
Abstract
Pancreatic cancer, with high morbidity and mortality rates, is one of the most malignant tumors worldwide. Despite extensive research, the prognosis remains poor. Autophagy, a lysosomal-mediated, highly conserved degradation process that removes abnormal proteins and damaged organelles from the body, is upregulated in pancreatic ductal adenocarcinoma. Based on differences in the tumor microenvironment and tumor stage, the functions of autophagy in the pathophysiology and treatment of pancreatic cancer differ. In the initial phase, autophagy inhibits the transformation of precancerous lesions to cancer. However, in the progressive stage, autophagy promotes tumor growth. Autophagy is also one of the main mechanisms of drug resistance during treatment. Here, we describe the role of autophagy in pancreatic cancer progression and discuss relevant treatment strategies for this disease.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) originates from malignant pancreatic acinar epithelial cells and is the most common pancreatic cancer. Globally, PDAC is the twelfth most common malignant tumor and the seventh leading cause of cancer-related deaths, with a 5-year survival rate of only 9%.Citation1 PDAC metastasizes early and progresses rapidly; symptoms are irregular. PDAC is typically diagnosed during middle or late-stage cancer. Thus, most patients have lost the chance of surgical resection, making chemotherapy the main treatment for PDAC. However, frequent drug resistance of tumor cells makes late chemotherapy less effective than early chemotherapy.Citation2 The oncogene, KRAS, and tumor suppressor gene, TP53, are the most frequently altered genes in PDAC patients. Despite numerous studies on the therapeutic effects of inhibitors of the KRAS pathway, the results are not satisfactory. Therefore, it is necessary to further study targeted treatment of PDAC.
Autophagy is one mechanism of resistance in tumor therapy. It can promote cell survival in harsh environments, though unregulated autophagy can promote apoptosis.Citation4 There are three types of autophagy: macroautophagy, chaperone-mediated autophagy, and microautophagy.Citation3 Macroautophagy (hereafter referred to as autophagy) is the main autophagy process that is activated during cell nutrient insufficiency and hypoxia where it provides cells with metabolites after the encapsulated cargo is degraded.
Autophagy plays a complex role in tumor cells; studies have shown that autophagy can both inhibit cancer onset and promote advanced tumor growth.Citation4 Increasing evidence shows that cancer cells are more dependent on autophagy than normal cells, and this dependence may be further strengthened during the treatment process.Citation5 An increasing number of research groups are examining autophagy as a target for cancer treatment.
In this review, we introduce autophagy and focus on its role in the onset, development, and treatment of pancreatic cancer. Research on autophagy is developing rapidly, and the knowledge we have gained is not only helpful for us to understand the basic molecular mechanism of autophagy, but also can provide a basis for clinical decision-making of targeted therapy related to autophagy.
Process of Autophagy
Autophagy is a highly conserved cell stress response. Dr. Yoshinori Ohsumi discovered the mechanisms that regulate autophagy using yeast.Citation6 Five events initiate autophagy (): (1) initiation, (2) double-membrane nucleation and phagophore formation, (3) elongation of phagophores and encapsulation of cytoplasmic degradation targets, (4) autophagosome fusion with lysosomes to form the autolysosome, and (5) degradation of the autolysosome cargo.Citation7
Figure 1 Regulation of autophagy signaling pathways. Autophagy is a complex degradation process involving the following key steps: (A) initiation; (B) nucleation; (C) maturation; (D) fusion; (E) degradation. During initiation, low ATP, hypoxia, and amino acid deficiency lead to AMPK activation or mTOR inhibition, and the ULK complex forms. The ER membrane breaks off to form phagophores. In the starvation state, JNK1-mediated phosphorylation of BCL2 is blocked. BECN1 separates from BCL2 to form the Class III PI3K complex. BECN1 in the Class III PI3K complex interacts with the ER and participates in double-membrane nucleation to form a phagophore, which contains abnormal proteins and damaged organelles. ATG5 is conjugated with ATG12 and forms a complex with ATG16L, which is involved in phagophore elongation. After LC3 processing, it is inserted into the extended phagophore to form a mature autophagosome. Then, the autophagosome fuses with a lysosome to form the autolysosome and degrade the contents.
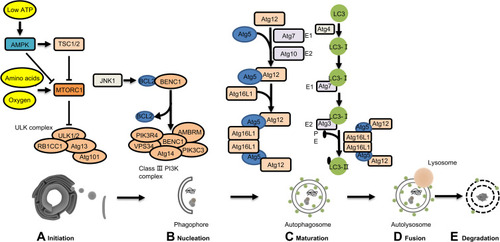
The mammalian target of rapamycin (mTORC1) is a serine-threonine kinase responsible for transducing autophagy signals;Citation8 mTORC1 binds and phosphorylates Unc-51-like kinase 1 (ULK1). When a cell is deprived of amino acids, ULK1 is dephosphorylated and initiates autophagy; under low ATP conditions, AMP-activated protein kinase (AMPK) promotes autophagy.Citation8 In addition, suppressed mTORC1 or activated AMPK can transduce the downstream pre-initiation complex ULK1/2-Atg13-FIP200-Atg101, which initiates autophagy.Citation9
The class III phosphatidylinositol 3-kinase (PtdIns3K) complex nucleates autophagosomes; the complex includes phosphatidylinositol 3-kinase catalytic subunit type (3PIK3C3), phosphoinositide 3-kinase regulatory subunit 4 (PIK3R4), vesicular protein sorting 34 (VPS34), activating molecule in Beclin1-regulated autophagy protein 1 (AMBRA1), Beclin1 (BECN1), and ATG14. BECN1 regulates PtdIns3K complex formation during phagophore assembly. B-cell lymphoma 2 (BCL2) prevents BECN1 from binding to VPS34 and inhibits autophagy. During the cell starvation state, JNK1-mediated phosphorylation of BCL2 is inhibited, and autophagy is promoted.Citation10 VPS34 combined with BECN1 phosphorylates phosphatidylinositol (PI) to phosphatidylinositol 3-phosphate (PI3P).Citation11 PI3P recruits other ATG proteins and elongates the phagophore. The phagophore disassembles if any of these processes are inhibited, preventing autophagy.
Two ubiquitin-like (UBL) systems regulate phagophore elongation: Atg12-Atg5 UBL and microtubule-associated light-chain 3 B (LC3B)-phosphatidylethanolamine (PE) UBLCitation12 by E1-E2-E3 tertiary enzyme chain reaction.Citation8 The E1-like enzyme Atg7 and the E2-like enzyme Atg10 form a covalent bond between Atg12 and Atg5. The Atg12-Atg5 conjugate recruits Atg16L1 to form a complex-Atg12-Atg5-Atg16L1, which functions as E3-like enzymes for LC3B-PE UBLs. LC3 is cleaved by the cysteine protease ATG4B to generate LC3B-I. LC3B-1 then conjugates with the glycine residue on PE. A diffuse form of LC3-I is converted into the lipidated form LC3-II by the E1-like enzyme ATG7 and the E2-like enzyme ATG3.
LC3-II inserted into the phagophore lengthens the membrane and forms a bilayer structure in the autophagosome. LC3 is found on both the inner and outer autophagosome surface membrane and is recruited into the phagophore by Atg5-Atg12.Citation10
Rab7 is a member of the Ras-related protein in the brain (Rab) family.Citation13 Rab7 and the homotypic fusion and protein sorting (HOPS)/vacuole protein sorting class C (Vps class C) complex interact to mediate membrane binding and fusion.Citation14 In addition, the pleckstrin homology domain-containing family M member 1 (PLEKHM1), RAB7 effector, also binds to components of the HOPS complex. PLEKHM1 interacts with LC3 /GABARAP family proteins, which regulate the fusion of autophagosomes and lysosomes.Citation15 After the autolysosome forms, it degrades the materials isolated inside,Citation16 providing cells with nutrients to maintain homeostasis. However, the role of autophagy in PDAC remains to be further elucidated.
PDAC and Autophagy
Histological and cytological studies have shown that autophagy increases when the pancreas is afflicted with intravascular pancreatic neoplasia (PanIN).Citation17 In addition, PDAC cells have been shown to rely on autophagy to obtain nutrients and energy to survive in vitro.Citation18 Here, we review the role of autophagy in the onset, development, and treatment of PDAC.
Role of Autophagy in the Onset of PDAC
PDAC is induced by various factors, including inflammation, gene mutations, and impaired mitochondrial function. Here, we review the role of autophagy in the onset of PDAC.
Autophagy regulates inflammation during PDAC pathogenesis. Studies have shown that knockout of autophagy-related genes (ATG5 or ATG7) or proteins encoding lysosomal function LAMP2 leads to severe acinar cell degeneration, pancreatic atrophy, fibrosis, and inflammation.Citation19 ATG7-deficient mice were found to have decreased autophagic flux, increased endoplasmic reticulum stress, pancreatic cell degeneration, and acinar-ductal metaplasia (ADM) formation compared to the control.Citation20 ADM is a precursor to pancreatic intraepithelial neoplasia (PanIN), a common precancerous lesion of PDAC.Citation21 This indicates that autophagy inhibition increases a patient’s susceptibility to PDAC.
Mutations in the Kirsten rat sarcoma virus oncogene are a major cause of PDAC.Citation22 When the oncogene KRAS is activated, the metabolic demands of pancreatic cells increase. Overexpressed vacuolar membrane protein 1(VMP1) interacts with BECN1 and promotes pancreatic cell autophagy to provide nutrients for cell metabolism.Citation23 Administering chloroquinine, an autophagy inhibitor, was found to effectively reverse VMP1 overexpression and induce PanIN formation in Pdx1-Cre; KrasG12D; vmp1 mice.Citation23
Moreover, 75% of human PDAC cells have P53 deletions.Citation22 Rosenfeldt et alCitation24 found that in KRAS mutation-driven PDAC cells, autophagy had both tumor-promoting and tumor-inhibiting effects, depending on the presence of the P53 gene. They also found that KARS mice lacking ATG5 or ATG7 developed low-grade malignant peripancreatic lesions, which did not progress to high-grade malignant PanIN and PDAC. Conversely, in mice with carcinogenic KRAS and P53 deletion, PDAC pathogenesis was accelerated. However, Yang et alCitation25 proved that chloroquine treatment or RNAi inhibition of autophagy inhibited PDAC cell growth, independent of P53.
The different results may be related to Rosenfeldt’s use of the P53 homozygous deletion model. The pancreas in these mice develop without functional P53, which is different from the gradual progression from PanIN to PDAC in other models and humans. In human PDAC cells, there is typically only one deletion of the P53 allele. Moreover, inhibiting autophagy prevents oncogenic KARS mutations from progressing to PDAC.Citation25 Thus, according to the above studies, manipulating autophagy to treat PDAC may depend on the KRAS oncogene status.
PDAC cells feature increased mitochondrial destruction compared with normal cells.Citation26 It is essential for cell homeostasis to maintain healthy mitochondria, including quantity and quality. Therefore, mitochondrial autophagy is increased in PDAC cells and it selectively degrades damaged mitochondria. Mitochondrial autophagy is regulated by PTEN-induced kinase 1 (PINK1), Parkin RBR E3 ubiquitin-protein ligase (PRKN/PARK2), and BCL2 interacting protein 3 (BNIP3L/NIX).Citation26 Xie et al showed that PINK1/ PRKN deficiency accelerates the onset of PDAC.Citation26 However, KRAS-mediated BNIP3L overexpression can increase the cell’s glucose metabolism and antioxidant capacity, promoting PDAC onset ().Citation26
Figure 2 Role of autophagy regulatory factors in pancreatic tumorigenesis in mice with mutant KRAS.
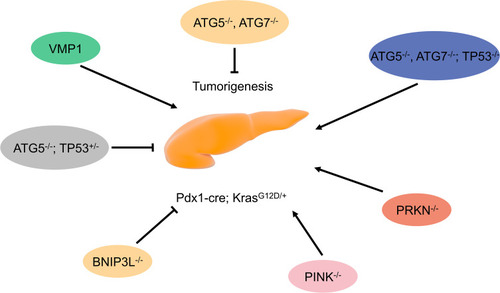
In conclusion, autophagy can inhibit PDAC pathogenesis, but can provide energy for cells, promoting PDAC onset when pancreatic cells have carcinogenic KRAS mutations.
Role of Autophagy in the Development of PDAC
Autophagy promotes pathogenesis by regulating cell proliferation, invasion, migration, metabolism, and immune escape. Studies have shown that the use of autophagy inhibitor chloroquine, or ATG5 and ATG7 inhibition by shRNA can inhibit human PDAC cell line growth and reduce tumor mass in PDAC mouse models.Citation17 We discuss the complex regulatory network of autophagy and its role in PDAC development in the subsequent sections.
Autophagy and Activation of Pancreatic Stellate Cells
An important feature of PDAC is that fibrosis of the extracellular matrix increases as PDAC progresses,Citation27 leading to low tumor vascular density, severe hypoxia, and limited access to nutrients. As a result, cancer cells activate pancreatic stellate cells (PSCs), increase the expression of autophagic proteins, and secrete non-essential amino acids (NEAA), such as alanine, to meet the metabolic needs of growing cancer cells.Citation27 In addition, increased autophagy causes the PSCs to change from a dormant state to an active state. Activated PSCs secrete extracellular matrix (ECM) proteins and cytokines to increase the aggressiveness of tumors,Citation28 closely correlated to PDAC patients’ short survival time. Administering autophagy inhibitors to PSCs reduced ECM protein and cytokine production and decreased the proliferation and invasiveness of PDAC cells in mice.Citation28 This suggests that autophagy can promote PDAC progression by regulating non-tumor cells.
Autophagy and Cell Proliferation, Invasion, and Metastasis
PDAC cells are characterized by dysregulated proliferation and the ability to invade surrounding tissues, which is a reason for poor prognosis and high mortality. Gorgulu et alCitation29 showed that in ATG5-deficient PDAC mice, tumor cell proliferation and migration increased. However, in such mice, the tumor formation was prevented. Interestingly, knockout of ATG5 in pancreatic cancer cell line increased their proliferation and migration. In human PDAC samples, lower ATG5 expression levels are associated with tumor migration and shorter patient survival times. This suggests that the expression level of ATG5 should be considered when using autophagy inhibitors to treat PDAC, so as to avoid the occurrence of drug resistance.
Transforming growth factor-β (TGF-β) promotes proliferation, invasion, and migration in late stage tumors.Citation30 He et alCitation30 found that TGF-β can influence TFEB expression in SMAD4-positive PDAC cells, thereby promoting TFEB-mediated autophagy. The increase in TFEB-mediated autophagy was negatively correlated with prognosis in patients with PDAC. Chen et alCitation31 found that TGF-β induced autophagy effects on PDAC proliferation and invasiveness, dependent on SMAD4 expression. TGF-β-induced autophagy enhanced SMAD4-negative PDAC cell migration and inhibited cell growth, while the opposite effect was observed in SMAD4-positive PDAC cells. This suggests that different genetic backgrounds may differentially affect autophagy in PDAC.
Ubiquitin-like protein 4A (UBL4A) can act on lysosomal-associated membrane protein 1 (LAMP1) to inhibit autophagy and thus inhibit PDAC cell proliferation and metastasis.Citation32 High UBL4A expression in PDAC is associated with good prognosis and prolonged patient survival.Citation33 The RNA-binding protein QKI can activate surrounding fibroblasts to proliferate, invade, and metastasize, increasing autophagy in PDAC cells.Citation33 In addition, MAPK can affect the interaction between PDAC and stroma and induce autophagy to increase PDAC cell proliferation, invasion, and migration.Citation34 These results suggest that cell autophagy in tumor microenvironments may promote PDAC cell invasion and migration.
Autophagy and Metabolism
Low vascular density during PDAC leads to severe hypoxia and limited nutrient utilization. Therefore, PDAC cells must alter their metabolic pathways to sustain indefinite proliferation. As mentioned above, the changes in mTORC1 activity and AMPK signal transduction enhancement increase autophagy during cell starvation. Additionally, TEFB overexpression can regulate Ras-related GTP binding D (RagD) expression, promote mTORC1 recruitment to the lysosome, and enhance tumor growth.Citation35 TFEB stabilizes lysosomes and supports the nutritional requirements of PDAC cell proliferation which is vital for sustaining tumor growth.Citation35 Thus, there is a complex relationship between autophagy and PDAC metabolism, which may be used to develop new treatments for PDAC. We focused on the regulatory mechanisms of autophagy in glucose metabolism, amino acid metabolism, and oxidative stress ().
Figure 3 Autophagy maintains the metabolism and function of PDAC cells. Autophagy pathways are modulated by different metabolic conditions (eg, oxidative stress, low glucose, and low amino acids) in which cellular components are degraded. In this process, bioenergy intermediates are reused, thereby promoting cell survival.
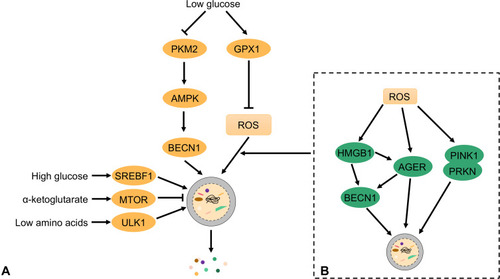
Glucose is an important nutrient for PDAC. Under aerobic conditions, PDAC uses aerobic glycolysis to produce lactic acid rather than oxidative phosphorylation (OXPHOS).Citation36 When PDAC cells are deprived of glucose, they produce large amounts of ROS to activate autophagy and the supply needed for growth.Citation37 In contrast, glutathione peroxidase 1 (GPX1) inhibits autophagy by reducing ROS production, sensitizing PDAC cells to hanger-induced cell death.Citation38 Although this finding indicates that GPX1 acts as an autophagy inhibitory protein in PDAC, the role of other members of the GPX family in PDAC remains unknown. Pyruvate kinase M2 (PKM2) is an important regulator of glycolysis and is downregulated in PDAC cells with low glucose levels; however, in PDAC cells with reduced glucose, low PKM2 expression upregulates AMPKα1 expression and induces autophagy to promote cell survival.Citation39 Bryant et al found that KRAS and MAPK inhibition increases autophagic flux in KRAS mouse models and human PDAC cells and drives PDAC to become acutely dependent on autophagy.Citation40,Citation41 Thus, the combined inhibition of KRAS and autophagy could play a significant role in PDAC treatment.Citation40
In PDAC cells, reprogramming energy metabolism plays an important role in cell proliferation and tumor growth. We have discussed glucose metabolism above, but more recently amino acid metabolism, particularly glutamine metabolism reprogramming in PDAC cells, has attracted a lot of research interest.
Autophagy supplies glutamine to PDAC cells through micropinocytosis.Citation39 Glutamine is broken down into ammonia and glutamic acid in the mitochondria. The latter is converted to the TCA cycle-intermediates α-ketoglutarate (α-KG) and DMKGCitation42 which promote PDAC growth. Thus, autophagy supplies glutamine to PDAC cells and promotes PDAC growth.Citation43 However, in PDAC cells, α-KG can activate mTORC1 to reduce autophagy.Citation44 When chronic mTORC1 inhibition leads to amino acid deficiency, TFEB expression is increased to activate autophagy. This suggests a dynamic balance between glutamine metabolism and autophagy to ensure the growth of PDAC cells.
The branched-chain amino acids (BCAAs) leucine, isoleucine, and valine are three essential amino acids (EAAs) that cannot be synthesized in the cell and must be obtained from the diet.Citation42 Interestingly, leucine, which is a mTORC1 agonist, promotes PDAC growth. Solute carrier family 38 member 9 (SLC38A9) transports leucine from lysosomes into the cytoplasm. Wyant et alCitation45 found that in cells lacking SLC38A9, lysosomal leucine levels remained essentially unchanged, even though whole-cell leucine levels were reduced compared to the control. This revealed that the leucine utilization of PDAC cells depends on the interaction between autophagy and lysosomal proteolysis.
PSCs in tumor microenvironments also influence autophagy and amino acid metabolism in PDAC. PDAC cells induce PSC autophagy to supply alanine.Citation27 Alanine can be converted to pyruvate to provide a carbon source for the TCA cycle or promote lipid biosynthesis and synthesize non-essential amino acids such as serine and glycine. Thus, autophagy induced by cancer cells and PCS interaction supports the metabolic demands of PDAC cells and promotes tumor growth. In the progressive stage of PDAC, the cells are in a state of oxidative stress. Tumor cells produce a large amount of ROS which damages DNA and accelerates PDAC development. Elevated ROS levels can inhibit mTORC1 and activate autophagy.Citation46 High mobility group box 1 (HMGB1) is a novel BECN1 binding protein expressed during autophagy.Citation47 In PDAC cells, the induction of autophagy depends on the REDOX state of HMGB1. When HMGB1 is reduced, it binds to advanced glycation end products (RAGE) receptors, induces BECN1 dependent autophagy, and promotes pancreatic tumor cell line survival.Citation48 The zinc chelator, TPEN, can disrupt the mitochondrial function of PDAC cells and cause oxidative stress. Meanwhile, TPEN inhibits lysosomal activity and inhibits autophagy, leading to PDAC cell apoptosis.Citation49 In conclusion, there is a complex relationship between oxidative stress, ROS and autophagy.
We described the complex relationship between autophagy and PDAC metabolism, and the potential for combining metabolic pathway manipulation and autophagy to treat PDAC in the future. However, the specific regulatory mechanism between PDAC autophagy and metabolism requires further study.
Autophagy and Immune Response
Yang et al established a PDAC mouse model expressing AtG4BC47A to inhibit autophagy and found that macrophage infiltration of the tumor increased and partially mediated tumor regression.Citation50 This suggests that autophagy is an immune response by the PDAC cells. Furthermore, in PDAC cells, the major histocompatibility complex class I (MHC-1) can present endogenous antigens to CD8+ T cells to identify cancer cells for destruction.Citation51
Interestingly, PDAC cells express low levels of MHC-1 but are highly enriched in autophagy-related autophagosomes and lysosomes compared to human pancreatic ductal epithelial (HPDE) cells.Citation52 This autophagic MHC-1 degradation mediated by NBR1 was increased, and the antigenicity was significantly decreased.Citation51 This impaired antigen presentation and promoted immune evasion of the tumor. However, after autophagy inhibition, the PDAC cell MHC-1 expression was restored, which promoted CD8+T cell proliferation and activation and enhanced tumor cell cytotoxicity in vivo and in vitro.Citation52 Moreover, after inhibiting autophagy MDSCs, CD4+ T cells and CD103+ DCs also changed, but their specific roles in PDAC remain unclear.Citation52 Therefore, the relationship between autophagy and the immune responses of PDAC cells will be the focus of future research.
Autophagy as a Therapeutic Target for PDAC
PDAC is difficult to cure and often recurring. Currently, the main treatment methods are surgery, chemotherapy, and radiotherapy. However, frequent drug resistance in PDAC is a major cause of poor patient treatment outcomes. This may be because autophagy helps tumor cells respond to a variety of stresses, including hypoxia, low pH, and reduced nutrient supply. Therefore, it is vital to understand the mechanism of autophagy to improve PDAC treatment.
Currently, the gold standard for PDAC chemotherapy combines FOLFIRINOX and nab-paclitaxel with gemcitabine.Citation53 Some studies have found that increased autophagy may be responsible for developing resistance to chemotherapeutic drugs. Gemcitabine and 5-fluorouracil induce apoptosis and increase autophagy.Citation54,Citation55 Combining chloroquine and gemcitabine or 5-fluorouracil can significantly inhibit tumor growth.Citation55 Meanwhile, AMPK inhibition can enhance the toxicity of gemcitabine or 5-fluorouracil toxicity to the tumor.Citation54 Clinical studies have also shown that administering gemcitabine combined with hydroxychloroquine is more effective than gemcitabine alone.Citation56
SMAD4 gene depletion induces autophagy by increasing ROS levels.Citation57 SMAD4-depleted PDAC cells were resistant to radiotherapy and this resistance was found to be reversed by administering autophagy inhibitors.Citation57 This suggests that autophagy plays a role in the resistance of PDAC cells to radiotherapy.
In a recent study, inhibiting KRAS-RAF-MEK-ERK signal transduction increased autophagy.Citation40 After ERK inhibition, tumor cells were increasingly dependent on autophagy for nutrients. Furthermore, PDAC development in vivo and in vitro was demonstrably affected after administering a combination of an ERK inhibitor trametinib and chloroquine.Citation40 In pancreatic cancer cell lines, the death rate in cells was found to increase by administering a combination of trametinib and chloroquine, compared with chloroquine alone. After administering this treatment protocol, the graft in MIA-PaCa2 xenograft mice had regressed and a patient with metastatic pancreatic cancer who had failed to respond to all standard treatment protocol had a reduction in tumor marker cancer antigen 19–9 (CA19–9) and overall tumor burden.Citation58 These results provide evidence for a new combination regimen to treat PDAC, suggesting that autophagy plays an important role in treating pancreatic cancer.
Autophagy plays an important role in the success of PDAC treatment. The preclinical study of targeted autophagy treatment of PDAC has achieved gratifying results. However, results in clinical studies have been disappointing. Therefore, maximizing the effect of autophagy in the treatment of pancreatic cancer patients is a challenge for future research.
Conclusion
The understanding of autophagy’s regulatory mechanisms has deepened over the past decade with the discovery of new regulatory networks and transduction pathways. The pathophysiological role of autophagy in cancer, specifically pancreatic cancer, has also been explored. Changes in autophagy play an important role in the onset and development of pancreatic cancer. This is not only reflected in cancer cells but also in non-cancer cells in the tumor microenvironment. Moreover, autophagy functions in PDAC are regulated by oncogenes (eg, KRAS) and tumor suppressor genes (eg, TP53).
Autophagy plays a complex role in cancer, and our current knowledge remains very limited. Benign lesions in autophagy defective mice are prevented from transforming into pancreatic cancer. Because autophagy provides nutrients for pancreatic cancer growth, this finding in autophagy defective mice suggests that the autophagy process can be an effective intervention target to prevent and treat pancreatic cancer.
Targeted autophagy therapy for pancreatic cancer has achieved great success in preclinical studies, but its clinical application in humans has yielded disappointing results. This discrepancy may be due to the following. First, our understanding of the specific role of autophagy at the molecular level and how it affects tumors is currently very limited. Second, mouse models do not accurately replicate human pancreatic cancer pathology. Finally, autophagy is essential for cell homeostasis, and the systematic application of autophagy inhibitors may interfere with its normal function in tissues.
Autophagy is thought to be a mechanism by which tumor cells maintain their high metabolic level in a nutrient-poor environment. Autophagy also helps tumor cells cope with multiple stresses (ie, hypoxia, low pH, and reduced nutrient supply). Inhibition of autophagy may be an important direction of targeted therapy for pancreatic cancer in the future, and the development and use of autophagy inhibitors should be a priority. In particular, designing such autophagy inhibitors that do not penetrate the blood-brain barrier can reduce nerve toxicity. In addition, it is important to develop inhibitors that do not affect autophagy of non-cancerous cells around pancreatic cancer, but do affect tumor cell growth and reduce the ability of cancerous cells to metastasize.
Because genetic alterations can affect autophagy, it will be important to collect tumor or blood samples from PDAC patients prior to treatment with autophagy inhibitors to assess the dependence of pancreatic cancer cells on autophagy. Autophagy inhibition has a positive effect only when tumor changes occur. Clinically, autophagy inhibitors should be applied to patients with a definitive diagnosis of pancreatic cancer. In addition, because autophagy inhibitors are applied to the whole body, researchers should consider the long-term potential toxic effects on normal cells and tissues when developing and applying drugs. In conclusion, exploring the mechanism of autophagy inhibitors and elucidating their interactions with other anti-tumor drugs in pancreatic cancer will provide new methods and ideas for the clinical treatment of pancreatic cancer.
Abbreviations
PDAC, pancreatic ductal adenocarcinoma; mTORC1, mammalian target of rapamycin complex 1; ULK1, Unc-51-like kinase 1; AMPK, AMP-activated protein kinase; PtdIns3K, class III phosphatidylinositol 3-kinase; PIK3C3, phosphatidylinositol 3-kinase catalytic subunit type 3; VPS34, vesicular protein sorting 34; AMBRA1, activating molecule in Beclin1-regulated autophagy protein 1; BCL2, B-cell lymphoma 2; PI3P, phosphatidylinositol 3-phosphate; UBL, two ubiquitin-like; LC3B, light-chain 3B; PE, phosphatidylethanolamine; PanIN, pancreatic neoplasia; ADM, acinar-ductal metaplasia; PSCs, pancreatic stellate cells; NEAA, non-essential amino acids; OXPHOS, oxidative phosphorylation; MHC-1, major histocompatibility complex class I; CA19–9, cancer antigen 19–9.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest related to this work.
Acknowledgment
We would like to thank Editage (www.editage.cn) for English language editing.
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.2159031912902
- Kong C, Li Y, Liu Z, et al. Targeting the Oncogene KRAS Mutant Pancreatic Cancer by Synergistic Blocking of Lysosomal Acidification and Rapid Drug Release. Acs Nano. 2019;13(4):4049–4063. doi:10.1021/acsnano.8b0824630912923
- Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20(3):460–473. doi:10.1089/ars.2013.537123725295
- Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Gene Dev. 2016;30(17):1913–1930. doi:10.1101/gad.287524.11627664235
- Poillet-Perez L, White E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019;33(11–12):610–619. doi:10.1101/gad.325514.11931160394
- Levine B, Klionsky DJ. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: breakthroughs in baker’s yeast fuel advances in biomedical research. Proc Natl Acad Sci U S A. 2017;114(2):201–205. doi:10.1073/pnas.161987611428039434
- Kuo CJ, Hansen M, Troemel E. Autophagy and innate immunity: insights from invertebrate model organisms. Autophagy. 2018;14(2):233–242. doi:10.1080/15548627.2017.138982429130360
- Cao W, Li J, Yang K, Cao D. An overview of autophagy: mechanism, regulation and research progress. Bull Cancer. 2021;108(3):304–322. doi:10.1016/j.bulcan.2020.11.00433423775
- Kocaturk NM, Akkoc Y, Kig C, Bayraktar O, Gozuacik D, Kutlu O. Autophagy as a molecular target for cancer treatment. Eur J Pharm Sci. 2019;134:116–137. doi:10.1016/j.ejps.2019.04.01130981885
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12. doi:10.1002/path.269720225336
- Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176(1–2):11–42. doi:10.1016/j.cell.2018.09.04830633901
- Singh SS, Vats S, Chia AY, et al. Dual role of autophagy in hallmarks of cancer. Oncogene. 2018;37(9):1142–1158. doi:10.1038/s41388-017-0046-629255248
- Nakamura S, Yoshimori T. New insights into autophagosome-lysosome fusion. J Cell Sci. 2017;130(7):1209–1216. doi:10.1242/jcs.19635228302910
- Stroupe C. This Is the End: regulation of Rab7 Nucleotide Binding in Endolysosomal Trafficking and Autophagy. Front Cell Dev Biol. 2018;6:129. doi:10.3389/fcell.2018.0012930333976
- McEwan DG, Popovic D, Gubas A, et al. PLEKHM1 Regulates Autophagosome-Lysosome Fusion through HOPS Complex and LC3/GABARAP Proteins. Mol Cell. 2015;57(1):39–54. doi:10.1016/j.molcel.2014.11.00625498145
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi:10.1016/j.cell.2007.12.01818191218
- Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25(7):717–729. doi:10.1101/gad.201611121406549
- Maertin S, Elperin JM, Lotshaw E, et al. Roles of autophagy and metabolism in pancreatic cancer cell adaptation to environmental challenges. Am J Physiol-Gastr L. 2017;313(5):G524–G536. doi:10.1152/ajpgi.00138.2017
- Gukovskaya AS, Gukovsky I, Algul H, Habtezion A. Autophagy, inflammation, and immune dysfunction in the pathogenesis of pancreatitis. Gastroenterology. 2017;153(5):1212–1226. doi:10.1053/j.gastro.2017.08.07128918190
- Antonucci L, Fagman JB, Kim JY, et al. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci U S A. 2015;112(45):E6166–74. doi:10.1073/pnas.151938411226512112
- Wang L, Xie D, Wei D. Pancreatic acinar-to-ductal metaplasia and pancreatic cancer. Methods Mol Biol. 2019;1882:299–308. doi:10.1007/978-1-4939-8879-2_2630378064
- Mann KM, Ying H, Juan J, Jenkins NA, Copeland NG. KRAS-related proteins in pancreatic cancer. Pharmacol Ther. 2016;168:29–42. doi:10.1016/j.pharmthera.2016.09.00327595930
- Loncle C, Molejon MI, Lac S, et al. The pancreatitis-associated protein VMP1, a key regulator of inducible autophagy, promotes Kras(G12D)-mediated pancreatic cancer initiation. Cell Death Dis. 2016;7:e2295. doi:10.1038/cddis.2016.20227415425
- Rosenfeldt MT, O’Prey J, Morton JP, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300. doi:10.1038/nature1286524305049
- Yang A, Rajeshkumar NV, Wang XX, et al. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with p53 Alterations. Cancer Discov. 2014;4(8):905–913. doi:10.1158/2159-8290.Cd-14-036224875860
- Xie YC, Liu J, Kang R, Tang DL. Mitophagy in Pancreatic Cancer. Front Oncol. 2021;11;2548.
- Sousa CM, Biancur DE, Wang XX, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536(7617):479. doi:10.1038/nature1908427509858
- Endo S, Nakata K, Ohuchida K, et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology. 2017;152(6):1492. doi:10.1053/j.gastro.2017.01.01028126348
- Gorgulu K, Diakopoulos KN, Ai J, et al. Levels of the Autophagy-Related 5 Protein Affect Progression and Metastasis of Pancreatic Tumors in Mice. Gastroenterology. 2019;156(1):203–217e20. doi:10.1053/j.gastro.2018.09.05330296435
- He R, Wang M, Zhao C, et al. TFEB-driven autophagy potentiates TGF-beta induced migration in pancreatic cancer cells. J Exp Clin Cancer Res. 2019;38(1):340. doi:10.1186/s13046-019-1343-431387632
- Liang C, Xu J, Meng Q, et al. TGFB1-induced autophagy affects the pattern of pancreatic cancer progression in distinct ways depending on SMAD4 status. Autophagy. 2020;16(3):486–500. doi:10.1080/15548627.2019.162854031177911
- Chen H, Li L, Hu J, et al. UBL4A inhibits autophagy-mediated proliferation and metastasis of pancreatic ductal adenocarcinoma via targeting LAMP1. J Exp Clin Cancer Res. 2019;38(1):297. doi:10.1186/s13046-019-1278-931288830
- Chu L, Hu Y, Jiang YH, Xu C, Liu WC, Lu ZF. Effects of RNA binding protein QKI on pancreatic cancer ductal epithelial cells and surrounding activation fibroblasts. J Cell Biochem. 2019;120(7):11551–11561. doi:10.1002/jcb.28435
- Yan Z, Ohuchida K, Fei S, et al. Inhibition of ERK1/2 in cancer-associated pancreatic stellate cells suppresses cancer-stromal interaction and metastasis. J Exp Clin Cancer Res. 2019;38(1):221. doi:10.1186/s13046-019-1226-831133044
- Di Malta C, Siciliano D, Calcagni A, et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017;356(6343):1188–1192. doi:10.1126/science.aag255328619945
- Yang J, Ren B, Yang G, et al. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life Sci. 2020;77(2):305–321. doi:10.1007/s00018-019-03278-z31432232
- Biancur DE, Kimmelman AC. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Bba Rev Cancer. 2018;1870(1):67–75. doi:10.1016/j.bbcan.2018.04.011
- Meng QC, Xu J, Liang C, et al. GPx1 is involved in the induction of protective autophagy in pancreatic cancer cells in response to glucose deprivation. Cell Death Dis. 2018;1:9.
- Li X, Deng SC, Liu ML, et al. The responsively decreased PKM2 facilitates the survival of pancreatic cancer cells in hypoglucose. Cell Death Dis. 2018;9:487.29706630
- Bryant KL, Stalnecker CA, Zeitouni D, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25(4):628. doi:10.1038/s41591-019-0368-830833752
- Kim JH, Kim HY, Lee YK, et al. Involvement of mitophagy in oncogenic K-Ras-induced transformation: overcoming a cellular energy deficit from glucose deficiency. Autophagy. 2011;7(10):1187–1198. doi:10.4161/auto.7.10.1664321738012
- Xu R, Yang J, Ren B, et al. Reprogramming of Amino Acid Metabolism in Pancreatic Cancer: recent Advances and Therapeutic Strategies. Front Oncol. 2020;10:572722. doi:10.3389/fonc.2020.57272233117704
- Seo JW, Choi J, Lee SY, et al. Autophagy is required for PDAC glutamine metabolism. Sci Rep-Uk. 2016;6:251.
- Hosein AN, Beg MS. Pancreatic Cancer Metabolism: molecular Mechanisms and Clinical Applications. Curr Oncol Rep. 2018;20(7):56. doi:10.1007/s11912-018-0699-529752600
- Wyant GA, Abu-Remaileh M, Wolfson RL, et al. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell. 2017;171(3):642. doi:10.1016/j.cell.2017.09.04629053970
- Alexander A, Cai SL, Kim J, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107(9):4153–4158. doi:10.1073/pnas.091386010720160076
- Kang R, Livesey KM, Zeh HJ 3rd, Lotze MT, Tang D. Metabolic regulation by HMGB1-mediated autophagy and mitophagy. Autophagy. 2011;7(10):1256–1258. doi:10.4161/auto.7.10.1675321691146
- Tang D, Loze MT, Zeh HJ, Kang R. The redox protein HMGB1 regulates cell death and survival in cancer treatment. Autophagy. 2010;6(8):1181–1183. doi:10.4161/auto.6.8.1336720861675
- Yu Z, Yu Z, Chen ZB, et al. Zinc chelator TPEN induces pancreatic cancer cell death through causing oxidative stress and inhibiting cell autophagy. J Cell Physiol. 2019;234(11):20648–20661. doi:10.1002/jcp.2867031054150
- Yang AN, Herter-Sprie G, Zhang HK, et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov. 2018;8(3):276–287. doi:10.1158/2159-8290.Cd-17-095229317452
- Yamamoto K, Venida A, Perera RM, Kimmelman AC. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy. 2020;16(8):1524–1525. doi:10.1080/15548627.2020.176997332459143
- Yamamoto K, Venida A, Yano J, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020;581(7806):100–105. doi:10.1038/s41586-020-2229-532376951
- Chiorean EG, Coveler AL. Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther. 2015;9:3529–3545. doi:10.2147/DDDT.S60328
- Zhu J, Chen Y, Ji Y, et al. Gemcitabine induces apoptosis and autophagy via the AMPK/mTOR signaling pathway in pancreatic cancer cells. Biotechnol Appl Biochem. 2018;65(5):665–671. doi:10.1002/bab.165729575133
- Hashimoto D, Blauer M, Hirota M, Ikonen NH, Sand J, Laukkarinen J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur J Cancer. 2014;50(7):1382–1390. doi:10.1016/j.ejca.2014.01.01124503026
- Karasic TB, O’Hara MH, Loaiza-Bonilla A, et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019;5(7):993–998. doi:10.1001/jamaoncol.2019.068431120501
- Wang F, Xia XJ, Yang CY, et al. SMAD4 gene mutation renders pancreatic cancer resistance to radiotherapy through promotion of autophagy. Clin Cancer Res. 2018;24(13):3176–3185. doi:10.1158/1078-0432.Ccr-17-343529602802
- Kinsey CG, Camolotto SA, Boespflug AM, et al. Protective autophagy elicited by RAF -> MEK -> ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019;25(4):620. doi:10.1038/s41591-019-0367-930833748