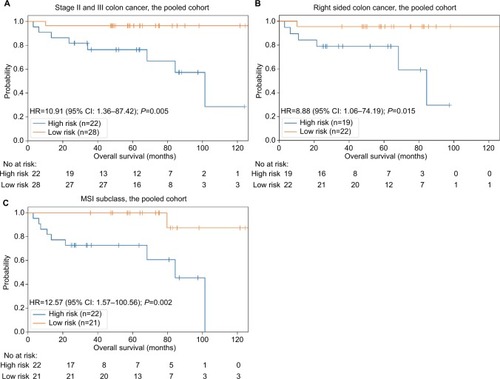
As hypermutations are more likely to occur on the right side of colon cancer,Citation14 we selected right-side colon cancer patients and employed the 4-gene signature to predict outcomes. The results showed markedly worse survival among patients in the high-risk group (HR =8.88, 95% CI: 1.06–74.50, P=0.015; ).
MSI status is important for both prognosis and therapy choice among patients with hypermutated tumors. Thus, we segregated patients into MSI and microsatellite stable (MSS) subgroups, and the results showed that our signature can well predict outcomes in the MSI group (HR =12.57, 95% CI: 1.57–100.69, P=0.002; ), although the difference was not significant in the MSS group (P=0.082).
Comparison of prognostic biomarkers in hypermutated CRC
We established a prognostic predictor based on four genes (DOCK2, ACVR2A, APC, and POLE) for hypermutated CRC. Previous studies have also proposed prognostic risk stratifications based on MSI/POLE or MSI/BRAF. Hypermutation, MSI status, and tumor mutational burden (TMB) are correlated with the response to immune checkpoint inhibitors, and we therefore conducted a performance comparison for these risk factors/groups using the pooled cohort.
Our 4-gene risk classifier stratifies patients with hypermutated CRC into high-risk and low-risk groups. Worse outcomes were significantly associated with the high-risk group. MSI/POLE status divided patients into three groups: POLE, MSI, and POLE/MSI. Although the MSI group accounted for the majority of patients, with better survival than those of the POLE group (75% vs 40%, 5-year survival rate), the difference was not significant (). No death occurred in two patients in the POLE/MSI group. MSI/BRAF status also classified patients into three groups, MSI, MSS/ BRAF-wild-type, and MSS/BRAF-p.V600E, and there was no prognostic difference between the first two groups, with only one patient in the last group (). To investigate the association between TMB and prognosis, we divided patients into high-TMB and low-TMB groups based on the median (40 mut/Mb). High-TMB patients showed a worse, but not significant, outcome (). All patients with a POLE driver mutation were sorted into the high-TMB group, which was confirmed by the poor prognosis of the POLE group. Lastly, consistent with the results of the multivariate Cox regression analysis, TNM staging confirmed that the prognosis of hypermutated CRC does not comply with the TNM staging system ().
Discussion
In our study, patients with hypermutated CRC comprised 13.30%, which agrees with previous TCGA studies (in which hypermutated CRC accounted for 15.6%,Citation16 16.9%,Citation17 and 15%).Citation18 To predict the prognosis of hypermutated CRC, we constructed a prognostic mutation signature of four genes (DOCK2, ACVR2A, APC, and POLE) that separated patients into two risk groups. The high-risk group was significantly associated with worse survival (HR =8.62, P=0.001). Overall, the proportion of hypermutated CRC suggested that hypermutation should not be underestimated in CRC. Furthermore, this special subgroup of CRC patients deserves greater attention because there are currently many novel therapeutic options. For example, Campbell et alCitation12 recently reported dramatically different survival rates among 217 patients with hypermutated cancer. In addition, the abundance of immune checkpoint inhibitors that target programmed cell death 1 (PD-1) or their ligands (PD-L1) brings new hope to hypermutated tumor patients. Many studies have proven that hypermutation and MSI status predict the clinical benefit of immune checkpoint inhibitors, which can lead to durable remission in some patients with conventional therapy failure.Citation19,Citation20 Thus, establishing a prognostic biomarker will help physicians screen patients who have poor prognoses but good responses to immunotherapy. Regardless, not only are the clinicopathological features of hypermutated tumors poorly understood, but prognostic tools are also lacking due to a lack of understanding of this type of disease. Recently, sequencing of individual cancer genomes has prompted scientists to search for biomarkers based on gene mutation signatures. However, most previous efforts treated hypermutation and non-hypermutation as a whole or focused on non-hypermutated CRC.Citation21 To the best of our knowledge, no well-known study has been conducted on hypermutated CRC, highlighting the value of our work.
The genes in our signature include DOCK2, ACVR2A, APC, and POLE. APC and POLE play important roles in the development and progression of CRC, and DOCK2 and ACVR2A are worth discussing further. DOCK2 is a gene frequently mutated in CRC and esophageal cancer,Citation22 and data for the pooled cohort showed that DOCK2 is more frequently mutated in hypermutated CRC (38.0%) than in non-hypermutated CRC (3.9%). Germline deficiency of DOCK2 leads to life threatening, invasive bacterial and viral infections,Citation23,Citation24 and transgenic mice experiments confirmed that DOCK2 is required for recruitment and infiltration of immune cells into the colon mucosa during bacterial infection.Citation24,Citation25 Several recent studies have reported that the gut microbiome influences the efficacy of anti-PD-1/L1 immunotherapy.Citation26,Citation27 Accordingly, DOCK2 may participate in the immune response initiated by gut microbes, and its deficiency is most likely involved in an immune evasion mechanism of high-risk hypermutated CRC. ACVR2A has two 8-bp polyadenine tracts, a hot spot for mutation in MSI CRC.Citation28,Citation29 ACVR2A is a member of the TGF-β superfamily, which plays a key role during CRC progression, and a previous study showed that ACVR2A is the most frequently mutated gene of hypermutated CRC. Further study of ACVR2A is urgently needed.
We performed subgroup analysis to verify the universality of our 4-gene signature. Due to the different treatment strategies and clinical outcomes in colon and rectal cancer, stage II and III colon cancer patients were selected as the first subgroup, and the results showed that our signature is sufficiently powerful in this subgroup. Nonetheless, the effectiveness of our 4-gene signature in rectal cancer needs to be further investigated. We then focused on the subgroup of high microsatellite instability (MSI-H) patients because these CRC patients are considered to have different clinical outcomes and therapeutic choices; for example, patients with MSI-H CRC usually do not respond to 5-fluorouracil chemotherapy, unlike non-hypermutated CRC patients. We further classified MSI-H patients into two risk groups using our 4-gene signature and observed that the low-risk group showed markedly good survival. Although recent studies have reported that MSI-H patients have a good response to immune checkpoint inhibitors, there are still some patients for whom benefit may not actually be gained or for whom the response may not be translated to OS. Using our 4-gene signature in MSI patients to identify different risk groups is promising in clinical practice that selecting patients who will actually gain benefits from immunotherapy, with an effect on OS. The final subgroup analysis focused on the primary locations, as hypermutation is more common in right-side colon cancer.Citation16 This subgroup analysis further verified that our 4-gene signature is a powerful tool for patients with hypermutated CRC.
Using gene mutation status to construct a prognostic signature is currently a hot topic in cancer research. A powerful prognostic signature will identify different risk group of patients to receive different intensity of therapies. Our 4-gene signature classifies hypermutation CRC patients into two groups and we suggest that high-risk group patients should receive more aggressive treatments than the low-risk group patients. A previous study stratified patients into three groups based on the MSI/BRAF status: MSI/BRAF-wild-type or mutant (best prognosis), MSS/BRAF-wild-type (intermediate prognosis), and MSS/BRAF mutant (worst prognosis).Citation30 Although the MSI/BRAF status can as a whole serve as a prognostic biomarker for CRC, it was not suitable for hypermutated CRC according to our study. TMB has also been recommended as a critical criterion correlated with the objective response rate of anti-PD-1/L1 immunotherapy. However, the threshold in different kinds of tumor remains controversial, and the power for predicting patient prognosis needs to be developed. Compared with the above biomarkers, our 4-gene risk classifier exhibits good performance for predicting clinical outcome in patients with hypermutated CRC.
However, there are some limitations to our study; the most important one is the limited number of patients in our study group due to the small percentage of hypermutated CRC. Fortunately, gene sequencing technology is gradually maturing and becoming faster and less expensive. We will continue to collect cases of hypermutated CRC to further verify our signature. Furthermore, although we believe that the 4-gene signature is promising in selecting patients who will gain benefits from immunotherapy, its significant value still needs to be verified in prospective studies that we are devoting ourselves to.
Conclusion
We constructed a 4-gene signature for patients with hypermutated CRC, which can classify patients into different risk groups and predict prognosis. This signature is a powerful tool in stage II and III colon cancer and MSI-H CRC. Future prospective studies are needed to confirm the power of the 4-gene signature in patients receiving immunotherapy.
Acknowledgments
This study was supported by the Fundamental Research Funds for the Central Universities, National Human Genetic Resources Sharing Service Platform (2005DKA21300), and National High Technology Research and Development Program of China (2012AA02A204). The abstract of this paper was presented at the MAP Conference named “A novel 4-gene prognostic signature for hypermutated colorectal cancer (CRC)” as a poster presentation with interim findings. The poster’s abstract was published in “Poster Abstracts” in ESMO’s official journal Annals of Oncology.
Supplementary material
Figure S1 Power of different prognosis biomarkers.
Notes: (A) MSI/POLE status divides patients into three groups: POLE, MSI, and POLE/MSI. The difference of patients’ survival is not significant. (B) MSI/BRAF status divides patients into three groups: MSI, MSS/BRAF-wild-type, and MSS/BRAF-V600E. The difference of patients’ survival is not significant. (C) Patients were divided into high TMB and low TMB groups by median(as 40 mut/Mb). The difference of patients’ survival is not significant. (D) Hypermutated patients were not comply the traditional TNM stages.
Abbreviations: MSI, microsatellite instability; MSS, microsatellite stable; TMB, tumor mutational burden.
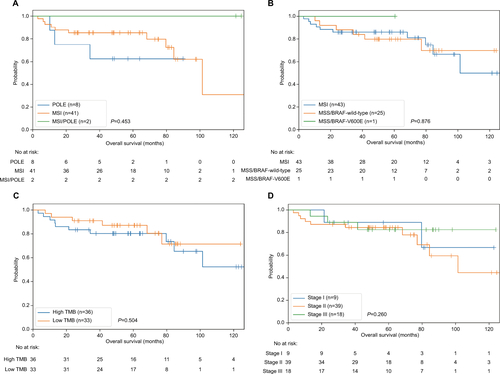
Table S1 List of 524 genes for panel sequencing
Table S2 Baseline values of all initial selected patients
Table S3 Chemotherapy of patients in the final analysis
Disclosure
The authors report no conflicts of interest in this work.
References
- WolfAMDFonthamETHChurchTRColorectal cancer screening for average-risk adults: 2018 guideline update from the American cancer SocietyCA Cancer J Clin201868425028129846947
- AkbaniRAkdemirKCAksoyBAGenomic classification of cutaneous melanomaCell201516171681169626091043
- GovindanRDingLGriffithMGenomic landscape of non-small cell lung cancer in smokers and never-smokersCell201215061121113422980976
- Cancer Genome Atlas Research NetworkComprehensive molecular characterization of urothelial bladder carcinomaNature2014507749231532224476821
- RehderDSNelsonRWBorgesCRGlycosylation status of vitamin D binding protein in cancer patientsProtein Sci200918102036204219642159
- de La ChapelleAHampelHClinical relevance of microsatellite instability in colorectal cancerJ Clin Oncol201028203380338720516444
- IonovYPeinadoMAMalkhosyanSShibataDPeruchoMUbiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesisNature199336364295585618505985
- AaltonenLAPeltomäkiPLeachFSClues to the pathogenesis of familial colorectal cancerScience199326051098128168484121
- JiricnyJThe multifaceted mismatch-repair systemNat Rev Mol Cell Biol20067533534616612326
- LeeVMurphyALeDTDiazLAMismatch repair deficiency and response to immune checkpoint blockadeOncologist201621101200121127412392
- LeeSYChungHDevarajBMicrosatellite alterations at selected tetranucleotide repeats are associated with morphologies of colorectal neoplasiasGastroenterology201013951519152520708618
- CampbellBBLightNFabrizioDComprehensive analysis of hypermutation in human cancerCell201717151042105629056344
- JoverRNguyenTPPérez-CarbonellL5-fluorouracil adjuvant chemotherapy does not increase survival in patients with CpG island methylator phenotype colorectal cancerGastroenterology201114041174118121185836
- WangKLiMHakonarsonHANNOVAR: functional annotation of genetic variants from high-throughput sequencing dataNucleic Acids Res20103816e16420601685
- SalipanteSJScrogginsSMHampelHLTurnerEHPritchardCCMicrosatellite instability detection by next generation sequencingClin Chem20146091192119924987110
- Cancer Genome Atlas NetworkComprehensive molecular characterization of human colon and rectal cancerNature2012487740733033722810696
- TianSRoepmanPPopoviciVA robust genomic signature for the detection of colorectal cancer patients with microsatellite instability phenotype and high mutation frequencyJ Pathol2012228458659522926706
- AroraSYanHChoIGenetic variants that predispose to DNA double-strand breaks in lymphocytes from a subset of patients with familial colorectal carcinomasGastroenterology201514971872188326344056
- DudleyJCLinMTLeDTEshlemanJRMicrosatellite instability as a biomarker for PD-1 blockadeClin Cancer Res201622481382026880610
- LaDPD-1 blockade in tumors with mismatch-repair deficiencyN Engl J Med2015373201979
- ShoSCourtCMWinogradPRussellMMTomlinsonJSA prognostic mutation panel for predicting cancer recurrence in stages II and III colorectal cancerJ Surg Oncol20171168996100428767131
- DulakAMStojanovPPengSExome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexityNat Genet201345547848623525077
- DobbsKDomínguez CondeCZhangSYInherited DOCK2 deficiency in patients with early-onset invasive infectionsN Engl J Med2015372252409242226083206
- LiuZManSMZhuQDOCK2 confers immunity and intestinal colonization resistance to Citrobacter rodentium infectionSci Rep2016612781427291827
- MiaoSZhangRYWangWOverexpression of dedicator of cytokinesis 2 correlates with good prognosis in colorectal cancer associated with more prominent CD8+ lymphocytes infiltration: a colorectal cancer analysisJ Cell Biochem2018119118962897030076747
- RoutyBLe ChatelierEDerosaLGut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumorsScience20183596371919729097494
- HamptonTGut microbes may shape response to cancer immunotherapyJAMA2018319543043129411013
- HempenPMZhangLBansalRKEvidence of selection for clones having genetic inactivation of the activin A type II receptor (ACVR2) gene in gastrointestinal cancersCancer Res200363599499912615714
- JungBDoctoleroRTTajimaALoss of activin receptor type 2 protein expression in microsatellite unstable colon cancersGastroenterology2004126365465914988818
- LochheadPKuchibaAImamuraYMicrosatellite instability and BRAF mutation testing in colorectal cancer prognosticationJ Natl Cancer Inst2013105151151115623878352