
Abstract
Objective: CD166 is known as a tumor stem cell specific marker, associating with tumor metastasis. The purpose of this study was to further discuss CD166 gene on cell proliferation, invasion, metastasis, and the epithelial-mesenchymal transition (EMT) in CNE-2R cell line of nasopharyngeal carcinoma (NPC).
Materials and methods: CNE-2R cells were transfected with lentivirus CD166-shRNA, and quantitative reverse transcription polymerase chain reaction (RT-qPCR), and Western blotting were used to confirm the silencing effects. The wound healing test and transwell test were carried out to assess cell invasive and migratory abilities in vitro. With the establishment of xenograft nude mouse model, Western blotting and immunohistochemistry were undertaken to detect the expression level of E-cadherin, N-cadherin, and vimentin. In vivo metastasis detection was carried out by injecting tumor cells into nude mice via the tail vein.
Results: The invasive and migratory abilities of CNE-2R cells were significantly reduced after CD166 was downregulated. In addition, silencing of CD166 of CNE-2R cells increased the expression of E-cadherin, while down-regulated the expression of N-cadherin and vimentin. Immunohistochemistry of tumors showed consistent results with in-situ tumor formation experiment. Additionally, the growth of transplanted tumor was inhibited. In addition, in vivo metastasis test proved that knockdown of CD166 suppressed pulmonary metastasis and liver metastasis according to hematoxylin and eosin (H&E) staining. Expression of E-cadherin increased, while expression of N-cadherin and vimentin decreased, as revealed by Western blotting of metastatic lung tumors.
Conclusion: Silencing of CD166 in CNE-2R cells evidently inhibited proliferation, invasion, metastasis, and EMT process in vivo and in vitro.
Introduction
Nasopharyngeal carcinoma (NPC) is an uncommon malignant tumor of the head and neck, while it, with a high incidence, has been reported in southern Asia, especially southern China. Citation1,Citation2 The rapid evolution of intensity-modulated radiation therapy (IMRT) is taken the foremost treatment for NPC at present. Citation3 Several NPC patients were effectively treated with IMRT; however, the high rate of distant metastasis leads to treatment failure for NPC. Citation4 Thus, it is essential to further explore the basic mechanism of radiation resistance in cell migration and invasion to promote the survival rate of radiation-resistant NPC.
Cells can transform from epithelial to mesenchymal states in a dynamic manner during embryonic process, and then cells are allowed to migrate and invade due to the change of mesenchymal state in the epithelial-mesenchymal transition (EMT). Citation5 EMT plays a major role in differentiating multiple tissues and organs, and it is also associated with tissue repair and organ fibrosis, in addition to accelerate progression of metastasis carcinoma. Citation6,Citation7 The decreased expression of E-cadherin is correlated with EMT, playing an important role during tumor progression. Citation8 Vimentin has a high expression in different types of cancer, promoting tumor growth and invasion. Citation9 The hallmark of EMT is the loss of epithelial surface markers, most notably E-cadherin, and the acquisition of mesenchymal markers, including vimentin and N-cadherin. Citation10 Previous studies have shown that CD166 is associated with the adjustment of EMT in colorectal cancer and lung cancer. Citation11,Citation12 However, the role of the CD166 gene in NPC has been rarely studied.
The occurrence of radio-resistance drugs is a complex process, and changes at the genetic level may certainly lead to some changes in protein levels. Our research group identified differentially secreted proteins in the cultured NPC radio-resistant cell line CNE-2R and its parental cell line CNE-2 by proteomics-related identification technique in vitro. Besides, CD166 is positively expressed in CNE-2R, while is negatively expressed in CNE-2; higher expression could also be observed in patients’ serum being radio-resistant NPC than that in radiation-sensitive patients;Citation13 we found that the sensitivity of radiation of NPC cells with positive expression of CD166 is not comparable to that of NPC cells with negative expression. Citation14 It is suggested that the expression of CD166 in the cell membrane is associated with radio-sensitivity of NPC cells.
In this study, CD166 was silenced by lentivirus-mediated RNA interference technique of CNE-2R cells. We aimed to indicate whether CD166 was connected to cell migration, invasion, and EMT, in addition to search for radio-resistant NPC biomarkers.
Materials and methods
Ethics statement
The procedures involving animal use were approved by Ethics Committee of the Affiliated Tumor Hospital of Guangxi Medical University (Nanning, China) (approval number: LW2018045). All animal experiments were conducted in accordance with the Laborayary animal – Guideline for ethical reiew of animal welfare.
Cell culture and infection
Our group generated the radio-resistant human NPC cell line CNE-2R, by fractional exposure of CNE-2 to radiation. Citation15 CNE-2, a human NPC cell line with low differentiation, was purchased from Fudan University Shanghai Cancer Center (Shanghai, China). The cell line was tested through short tandem repeat (STR)-DNA profiling. CNE-2R cells were cultured in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, USA), supplemented with 10% fetal bovine serum (FBS; Biological Industries, Cromwell, CT, USA), 1% penicillin/streptomycin in an incubator at 37°C in presence of 5% CO2. CD166 inhibitor and scrambled inhibitor were transferred into CNE-2R cells cultured in 6-well plates using lentivirus with the sequences (CD166-shRNA:ACAGATTGAACCTCTCAGAAA) and (NC:TTCTCCGAACGTGTCACGT), respectively, at a multiplicity of infection (MOI) of 20. The virus solution was then removed, and replaced by RPMI-1640 medium containing 10% FBS after 8 hrs, in which the infection efficiency was observed by inverted fluorescence microscope after 96 hrs of infection.
RT-qPCR analysis of mRNA expression of CD166 after lentivirus
Total RNA was abstracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Complementary DNA (cDNA) was amplified using ReverTra Ace qPCR RT Kit (Toyobo, Osaka, Japan). The protocol was conducted with the following cycling parameters: at 37°C for 15 mins, and at 98°C for 5 mins for 40 cycles. All quantitative reverse transcription polymerase chain reaction (RT-qPCR) examinations were performed using 96-microwell plates in a 7300 Real-Time PCR System (Applied Biosystems, Foster city, CA, USA). The RT-qPCR amplification was conducted in a 20 μl final reaction volume using THUNDERBIRD Probe and SYBR qPCR Mix (Toyobo, Osaka, Japan) according to the manufacturer’s instructions. Quantifications were normalized by taking β-actin as an internal reference and were calculated by using the 2ΔΔCt method. Primer sequences were as follows:
CD166 forward, 5’- ACTTGACGTACCTCAGAATCTCA −3’;
and reverse, 5’- CATCGTCGTACTGCACACTTT −3’;
β-actin forward, 5’-AGAGCTACGAGCTGCCTGAC-3’;
and reverse, 5’-AGCACTGTGTTGGCGTACAG-3’.
Western blot analysis
The protein was collected from cells of each group by radio-immunoprecipitation assay (RIPA) buffer (Beyotime Institute of Biotechnology, Shanghai, China) plus protease inhibitor, in which the xenograft tumors and lung metastasis tissues were lysed after grinding, and then the supernatant was collected. In addition, protein concentrations were detected with bicinchoninic acid (BCA) assay kit (Beyotime Institute of Biotechnology, Shanghai, China). Besides, 50 μg of total protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then cut and transferred onto polyvinylidene difluoride (PVDF) membranes. After that, the hybridized membranes were blocked in 5% fat-free milk in TBST (containing 0.05% Tween-20) for 1 hr and incubated with primary rabbit monoclonal antibody at 4°C overnight. Next, that was followed by the membranes, which were incubated in a horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (1:1000) for 1 hr. Moreover, β-actin or GAPDH antibody was used as an internal reference. The images were collected by using enhanced chemiluminescence (ECL) reagent. The primary antibodies used in this study were as follows: anti-CD166 antibody, E-cadherin, N-cadherin, vimentin, and β-actin; all of these antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA)
Wound healing detection
The migration capacities of cells were determined by scratch-wound assay. A Culture-Insert 3 Well was placed in 6-well plates; the cell suspension was regulated to 3×105 cells/ml, and 70 µl was applied to each well. After appropriate cell attachment (24 hrs), the Culture-Insert 3 Well was removed by using sterile tweezers; then the used well or dish was filled with serum-free medium, and cultured for 48 hrs at 5% CO2 at 37°C. The width of the wound was photographed with an inverted phase contrast microscope. All the examinations were performed in triplicate.
Transwell migration assay
The migration and invasive abilities were evaluated using 8 µm pore size 24-well transwell chambers. The concentration of cell was adjusted to 2×105 cells/ml with serum-free medium. Besides, 600 μl of 10% FBS-containing medium was placed to the lower chamber, in which 2×104 cells suspended in 100 µl serum-free medium were seeded into the upper chamber for 24 hrs. Non-migratory cells were cleared, while the migratory cells in the lower chamber were fixed with paraformaldehyde, then stained by Giemsa and dried at room temperature. An automated system for whole microscopic image acquisition was used, and 5 microscopic fields (20×) were randomly selected for cell counting, and then changes in the number of cells were calculated. For invasion assay, general chamber was replaced by Matrigel-coated transwell membranes. All tests were carried out in triplicate.
Subcutaneous xenograft detection in nude mice
Male BALB/C nude mice (age, 4-week-old) were purchased from Chongqing Tengxin Biotechnology Co. Ltd. (Chongqing, China). The nude mice were randomly assigned into 3 groups (n=5 for each group), including control, NC, and CD166-shRNA. For subcutaneous tumor formation in vivo, cells were suspended (1×106 cells/mL) in 200 μL of phosphate-buffered saline (PBS) and injected subcutaneously into the groin on the right side of the mice. Tumor growth was examined every 4 days. The nude mice were executed by cervical dislocation after 4 weeks of observation, and the tumor tissues were removed as well.
Metastasis experiment in nude mice
For metastasis in vivo, nude mice were also assigned into control, NC, and CD166-shRNA groups; 5×105 cells suspended in 100 μL of PBS were injected into the tail vein of each nude mouse. After 6 weeks of injection, the mice were sacrificed and lungs and livers were removed.
Hematoxylin and eosin (H&E) staining
The harvested lung and liver tissues were fixed with 4% paraformaldehyde dehydrated, and then embedded in paraffin; paraffin-embedded sections were stained with H&E.
Immunohistochemistry (IHC) analysis0
For IHC, paraffin-embedded sections were toasted in an incubator at 60°C for 3 hrs, and then deparaffinized in xylene for 30 mins and rehydrated in a decreasing progressively concentration 100% ethyl alcohol, 90% ethyl alcohol, 80% ethyl alcohol, and 70% ethyl alcohol for 10 mins. Antigen retrieval was conducted by high temperature and high pressure in citric acid-sodium citrate buffer (pH 6.0) for 3 mins. Endogenous peroxidase activity was blocked by 3% hydrogen peroxide for 15 mins, and then the sections were incubated with standard goat serum for 10 mins, and incubated with primary rabbit monoclonal antibody at 4°C overnight. Next, the sections were exposed to HRP-conjugated secondary antibody, and then incubated with Streptomycin avidin-peroxidase. After incubating with DAB (3,3’-diaminobenzidine tetrahydrochloride) to bind non-specific antigen, the sections were counterstained with the use of hematoxylin, as negative control, and the primary antibody was replaced by PBS. Eventually, the sections were dehydrated with gradient ethanol, transparent with xylene, and sealed with neutral gum. Chemical staining was assessed and marked by two pathologists independently. Differences were resolved by consensus as well. The intensity of staining score ranked 0–3: 0 point (no staining), 1 point (light yellow), 2 points (yellow), and 3 points (brownish yellow). The percentage score of stained cells was considered as 0 point (no staining), 1 point (<30% positive expression), 2 points (30~60% positive expression), and 3 points (>60% positive expression). The staining intensity was added to the percentage score as the total staining score for each tissue sample (range, 0–6). Besides, 0–3 points were considered as low expression, and 4–6 points were classified as high expression.
Results
Effective silencing of CD166 in CNE-2R cells
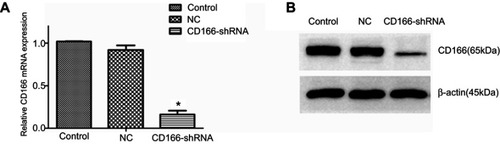
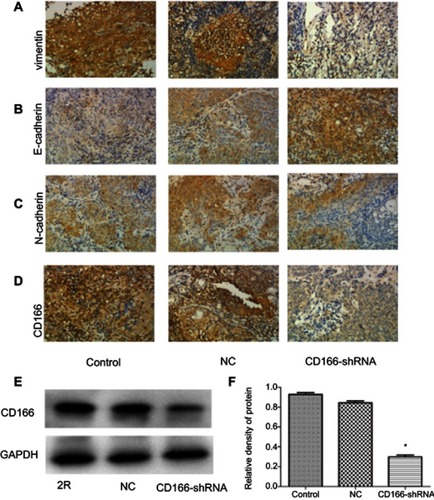
The lentivirus was successfully established and infected into the CNE-2R cells. The percentage of green fluorescence protein (GFP)-positive cells was above 90% after 96 hrs in both CD166-shRNA group and NC group ( and ). The efficiency of shRNA and lentivirus-mediated downregulation on the expression of CD166 was detected by RT-qPCR and Western blotting both at gene and protein levels. As shown in , either the mRNA or protein expression levels of CD166 was notably declined in the shRNA-transduced group compared with the NC group and control group ( and ) (*P<0.01).
Figure 1 Evaluation of the lentivirus transduction rate, which was calculated by cellular enumeration under an inverted fluorescence microscope (magnification, ×200). (A) NC group; (B) CD166-shRNA group.
Figure 2 CD166 was downregulated in the CNE-2R cell lines. (A) The analysis of CD166 mRNA expression in respective groups was conducted by RT-qPCR. (B) Western blot analysis showing CD166 expression in different groups. *P<0.01, the CD166-shRNA group compared with the NC group and control group.
Silencing of CD166 attenuated migration and invasion abilities in CNE-2R cells in vitro
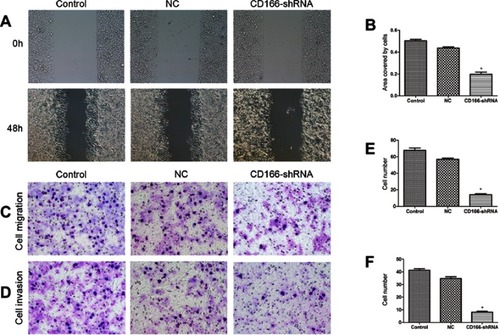
The consequence of the wound-healing experiment indicated that migration ability of the CD166-shRNA group was markedly lower than that of control group and NC group with the scratch-healing rate of 15.28%±1.42% compared with 47.44%±2.84% and 39.18%±1.07% (*P<0.001, and ). Transwell assay was carried out to suggest that the migration ability of CD166-shRNA group was evidently poorer than that of NC group and control group ( and ). Additionally, the number of invasive cells was evidently less in the CD166-shRNA group (8.20±1.64) compared with NC group (34.80±3.30) and control group (41.20±2.80) (*P<0.01, and ). Taken together, we found that down-regulation of CD166 expression could suppress cell proliferation and metastasis.
Figure 3 Silencing of CD166 inhibited the migration and invasion of CNE-2R cells. Notes: (A and B) Wound-healing assay to compare cell migration and invasion in the CD166-shRNA group, NC group, and control group (magnification, ×100). (C and E) The effects of silencing of CD166 on cell migration detected by transwell assay, (D and F) Effects of silencing of CD166 on cell invasion detected by transwell assay, (magnification ×200). *P<0.05, the CD166-shRNA group compared with the NC group and control group.
Silencing of CD166 inhibited EMT in CNE-2R cells
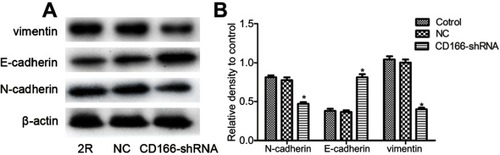
EMT plays a critical role in cell invasion and metastasis. As illustrated in , Western blotting suggested that knockdown of CD166 increased the E-cadherin expression, while restrained N-cadherin and vimentin expression, indicating that silencing of CD166 suppressed the invasion ability of EMT in CNE-2R cells ().
Figure 4 Silencing of CD166 decreased EMT in CNE-2R cells. (A) After CD166 was silenced in CNE-2R cells, E-cadherin was upregulated, and N-cadherin was downregulated as proved by Western blot analysis. (B) Schematical representation of N-cadherin, E-cadherin, and vimentin expression. *P<0.05, the CD166-shRNA group compared with the NC group and control group.
Silencing of CD166 inhibited EMT in xenograft nude mouse model
The xenograft nude mouse model was established to demonstrate the effects of CD166 on tumor growth in vivo (). The EMT-related markers were tested by using IHC. The results indicated that the tumor size of CD166-shRNA group was remarkably lower than that of CNE-2R group and NC group ( and ). Expression of CD166 in vivo was detected by IHC and Western blotting, indicating that the corresponding expression of CD166 protein in nude mice injected with CD166-shRNA-infected cells was evidently less than that of the CNE-2R group and NC group (). The expression of E-cadherin, N-cadherin, and vimentin was examined by IHC to verify the level of EMT. Vimentin and N-cadherin had a higher expression level in both CNE-2R group and NC group compared with CD166 group, while E-cadherin was less expressed in CNE-2R group and NC group compared with CD166 group (P<0.05, ).
Figure 5 Effects of silencing of CD166 in xenograft nude mouse model. (A) Nude mice used in the experiment. (B) Tumor obtained from nude mice. (C) Growth curve of xenograft tumors of nude mice.
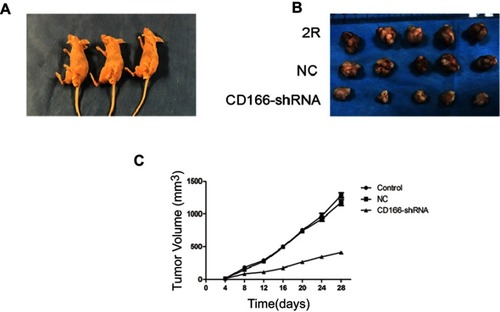
Figure 6 Expression of CD166 and EMT-related protein of nude mice xenograft model with the downregulation of CD166. (A–C) IHC suggested that E-cadherin was upregulated, while N-cadherin and vimentin were downregulated (magnification, ×200). (D) CD166 expression of nude mouse xenograft model identified by IHC. (E and F) CD166 expression of nude mouse xenograft model identified by Western blotting. *P<0.05, the CD166-shRNA group compared with the NC group and control group.
Silencing of CD166 suppressed metastasis in vivo
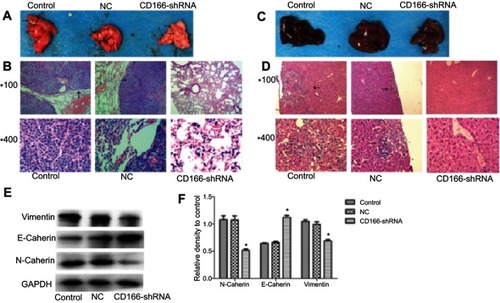
Nude mice injected with tumor cells through the tail vein were sacrificed 6 weeks later, and the metastatic tumor nodules on the surface of the lung in the control group and the NC group could be obviously observed by naked-eye ( and ). H&E staining showed that the pulmonary and liver metastasis ability of the CD166 group was significantly weaker than that of the NC group and the control group (P<0.05, and ). Western blot analysis indicated that knockdown of CD166 increased the E-cadherin expression, while restrained the expression of N-cadherin and vimentin (*P<0.05, and ), indicating that silencing of CD166 suppressed the metastasis in vivo.
Figure 7 The effects of silencing of CD166 on tumor cell metastasis in vivo. (A and C) Representative metastatic lung tissue and liver tissue of the three groups, (B and D) H&E staining morphology of the lung metastatic tumors (magnifications, ×100 and ×400). (E and F) E-cadherin was upregulated, whereas N-cadherin and vimentin were downregulated in the metastatic lung tissue detected by Western blot analysis. *P<0.05, the CD166-shRNA group compared with the NC group and control group.
Discussion
Radiotherapy is currently recommended as a principal treatment for NPC. However, distant metastasis is a main failure cause of NPC radiation therapy, especially for patients who are not sensitive to radiation. Therefore, the possible mechanism of tumor metastasis should be further elucidated.
CD166, also known as ALCAM, located on the human chromosome 3q13.1-q13.2, is one of the ligands of the lymphocyte antigen CD6 and is also one of the members of the immunoglobulin superfamily. Citation16 CD166/ALCAM is associated with the growth and development of cells in several tissues. Studies have shown that CD166/ALCAM had a high level of expression with the appearance of various tumor cells, which was related to the development of tumors. CD166/ALCAM has been verified to have a supervisory role in growth of various tumors and characteristics of metastasis-associated tumor cells in vitro; besides, increased expression of CD166/ALCAM has been observed in a variety of tumors, such prostate cancer,Citation17,Citation18 esophageal cancer,Citation19,Citation20 breast cancer,Citation21–Citation23 lung cancer,Citation24,Citation25 colorectal cancer,Citation26,Citation27 and malignant melanoma,Citation28,Citation29 which was associated with the malignant progression of tumors.
In our previous study, CD166/ALCAM was found to be highly expressed in CNE-2R, while rarely expressed in CNE-2. Citation13 The serum level of CD166/ALCAM in patients with radiation-resistant tumors was higher than that of radiation-sensitive patients; at the cell membrane level, it has been proven that CNE-2 and CNE-2R cells with high expression of CD166/ALCAM had a lower radio-sensitivity than those of CNE-2 and CNE-2R with negative expression. In addition, the expression of CD166/ALCAM was correlated with the radio-sensitivity of NPC. Citation14 In the present study, we applied lentivirus-mediated shRNA to downregulate CD166/ALCAM in CNE-2R cell line. The date of wound healing assay and transwell assay showed that the motility and invasive capability of CNE-2R cells were decreased, demonstrating that CD166/ALCAM could promote proliferation and metastasis of CNE-2R cells.
Cadherins are a family of cell–cell adhesion molecules and are divided into subclasses with distinct adhesive specificities and tissue distribution. E-cadherin-mediated cell–cell adhesion prevents invasiveness of human carcinoma cells,and loss of E-cadherin expression is related to invasive ability of different kinds of cancer. Citation30–Citation33 N-cadherin,another adhesion molecule, is able to increase invasive capability in cancers. Citation34,Citation35 Vimentin is involved in cell–cell adhesion, migration, invasion, signal transduction, cytoskeletal rearrangement, and cell morphology and plasticity regulation. Citation9,Citation12,Citation36 In the present study, silencing of CD166/ALCAM increased the E-cadherin expression, restrained N-cadherin, and vimentin expression in vitro and vivo. which indicated that EMT level of CNE-2R cells was significantly decreased, and the cell migration and invasion abilities were also reduced after silencing of CD166/ALCAM. Additionally, according to in vivo metastasis studies, liver and lung metastases were notably reduced after CD166/ALCAM knockdown. Moreover, the EMT level was obviously declined in the metastatic lung tissue. A previous research study demonstrated that CD166/ALCAM can be taken as a special marker for colon cancer stem cells into account, that is related to tumor’s chemical resistance and radiation resistance, and also can initiate tumor formation as well as activation of EMT. Citation37 In conclusion, silencing of CD166/ALCAM could suppress metastasis and proliferation of CNE-2R cells. However, the intrinsic mechanism that how CD166/ALCAM can promote the ability of NPC to resist against radiation and undergo cell migration is still complex, and further experiments are therefore required to explore the expression of CD166/ALCAM and its specific mechanism to EMT, and migration in radiation-resistant NPC.
Conclusion
In this study, it was revealed that downregulation of CD166 inhibited cell proliferation, migration and invasion. Besides, EMT of CNE-2R cells evidently suppressed with silencing of CD166.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that there are no conflicts of interest regarding the publication of this article.
Acknowledgments
The present study was financially supported by the National Natural Science Foundation of China (Grant No. 81760544), the Natural Science Foundation of Guangxi Province (Grant No. 2016GXNSFAA380127),Innovation Project of Guangxi Graduate Education (Grant No. YCSW2018108), and the Key Laboratory of High-Incidence-Tumor-Prevention and Treatment Research Project (Grant No. GK201806).
References
- MLK, Wee JTS, Hui EP, Chan ATC. Nasopharyngeal carcinoma. Lancet (London, England). 2016;387(10022):1012–1024.
- Tang LL, Chen WQ, Xue WQ, et al. Global trends in incidence and mortality of nasopharyngeal carcinoma. Cancer Lett. 2016;374(1):22–30. doi:10.1016/j.canlet.2016.01.04026828135
- Qu S, Liang ZG, Zhu XD. Advances and challenges in intensity-modulated radiotherapy for nasopharyngeal carcinoma. Asian Pac J Cancer Prev. 2015;16(5):1687–1692. 25773811
- Zheng LS, Yang JP, Cao Y, et al. SPINK6 promotes metastasis of nasopharyngeal carcinoma via binding and activation of epithelial growth factor receptor. Cancer Res. 2017;77(2):579–589. doi:10.1158/0008-5472.CAN-16-128127671677
- Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45. doi:10.1016/j.cell.2016.06.02827368099
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi:10.1016/j.cell.2009.11.00719945376
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–142. doi:10.1038/nrm183516493418
- Petrova YI, Schecterson L, Gumbiner BM. Roles for E-cadherin cell surface regulation in cancer. Mol Biol Cell. 2016;27(21):3233–3244. doi:10.1091/mbc.E16-01-005827582386
- Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68(18):3033–3046. doi:10.1007/s00018-011-0735-121637948
- Serrano-Gomez SJ, Maziveyi M, Alahari SK. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer. 2016;15:18. doi:10.1186/s12943-016-0502-x26905733
- Oh BY, Kim SY, Lee YS, et al. Twist1-induced epithelial-mesenchymal transition according to microsatellite instability status in colon cancer cells. Oncotarget. 2016;7(35):57066–57076. doi:10.18632/oncotarget.1097427494849
- Kidd ME, Shumaker DK, Ridge KM. The role of vimentin intermediate filaments in the progression of lung cancer. Am J Respir Cell Mol Biol. 2014;50(1):1–6. doi:10.1165/rcmb.2013-0314TR23980547
- Chen ZT, Li L, Guo Y, et al. Analysis of the differential secretome of nasopharyngeal carcinoma cell lines CNE-2R and CNE-2. Oncol Rep. 2015;34(5):2477–2488. doi:10.3892/or.2015.425526352878
- Lin H, Chen ZT, Zhu XD, et al. Serum CD166: A novel biomarker for predicting nasopharyngeal carcinoma response to radiotherapy. Oncotarget. 2017;8(38):62858–62867. doi:10.18632/oncotarget.1639928968954
- Guo Y, Zhu XD, Qu S, et al. Identification of genes involved in radioresistance of nasopharyngeal carcinoma by integrating gene ontology and protein–protein interaction networks. Int J Oncol. 2012;40(1):85–92. doi:10.3892/ijo.2011.117221874234
- Bowen MA, Patel DD, Li X, et al. Cloning, mapping, and characterization of activated leukocyte-cell adhesion molecule (ALCAM), a CD6 ligand. J Exp Med. 1995;181(6):2213–2220. 7760007
- Kristiansen G, Pilarsky C, Wissmann C, et al. ALCAM/CD166 is up-regulated in low-grade prostate cancer and progressively lost in high-grade lesions. Prostate. 2003;54(1):34–43. doi:10.1002/pros.1016112481253
- Kristiansen G, Pilarsky C, Wissmann C, et al. Expression profiling of microdissected matched prostate cancer samples reveals CD166/MEMD and CD24 as new prognostic markers for patient survival. J Pathol. 2005;205(3):359–376. doi:10.1002/path.167615532095
- Tachezy M, Effenberger K, Zander H, et al. ALCAM (CD166) expression and serum levels are markers for poor survival of esophageal cancer patients. Int J Cancer. 2012;131(2):396–405. doi:10.1002/ijc.2637721858815
- Verma A, Shukla NK, Deo SV, Gupta SD, Ralhan R. MEMD/ALCAM: a potential marker for tumor invasion and nodal metastasis in esophageal squamous cell carcinoma. Oncology. 2005;68(4–6):462–470. doi:10.1159/00008698916024937
- Burkhardt M, Mayordomo E, Winzer KJ, et al. Cytoplasmic overexpression of ALCAM is prognostic of disease progression in breast cancer. J Clin Pathol. 2006;59(4):403–409. doi:10.1136/jcp.2005.02820916484444
- Hein S, Muller V, Kohler N, et al. Biologic role of activated leukocyte cell adhesion molecule overexpression in breast cancer cell lines and clinical tumor tissue. Breast Cancer Res Treat. 2011;129(2):347–360. doi:10.1007/s10549-010-1219-y20972617
- Piao D, Jiang T, Liu G, Wang B, Xu J, Zhu A. Clinical implications of activated leukocyte cell adhesion molecule expression in breast cancer. Mol Biol Rep. 2012;39(1):661–668. doi:10.1007/s11033-011-0783-521670959
- Zakaria N, Yusoff NM, Zakaria Z, et al. Human non-small cell lung cancer expresses putative cancer stem cell markers and exhibits the transcriptomic profile of multipotent cells. BMC Cancer. 2015;15:84. doi:10.1186/s12885-015-1584-325881239
- Satar NA, Fakiruddin KS, Lim MN, et al. Novel triplepositive markers identified in human nonsmall cell lung cancer cell line with chemotherapy-resistant and putative cancer stem cell characteristics. Oncol Rep. 2018;40(2):669–681. doi:10.3892/or.2018.646129845263
- Weichert W, Knosel T, Bellach J, Dietel M, Kristiansen G. ALCAM/CD166 is overexpressed in colorectal carcinoma and correlates with shortened patient survival. J Clin Pathol. 2004;57(11):1160–1164. doi:10.1136/jcp.2004.01623815509676
- Shafaei S, Sharbatdaran M, Kamrani G, Khafri S. The association between CD166 detection rate and clinicopathologic parameters of patients with colorectal cancer. Caspian J Intern Med. 2013;4(4):768–772. 24294471
- van Kilsdonk JW, Takahashi N, Weidle U, et al. Modulation of activated leukocyte cell adhesion molecule-mediated invasion triggers an innate immune gene response in melanoma. J Invest Dermatol. 2012;132(5):1462–1470. doi:10.1038/jid.2011.48722318386
- van Kempen LC, van Den Oord JJ, van Muijen GN, Weidle UH, Bloemers HP, Swart GW. Activated leukocyte cell adhesion molecule/CD166, a marker of tumor progression in primary malignant melanoma of the skin. Am J Pathol. 2000;156(3):769–774. doi:10.1016/S0002-9440(10)64943-710702391
- Blanco D, Vicent S, Elizegi E, et al. Altered expression of adhesion molecules and epithelial-mesenchymal transition in silica-induced rat lung carcinogenesis. Lab Invest. 2004;84(8):999–1012. doi:10.1038/labinvest.370012915195114
- Sanchez-Tillo E, Lazaro A, Torrent R, et al. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29(24):3490–3500. doi:10.1038/onc.2010.10220418909
- Wang H, Wu Q, Zhang Y, Zhang HN, Wang YB, Wang W. TGF-beta1-induced epithelial-mesenchymal transition in lung cancer cells involves upregulation of miR-9 and downregulation of its target, E-cadherin. Cell Mol Biol Lett. 2017;22:22. doi:10.1186/s11658-017-0053-129118814
- Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta. 1994;1198(1):11–26. 8199193
- Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 2000;148(4):779–790. 10684258
- Zuo J, Ishikawa T, Boutros S, Xiao Z, Humtsoe JO, Kramer RH. Bcl-2 overexpression induces a partial epithelial to mesenchymal transition and promotes squamous carcinoma cell invasion and metastasis. Mol Cancer Res. 2010;8(2):170–182. doi:10.1158/1541-7786.MCR-09-035420145039
- Ivaska J, Pallari HM, Nevo J, Eriksson JE. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp Cell Res. 2007;313(10):2050–2062. doi:10.1016/j.yexcr.2007.03.04017512929
- Hwang WL, Yang MH, Tsai ML, et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology. 2011;141(1):279–291, 291.e271–275. doi:10.1053/j.gastro.2011.04.008