Abstract
An extrafine formulation of the long-acting muscarinic antagonist glycopyrronium bromide (GB) is in development for chronic obstructive pulmonary disease (COPD), in combination with beclometasone dipropionate and formoterol fumarate – a “fixed triple”. This two-part study was randomized, double blind, placebo controlled in patients with moderate-to-severe COPD: Part 1: single-dose escalation, GB 12.5, 25, 50, 100 or 200 μg versus placebo; Part 2: repeat-dose (7-day), four-period crossover, GB 12.5, 25 or 50 μg twice daily (BID) versus placebo, with an open-label extension in which all patients received tiotropium 18 μg once daily. On the morning of Day 8 in all five periods, patients also received formoterol 12 μg. In study Part 1, 27 patients were recruited. All GB doses significantly increased from baseline forced expiratory volume in 1 second (FEV1) area under the curve (AUC0–12h) and peak FEV1, with a trend toward greater efficacy with higher GB dose. All adverse events were mild–moderate in severity, with a lower incidence with GB than placebo and no evidence of a dose–response relationship. In study Part 2, of 38 patients recruited, 34 completed the study. Adjusted mean differences from placebo in 12 h trough FEV1 on Day 7 (primary) were 115, 142 and 136 mL for GB 12.5, 25 and 50 μg BID, respectively (all P<0.001). GB 25 and 50 μg BID were superior (P<0.05) to GB 12.5 μg BID for pre-dose morning FEV1 on Day 8. For this endpoint, GB 25 and 50 μg BID were also superior to tiotropium. Compared with Day 7, addition of formoterol significantly increased Day 8 FEV1 peak and AUC0–12h with all GB doses and placebo (all P<0.001). All adverse events were mild–moderate in severity and there was no indication of a dose-related relationship. This study provides initial evidence on bronchodilation, safety and pharmacokinetics of extrafine GB BID. Overall, the results suggest that GB 25 μg BID is the optimal dose in patients with COPD.
Acknowledgments
This study was funded by Chiesi Farmaceutici SpA. Writing support was provided by David Young of Young Medical Communications and Consulting Ltd. This support was funded by Chiesi Farmaceutici SpA.
Disclosure
DS has received sponsorship to attend international meetings, honoraria for lecturing or attending advisory boards and research grants from various pharmaceutical companies including Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, Genentech, GlaxoSmithKline, Glenmark, Johnson and Johnson, Merck, NAPP, Novartis, Pfizer, Skypharma, Takeda, Teva, Therevance and Verona. MS, SC, SV, FM, AM and DA are employees of Chiesi Farmaceutici SpA, the study sponsor. The authors report no other conflicts of interest in this work.
Supplementary materials
Inclusion criteria
Subjects had to meet all of the following inclusion criteria (applicable for study Parts 1 and 2):
Male and female subjects, aged 40–75 years.
Written informed consent obtained before the first study-related activity.
Diagnosis of moderate–severe COPD, according to the Global Initiative for Chronic Obstructive Lung Disease guidelines (2009).
Able to understand the study procedures, the risks involved and the ability to be trained to use the devices correctly.
Body mass index between 18 and 35 kg/m2.
Current or ex-smokers with a smoking history of ≥10 pack-years (eg, ≥20 cigarettes per day for 10 years and 40 cigarettes per day for 5 years).
Vital signs within the following ranges:
90 mmHg ≤ systolic blood pressure ≤160 mmHg,
50 mmHg ≤ diastolic blood pressure ≤90 mmHg,
50 beats per min ≤ heart rate ≤100 beats per min.
Twelve-lead electrocardiogram (ECG) with comput-erized protocol interpretation considered as normal: 120 ms ≤ PR ≤200 ms; QRS ≤120 ms; QTcF ≤450 ms. Minor deviations were acceptable, provided that they were not judged clinically significant by the cardiologist.
Post-bronchodilator forced expiratory volume in 1 second (FEV1) between 40% and 80% predicted values (40% ≤ FEV1 ≤80%), documented at the screening visit.
Post-bronchodilator FEV1/forced vital capacity: ≤70 (absolute value) documented at the screening visit.
Airway reversibility of at least 100 mL within 30–45 min after inhalation of ipratropium 80 μg (for study Part 2: historical reversibility was acceptable for subjects who performed study Part 1). If the reversibility criteria were not met and if the investigator deemed it appropriate, the testing could be repeated once. This requirement had to be met after retesting during the run-in period at least 24 h prior to randomization.
Exclusion criteria
Subjects meeting at least one of the following criteria could not be enrolled (applicable for study Parts 1 and 2):
History of chronic or seasonal allergy.
Blood eosinophil count above 0.6×109/L.
Clinically relevant findings on physical examination, laboratory or ECG parameters at screening. (Note that one patient had a prolonged QTc at screening and was allowed to enter the study by the investigator, who considered this to be not clinically relevant. The subject was later withdrawn from the study, as the study was designed to exercise caution in subjects who may be predisposed to an increased risk of cardiovascular events.)
Occurrence of one of the following 24 h Holter ECG abnormalities at screening:
More than 200 ventricular ectopics in 24 h;
Ventricular tachycardia;
Second-degree heart block;
Sustained cardiac arrhythmias (atrial fibrillation, supraventricular tachycardia, complete heart block);
Any symptomatic arrhythmia (except isolated extrasystoles);
Sinus pauses ≥2.5 s.
Significant disease not related to COPD (eg, myocardial infarction, stroke within the preceding 6 months).
Respiratory tract infection (including upper tract) 4 weeks prior to the screening visit, requiring change of treatment.
Subjects requiring oxygen therapy on a daily basis for chronic hypoxemia.
Subjects who had been hospitalized in the 6 weeks prior to the screening visit.
Having received an investigational product within the last 8 weeks before the screening visit.
Inability to comply with the study procedures or with the study treatment intake, including inability to be trained with the Vitalograph® Aerosol Inhalation Monitor.
History of cystic fibrosis, bronchiectasis, alpha-1 anti-trypsin deficiency or any other significant lung disease that was considered to be clinically significant by the investigator.
Intolerance/hypersensitivity or any contraindication (eg, history of glaucoma) to treatment with an M3 antagonist or any of the excipients contained in the formulations used in the study.
History of alcohol or substance abuse that, in the opinion of the investigator, could be of clinical significance.
Subjects who had undergone major surgery in the 12 weeks before the screening visit.
Subjects treated with oral (slow-release included) or parenteral steroids 8 weeks prior to the screening visit.
Subjects treated with oral β2-adrenergics, antihistamines or theophyllines 1 month prior to the screening visit or enzyme-inducing or -inhibiting drugs 2 months before the first administration.
Subjects treated with tiotropium in the 10 days prior to the screening visit.
Pregnant or lactating women (pregnancy defined as the state of a female after conception and until the termination of the gestation, confirmed by a positive serum human chorionic gonadotropin laboratory test [.5 mIU/mL]). Serum pregnancy test was to be done at screening for verification. (b) Women of childbearing potential (ie, all women physiologically capable of becoming pregnant), including women whose career, lifestyle or sexual orientation precluded intercourse with a male partner, unless they were postmenopausal (defined as 12 months of natural [spontaneous] amenorrhea) or were using an acceptable method of contraception. (c) Male subjects had to be sterile or they or their partner had to be willing to use an approved method of contraception from the time of dose administration until 30 days after the last dose of study medication. Subjects were not allowed to donate sperm for 30 days after the last dose of study drug. A reliable method of contraception for male and female subjects (or their partner) could be one or more of the following ones:
Surgical sterilization (ie, bilateral tubal ligation or hysterectomy for females, vasectomy for males);
Hormonal contraception (implantable, patch, or oral);
Double-barrier methods (any double combination of intrauterine device, male or female condom, diaphragm, sponge, cervical cap, condom or spermicide).
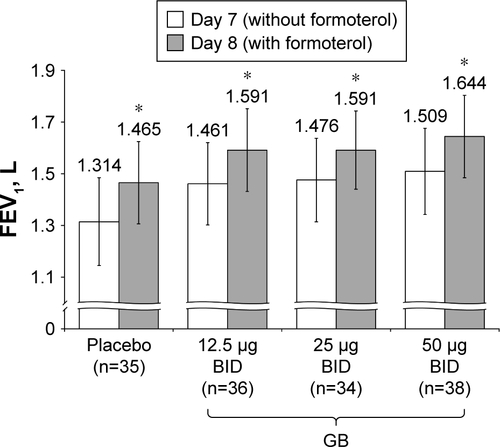
Figure S1 Study Part 2: Difference in 12 h trough FEV1 between Day 7 and Day 8, following administration of formoterol.
Notes: *P<0.001 versus Day 7. Data plotted are mean and 95% CI.
Abbreviations: BID, twice daily; CI, confidence interval; FEV1, forced expiratory volume in 1 second; GB, glycopyrronium bromide; n, number of randomized patients.