Abstract
Emphysema is mainly caused by cigarette smoking and is characterized by the loss of alveolar integrity and an enlargement of the alveolar space. However, mechanisms involved in its development are not fully understood. Alveolar cell apoptosis has been previously investigated in the lung of emphysematous subjects as a potential contributor to the loss of alveolar cell and has been found abnormally elevated. Though, mechanisms involved in the increased alveolar apoptosis that occurs in emphysema have now become a prolific field of research. Those mechanisms are reviewed here with special focus on how they affect cell viability and how they may be implicated in emphysema. Moreover, we suggest a model that integrates all those mechanisms to explain the increased alveolar apoptosis observed in emphysema. This review also includes some reflections and suggestions on the research to come.
Introduction
According to the World Health Organization, chronic obstructive pulmonary disease (COPD) is believed to become the third cause of death by 2030 with cigarette smoking as the main cause.Citation1 COPD features two main phenotypes: chronic bronchitis and emphysema with different physiopathology and symptoms. Emphysema has retained attention for its micro- and macroscopical manifestations: the loss of alveolar integrity and an enlargement of the alveolar space.Citation2 This leads to a poor gas exchange at the alveolar level and to the retention of air caused by airways collapse due to the loss of elastic recoil (hyperinflation).Citation2 But what is literally destructing the lung parenchyma of emphysematous patients? Proteases have been blamed very early, as deficiency in α1-antitrypsin (A1AT), the most important antiprotease of the lung, causes noxious gas-independent emphysema at an early age.Citation3 Emphysema was first considered a neutrophilic disease, neutrophils being the major protease generator of all immune cells.Citation4 However, as research progressed, emphysema was revealed to be a much more complex disease also involving alveolar macrophages (AM)Citation5 and cytotoxic CD8+ T lymphocytes (CTL).Citation6 Therefore, emphysema is now considered a complex inflammatory disease with only partly understood physiopathology. But the question remains: why and how alveolar structure is disappearing? Recently, new hypothesis rose trying to answer this question, among those was “apoptosis”, a complex and well-regulated process that leads to cell death. In this review, we will focus on the recent developments made on the involvement of apoptosis in emphysema and on the possible molecular mechanisms involved in the initiation and progression of the disease.
What we know
Apoptosis
Mechanisms involved in the programmed cell death are very complex but quite well described. There are many reasons why a cell would undergo apoptosis, among them: growth factor deprivation, mitotic aberrations, loss of contact with the extracellular matrix, direct induction by immune cells, activation of death receptors by soluble death ligands, and heavy damages caused by various stresses. All these “stimuli” will lead to cell apoptosis but will trigger different intracellular pathways to achieve their goal. The two main apoptotic pathways are the extrinsic and the intrinsic pathways, both leading to DNA fragmentation and cell death.
The extrinsic pathway
The extrinsic pathway () is triggered mostly by death ligands such as tumor necrosis factor (TNF), Fas ligand (FasL), and TNF-related apoptosis-inducing ligand (TRAIL) through their respective receptors.Citation7 Briefly, the intracellular death domains of those receptors will, after autophosphorylation, recruit various adaptor molecules such as Fas-associated death domain (FADD). If all recruiting steps are encountered, procaspase-8/10 will be cleaved into their active form, caspase-8/10. Caspases (cystein-aspartase) are cystein proteases found in their pro-form (inactive form) in living cells. Following specific cleavage, they will acquire their protease activity and will start to cleave specific substrates and activate other caspases. Once cleaved, those substrates will transmit the apoptotic signal to the nucleus or mitochondria (caspase-3, 6, 7) or interfere with the anti-apoptotic protection (cellular FLICE-like inhibitory protein [cFLIP]). In addition, cleavage of the BH3-interacting domain death agonist (Bid) to truncated Bib (tBid) will activate the intrinsic pathway.
Figure 1 Apoptosis pathways.
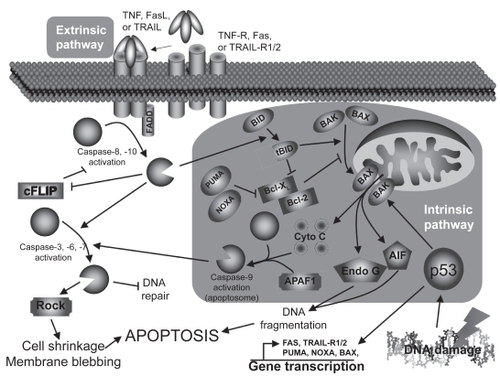
The intrinsic pathway
The intrinsic pathway () is mainly triggered by cellular stresses that cause DNA damages like oxidative stress or UV light.Citation8 Briefly, DNA damages will lead to the tumor suppressor p53 activation, stabilization, and to the acquisition of its transcription factor activity. Among its targets, p53 will either induce cell-cycle associated genes (eg, p21, 14-3-3σ) or apoptosis-related genes (eg, Bax [Bcl-2–associated X protein], NOXA [Latin for damage], PUMA [p53 upregulated modulator of apoptosis], Fas, TRAIL-R1/2) transcription, depending on the DNA damage severity. If damages are too severe, p53 will lead to apoptosis: Bax, NOXA, and PUMA will massively translocate to the mitochondria outer-membrane and facilitate the formation of channels that will allow pro-apoptotic factors to move to cytoplasm (DNAse, cytochrome c, and anti-apoptotic factors inhibitors). Cytochrome c will join Apaf-1 and caspase-9 to form the so-called apoptosome (activated caspase-9) that will activate caspase-3. As mentioned earlier, the extrinsic pathway can also activate the intrinsic pathway through Bid cleavage (tBid) by caspase-8/10, and help cytosolic Bax to translocate to the mitochondria and induce the release of the mitochondria-contained apoptosis factors.
Finalities
No matter how apoptosis is induced or which pathway is involved, the finalities will be very similar.Citation9 The most obvious observation is the dynamic membrane blebbing that precedes apoptotic bodies formation. Moreover, the membrane lipid phosphatidyl serine (PS) normally localized on the cytoplasmic side of the cell membrane will be externalized and used by scavenger cells to recognize apoptotic cells, leading to efficient cell clearance. DNA will also be affected, with features like chromatid condensation, its fragmentation into 180–200 bp fragments (endonuclease G [Endo G], apoptosis-inducing factor [AIF]), and redistribution of the genetic material into the forming apoptotic bodies (Rho kinase [ROCK]). All those changes will lead to a clean cell death without intracellular material loss and to the phagocytosis of apoptotic bodies by immune or adjacent structural cells.
Apoptosis and emphysema: cellular mechanisms
Presence of higher apoptosis level in the human emphysematous lung has been reported numerous times in previous years () and has been associated to decreased alveolar surface area,Citation10 but also to increased alveolar cell proliferation,Citation11 suggesting that regeneration processes of the emphysematous lung might be overwhelmed by its destruction or impared by unknown mechanisms. To explain this phenomenon, mechanisms involved in the regulation of cell survival and cell death have been studied in vivo (animal models, see ), ex vivo, and in vitro. Here, we are summarizing the main mechanisms and their impacts on alveolar apoptosis that occurs in emphysema.
Table 1 Characteristics of studies reporting elevated apoptosis in the emphysematous lung
Table 2 Animal models used to study apoptosis in emphysema
Protease-induced apoptosis
What are proteases?
Proteases are proteins able to cleave specific amino-acid sequences and are implicated in many biological processes. In the lung, as in other tissues, proteases activity is closely controlled by anti-proteases. Though, protease/anti-protease balance is an extremely important factor for tissue homeostasis.
What is known about proteases in apoptosis?
In addition to their role in protein turnover, cell migration, and lung immunity, it has been shown that proteases can also induce lung epithelial and endothelial cell apoptosis ().Citation12–Citation14 In fact, Suzuki and colleaguesCitation14 showed that leukocyte elastase (LE), mainly released by activated polymorphonuclear leucocytes, can induce small airway and alveolar epithelial cell apoptosis through the intrinsic pathway and by decreasing AKT phosphorylation (anti-apoptotic factor) following proteinase-activated receptor 1 (PAR-1) activation. Yang and colleaguesCitation12 also demonstrated a similar effect of neutrophil elastase (NE) and proteinase 3 (PR3), mainly secreted by activated neutrophils, on primary bovine arterial endothelial cells. On the other side, it has been demonstrated that the protease inhibitor α1-antitrypsin (A1AT) may act as a survival factor by preventing lung endothelial cell death induced by staurosporineCitation15 and cigarette smokeCitation16 through active caspase inhibition. Moreover, increasing evidences suggest that “tissues inhibitor of metalloproteinases-1” (TIMP-1) may act as a survival factor through CD63-mediated extracellular signal-regulated kinase (ERK) and AKT activation.Citation17 Taken together, these data suggest that protease/anti-protease imbalance can promote apoptosis both through direct activation of PAR-1 signaling and through the reduction of antiproteases’ ability to inhibit apoptotic processes.
Figure 2 Mechanisms by which leucocyte elastase (LE), neutrophil elastase (NE), and proteinase 3 (PR3) may induce alveolar epithelial and endothelial cell apoptosis. α-1 antitrypsin (A1AT), proteinase activated receptor 1 (PAR-1), matrix metalloproteinase (MMP), tissue inhibitor of matrix metalloproteinase (TIMP), extracellular-signal regulated kinase (ERK).
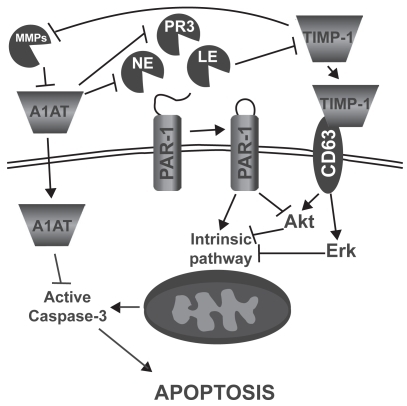
What is known about proteases in promoting apoptosis in emphysema?
Discovering that A1AT deficiency, the main inhibitor of NE, confers susceptibility to emphysema developmentCitation18 and that emphysema-like changes can be induced by elastolytic enzymes instillation in animal models (elastase emphysema)Citation19 were at the fundaments of the protease hypothesis of emphysema development. In fact, proteases such as NE,Citation20 cathepsins L,Citation21 and matrix metalloproteinases (MMP)-1,Citation22,Citation23 -2,Citation22,Citation24 -8,Citation22 -9,Citation22,Citation24 and -12Citation25 are increased in the emphysematous lung mainly due to a higher number of activated neutrophils and macrophages. The lung protease hyperactivity is believed to affect alveolar integrity by degrading elastic fibres responsible for the maintenance of alveolar structure and lung stretch. However, it seems that elastase-induced emphysema may not be attributable only to elastic fibre degradation. In fact, elastase activity is detectable for only 45 to 50 minutes after elastase instillation while emphysema phenotype and higher alveolar apoptosis remain for weeks,Citation26 probably due to the installation of an inflammatory state. Supporting that hypothesis, mice knockout for IL-1β and TNF receptors gene and submitted to elastase instillation showed fewer emphysematous lesions as well as alveolar cells undergoing apoptosis,Citation27 suggesting that elastase may promote disease progression and alveolar apoptosis through IL-1β and TNF-dependent mechanism. Moreover, in a mouse model of emphysema induced by local lung overexpression of interferon-γ, cathepsin S blockade, a protease involved in the antigen presentation process, limited the increase of apoptosis,Citation28 linking proteases once more with apoptosis in emphysema.
Decreased VEGF signaling
What is VEGF?
Vascular endothelial growth factor (VEGF) is a powerful angiogenic molecule with two receptors (VEGFR1 and 2) triggering opposite cell responses. VEGFR1 is mainly responsible for the inhibition of endothelial cell migration and proliferation, when VEGFR2 tends to promote those processes. VEGF is found in great amount in the lung and act as a key factor in the endothelial maintenance.Citation29
What is known about VEGF in apoptosis?
In regard to apoptosis and cell survival, it is known that VEGF, by itself, can inhibit serum deprivation-induced endothelial cell apoptosis in vitro.Citation30,Citation31 VEGF increases cell survival through phosphoinositide-3 kinase (PI3K)/Akt pathway activationCitation32 leading to caspase-9 and Bad inactivationCitation33,Citation34 and also by increasing the expression of the anti-apoptotic molecules Bcl-2, A1,Citation30 survivin, and XIAP.Citation35 VEGF is also able to prevent ceramide-induced HMEC (human microvascular endothelial cells) apoptosis through MAPK/ERK pathway activation and SAPK/JNK pathway inhibition.Citation31
What is known about decreased VEGF signaling in promoting apoptosis in emphysema?
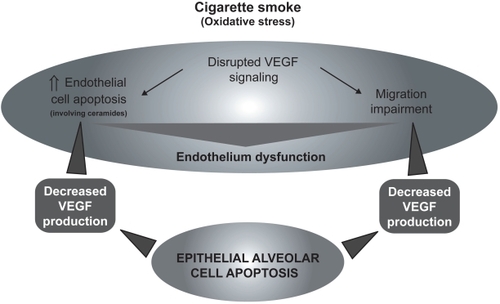
The role of VEGF signaling in the emphysema pathophysiology has raised significant interest in the past years.Citation36,Citation37 It has been shown that VEGF and VEGFR2 levels were decreased in the lung of emphysematous subjects compared to healthy controlsCitation38 and were related to increased alveolar cell apoptosis.Citation39 Cigarette smoke-exposed Sprague-Dawley rats also show decreased lung VEGF and VEGFR2.Citation38 Moreover, rats treated with the VEGF receptor blocker SU5416 developed emphysema-like phenotype, condition that may be prevented by the administration of the caspase inhibitor Z-Asp-CH2-DCB,Citation40 suggesting that decreased VEGF signaling may induce apoptotic process leading to emphysema. In addition, SU5416 treated rats presented evidences of alveolar oxidative stress (higher 8-hydroxyguanine and 4-hydroxynonenal staining).Citation41 In this model, oxidative stress and emphysema may be prevented by the administration of the superoxyde dismutase mimic M40419,Citation41 by N-acetylcystein (NAC),Citation42 and, interestingly, by the caspase inhibitor Z-Asp-CH2-DCB,Citation41 revealing that the apparition of oxidative stress is caspase-dependent. Taken together, these data suggest that decreased VEGF signaling may be responsible for endothelial cell apoptosis. However, the capillary bed appears essential for the growth and maintenance of alveolar septa.Citation43 Moreover, as demonstrated in human fetal lungCitation44,Citation45 and in adult lung,Citation46 VEGF is released principally by respiratory epithelial cells. Therefore, it may lead to a potential vicious circle that will cause endothelial and epithelial cell apoptosis ().
Figure 3 Cigarette smoke (oxidative stress)-mediated VEGF signaling disruption leading to endothelial cell death, migration impairment, and general endothelium dysfunction causing epithelial cells apoptosis.
Oxidative stress
What is oxidative stress?
Basically, oxidative stress happens when a cell is exposed to molecules with important oxidative power such as free radicals. If the oxidative defenses are strong enough, cellular damages will be negligible. However, if the oxidative aggression is too strong or if it persists for too long, cellular damages may then be very important.
What is known about oxidative stress in apoptosis?
It is well known that oxidative stress can trigger numerous cell responses, including the signaling cascade that will lead to apoptosis (through the intrinsic pathway), and many studies are available on the subject.Citation47 In vitro, exogenous hydrogen peroxide (H2O2) or compounds that promote endogenous production of reactive oxygen species (ROS) (eg, arsenic trioxide,Citation48 anthracyclines,Citation49 bleomycin,Citation50 N-(4-hydroxyphenyl) retinamideCitation51) are usually used to create oxidative stress.
In the majority of cell types, those treatments will trigger apoptosis through the intrinsic pathway as previously discussed ().Citation47 Another feature of oxidative stress-induced apoptosis is the activation of the mitogen-activated protein kinase (MAPK) pathways ERK 1/2, jun N-terminal kinase (JNK), and p38 MAPK). The JNK pathway seems to be pro-apoptotic as its inhibition promotes survival in primary rat alveolar epithelial cells treated with H2O2.Citation52 On the other side, ERK 1/2 and p38 MAPK tend to have important anti-apoptotic effects as their inhibition impairs cell survival.Citation52 However, activation and cellular effects of MAPK pathways are known to be different according to cell types studied.
Main defenses against oxidative stress are antioxidant molecules and enzymes. The major antioxidants found in the lung are superoxide dismutase (superoxide anion scavenger), (converts H2O2 to water), and glutathione peroxidase catalase (converts organic hydroperoxides to organic hydroxides).Citation53
What is known about oxidative stress in promoting apoptosis in emphysema?
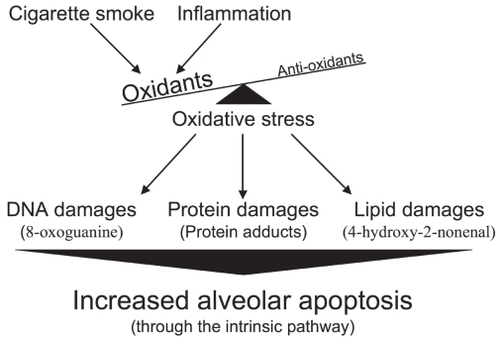
In the case of emphysema, oxidant molecules have two main origins. The first is of course cigarette smoke (active smoker), which contains free radicals with tremendous oxidative power such as superoxide (O2•−), hydroxyl radical (•OH), and hydrogen peroxide (H2O2).Citation54 It is important to note that nonemphysematous smokers are also exposed to the oxidative agents contained in cigarette smoke, suggesting that smokers who have developed emphysema (15%–20%) reacted differently or were more susceptible to this stress. The second source, and probably the most sustained, comes from the chronic inflammation inherent to emphysema that persists even after smoking cessation.Citation55 Activated macrophages and neutrophils, present in higher number in the emphysematous lung,Citation56–Citation59 are powerful producers of reactive oxygen species (ROS).Citation60,Citation61 It is well known that the lung antioxidant defenses of subjects with emphysema are overwhelmed, leading to important oxidative damages. In fact, compared to those of normal smokers and ex-smokers, the emphysematous lung presents more damaged proteinsand more peroxidated lipids.Citation62,Citation63 All those oxidative damages may then lead to the increased alveolar apoptosis observed in emphysema ().
Figure 4 Increased alveolar apoptosis mediated through oxidative stress-induced cellular damages.
TRAIL-mediated apoptosis
What is TRAIL?
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a member of the death ligand TNF family that also includes FasL. TRAIL has four receptors: TRAIL-R1 and R2 which contain functional death domains, TRAIL-R3 which contains a truncated death domain and acts as a decoy, and TRAIL-R4 which is unable to induce apoptotic signals but, as well as TRAIL-R1 and R2, can activate kinases that will lead to NF-κB activation.Citation64
What is known about TRAIL in apoptosis?
First interest on TRAIL has been brought by its ability to induce transformed/cancer cell apoptosis while having no such effects on normal cells.Citation65 Further research has shown that not all transformed/cancer cells were sensitive to its apoptotic signal.Citation66 Interestingly, DNA damage and ROS generating agents are able to suppress cellular resistance to TRAIL-mediated apoptosis in many cell lines, including the lung adenocarcinoma cell line A549.Citation67–Citation69 Moreover, in a mouse model of Alzheimer’s disease, a disease characterized by a progressive lost of neuronal cells, blockade of TRAIL-R2 prevents β-amyloid-induced neuronal cell apoptosis. Citation70 The main cells releasing TRAIL are activated macrophages, NK cells, and T lymphocytes.Citation71–Citation73
What is known about TRAIL in promoting apoptosis in emphysema?
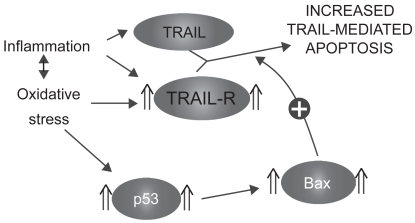
TRAIL receptors 1, 2, and 3 have been shown to be upregulated in the alveolar epithelial cells of emphysematous smokers and ex-smokers.Citation74 Moreover, their expression was closely related to the levels of the tumor suppressor p53 in the emphysematous lung parenchyma.Citation74 Interestingly, A549 cells (lung adenocarcinoma cell line) exposed in vitro to oxidative stress (H2O2) and TNF had higher expression of TRAIL-R1, 2, and 3 but also higher levels of p53,Citation74 suggesting that the modulation of the TRAIL system observed in the emphysematous lung may be attributable to oxidative stress and/or inflammatory cytokines. As TRAIL is released by activated inflammatory cells, oxidative stress and inflammation present in the emphysematous lung may sensitize alveolar cells to its apoptotic effects ().
Figure 5 Sensitization to TRAIL-mediated apoptosis.
Killer cells and autoimmunity
What are killer cells and autoimmunity?
Killer cells are immune cells specialized in the act of killing infected or unwanted cells. There are mainly two types of killer cells: cytotoxic CD8+ T lymphocytes (CTL), members of the adaptive immunity, and natural killer (NK) cells, members of the innate immunity. The main difference between these two cell types is the way they recognize cells that have to be killed. When a given cell is infected with intracellular pathogens, major histocompatibility complex (MHC) class I will present pathogen peptides at the cell surface allowing CTL to recognize it with their T cell receptor (TCR) and kill the infected cell.Citation75 On its side, with no TCR, NK cell cannot use by itself antigen-based recognition to identify cells to be killed. However, specific antigen-mediated cytotoxicity can be indirectly induced through antibody-dependent cellular cytotoxicity (ADCC) via the binding of FcγRIII (CD16) to the Fc portion of IgG bound to specific target cell membrane antigen.Citation76
Autoimmunity is an inappropriate immune response against antigens of the host (self-antigen) and can be triggered by a variety of mechanisms;Citation77 we describe here only those that may be relevant in emphysema. Autoimmune response can be triggered by modified self-antigen, as exposure of cells to agents that may affect protein integrity (eg, oxidants, proteases) can create molecules unknown to the immune system and cause autoimmune response. Moreover, molecules normally hidden from the immune system (located into the nucleus or the cytoplasm) may trigger autoimmune response if released. Failed clearance of apoptotic cells or high levels of necrosis may expose molecules normally sequestered to the cytoplasm or to the nucleus, as it is observed in systemic lupus erythematosus.Citation78
What is known about killer cells and autoimmunity in apoptosis?
When an immune response is initiated against a self-antigen, processes involved in the targeting and recognition by antibodies or killer cells are very similar to those raised against pathogens. First, the humoral response is involved: subjects with autoimmune diseases like systemic lupus erythematosus and rheumatoid arthritis are presenting autoantibodies in their serum.Citation79 Those antibodies secreted by activated B cells will, after recognition of their specific antigens, trigger local inflammatory response to eliminate the cells that bears autoantigen/antoantibody complexes.Citation80 As mentioned earlier, NK cell can recognize these cells and induce their apoptosis. In fact, following recognition, NK cell will induce target cell’s apoptosis mainly trough the release of granules that contain cytotoxic molecules like “pore forming protein” (perforin), granzymes, and granulisin. Death ligands such as FasL and TRAIL also play an important role in the NK-mediated cell apoptosis.Citation81 In addition to the antibody-mediated immune response, a specific cellular response can be mediated by CD4+ and CD8+ T lymphocytes. Once CD4+ T lymphocytes have encountered their antigen presented by an antigen-presenting cell (APC) like dendritic cells, they will proliferate (oligoclonal expansion) and be involved in the activation of B cells and alveolar macrophages through cell/cell contact or the release of inflammatory mediators.Citation82 On their side, activated CD8+ T lymphocytes will directly target cells that express the specific autoantigen and, with mechanisms similar to those used by NK cell, induce target cell apoptosis.Citation83 All these processes will lead to the deletion of a specific autoantigen-bearing cell population.
What is known about killer cells and autoimmunity in promoting apoptosis in emphysema?
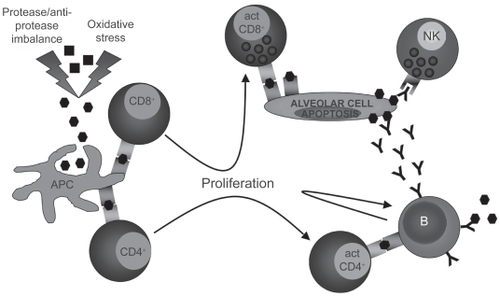
There are evidences that emphysema may have an autoimmune component. Autoreactive antibodies with avidity for pulmonary epithelial cells were found in the serum of about 70% of subjects with emphysema when only 10% of normal smokers or nonsmokers were positive.Citation84 Moreover, in vitro peripheral blood mononuclear cells cytotoxicity against pulmonary epithelial cells was stronger in presence of plasma from emphysematous subjects than from normal subjects,Citation84 suggesting that autoreactive antibodies may initiate immune cell-mediated apoptosis. Moreover, a model of autoimmune emphysema () revealed that immune response against lung endothelial cells can lead to alveolar endothelial and epithelial cell apoptosis and cause emphysema.Citation85 Antibodies against elastin protein have also been found in the serum of emphysematous subjectCitation86 as well as a higher number of B cells secreting antielastin antibodies in the emphysematous lung.Citation86 In the same study, they also found that peripheral blood CD4+ T lymphocytes from emphysematous subjects were releasing more gamma interferon (IFN-γ) and interleukin-10 (IL-10) than those from normal subjects upon in vitro elastin exposure.Citation86 Oligoclonal CD4+ T lymphocytes able to proliferate in vitro upon interleukin-2 stimulationCitation87,Citation88 as well as oligoclonal CD8+ T lymphocytesCitation89 were found in the lung of subjects with emphysema. Moreover, CD8+ T lymphocytes from the lung of COPD subjects expressed higher levels of perforin and cytotoxic activity than those from controls,Citation90.Citation91 phenomenon that is not observed in the peripheral blood.Citation92 All these data suggest the existence of a local antigen-driven cellular immunity in the lung of emphysematous subjects ().
Figure 6 Induction of an autoimmune response against immunogenic self-antigens after protease and oxidative stress-induced modifications. Antigen-presenting cell (APC). Self-antigen (▪), modified self-antigen (⬢), antibody against modified self-antigen (Y).
What we need to know
Which cell type is dying?
There are three major structural cell types in the alveolar tissue: Type I alveolar cells, Type II alveolar cells, and endothelial cells. Type I alveolar is the most abundant cell type found in the alveolar tissue. They are responsible for the maintenance of the alveolar structure and gas diffusion toward alveolar capillaries. Type II alveolar cells are the major source of surfactant proteins and are the progenitor of Type I alveolar cells. Endothelial cells are in charge of lung perfusion and, with Type I alveolar cells, of gas diffusion between alveoli and blood. As all these cell types are important to maintain alveolar tissue integrity, the accelerated loss of one or another will have direct effect on the whole alveolar tissue. The technique of choice to detect apoptotic cells in the emphysematous lung is terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) analysis. Unlike tissue lysat-based analyses that only give a general view of the tissue apoptosis level (eg, Ligation-mediated polymerase chain reaction [LM-PCR], caspase-3 activity, poly(ADP-ribose) polymerase [PARP]) cleavage), TUNEL allows a precise identification of the cells that undergo apoptosis. However, despite comparative TUNEL analyses of normal and emphysematous lung (), no real consensus has been established on which cell type seems to die predominantly from apoptosis in the emphysematous lung.
According to the different hypothesis that attempt to explain the increased alveolar apoptosis that occurs in emphysema, different cell types are targeted as the earliest cell type touched by apoptosis. The VEGF hypothesis points mainly at endothelial cellsCitation40 when the proteases imbalance,Citation14,Citation27 oxidative stress,Citation62 and TRAIL hypothesisCitation74 tend to point at epithelial cells. AutoimmunityCitation84,Citation85 seems to be touching both endothelial and epithelial cells. If we consider the conformation of the alveoli, epithelial alveolar cells are exposed directly to cigarette smoke and may be acting as a “cellular shield” for endothelial cells. It is then logic to assume that most of the damages induced by cigarette smoke will be first done to epithelial rather than endothelial cells. There is a lot of in vitro data on the effects of cigarette smoke on signaling pathways of alveolar epithelial and endothelial cells. However, no data are currently available on the crosstalk of alveolar epithelial cells exposed to cigarette smoke with endothelial cells. This would be extremely relevant to understand how damaged alveolar epithelial cells may affect the functions and the faith of endothelial cells, and would give important information on which cell type may be susceptible to undergo apoptosis first.
When does it die?
Until now, publications on apoptosis in emphysema compared the apoptosis levels in the lung of emphysematous subjects with well-developed disease with those of nonem-physematous subjects. From this, we can say that apoptosis levels are higher in emphysematous subjects despite smoking cessation and we can suppose that apoptosis is involved in the progression of the disease. We also know that alveolar epithelial cells’ proliferation is increased, but does not seem to compensate for cell loss (apoptosis/proliferation imbalance). However, we do not know if the apoptotic mechanisms that have been described in this review are involved in the initiation of the disease or only in the progression once the disease has started. When does apoptosis overcome proliferation? When do proteases overcome anti-proteases? When do VEGF and VEGFR2 levels go down? When do p53, Bax/Bcl-xL ratio, and TRAIL-R go up? And finally, when does autoimmunity get in play? Answering these questions is not an easy task. In order to do so, we would need to study for a long period of time smoking subjects that are susceptible to develop emphysema but that have not developed it yet. However, since there is no reliable method to identify these subjects yet, animal models and in vitro cell culture are the only tools that can help to understand mechanisms involved in emphysema initiation rather than on its progression.
What is responsible for cell death?
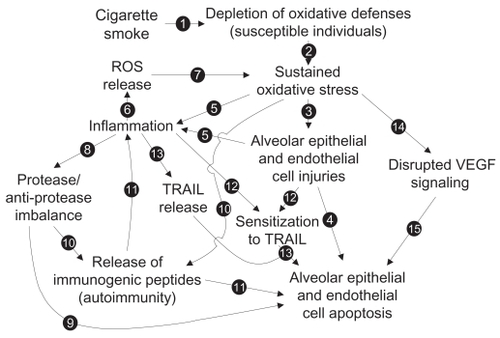
Looking for only one mechanism to explain the increased alveolar apoptosis would be an important mistake. Proteases, oxidative stress, decreased VEGF, autoimmunity, and TRAIL-mediated apoptosis are probably all involved at different levels in the increased apoptosis levels in emphysema. The elaboration of a complete model that would integrate all mechanisms and link them to each other may be extremely relevant to guide research on the increased apoptotic events observed in emphysema. Here, we propose a model () built from the studies we previously discussed. Emerging concepts or hypothesis such as alveolar apoptosis induced by neutrophils-derived defensins,Citation93,Citation94 link between innate immunity and oxidative stress through Toll-like receptor 4 (TLR4) tone,Citation95 and reduced or impared apoptotic cell clearance (efferocytosis),Citation96 even if very interesting and promising ways of research, were not included in the model.
Figure 7 Proposed model of the mechanisms involved in the increased alveolar apoptosis observed in emphysema (see text for details).
Global model to explain increased apoptosis in emphysema
As we know, 90% of emphysema cases are developed following cigarette smoke exposure. We also know that, among all its toxic power, cigarette smoke contains powerful oxidants. In some individuals with weaker oxidative defenses (genetic feature), chronic cigarette smoke exposure will lead to oxidative defense depletion (see Arrow [1] in ) and the establishment of a sustained oxidative stress [2] leading to lethal injuries to alveolar cells [3] and an increase in alveolar cell apoptosis [4]. Oxidative stress can also be responsible for the initiation of an inflammatory process, either from direct immune cell activation or through the release of inflammatory mediators by injured alveolar cells [5]. In the present model, the following steps are very important to establish the chronic state of the disease. Activated inflammatory cells, mainly neutrophils and macrophages, are well known to be powerful reactive oxygen species (ROS) producers [6]. ROS released from inflammatory cells may then be involved in the establishment of a cigarette smoke-independent lung oxidative stress [7], as oxidative stress remains in the lung of emphysematous subjects after smoking cessation. Activated neutrophils and macrophages can also release great amount of proteases that then lead to lung protease/anti-protease imbalance [8] and induce alveolar epithelial and endothelial apoptosis [9]. Protease hyperactivity and oxidative stress could also be responsible for the release of immunogenic peptides [10] and lead to the development of antigen-driven immune response (antigen-specific activation of B cells and CD4+ and CD8+ T cells) and then promote alveolar cell apoptosis [11]. Inflammatory mediators, like TNF, and oxidative stress (cell injuries) can also modulate elements involved in alveolar cell sensitivity to TRAIL-mediated apoptosis (TRAIL receptors, p53, and Bax levels) [12] and switch alveolar cells from a TRAIL-mediated apoptosis resistant state to a sensitive state and lead to increased alveolar cell apoptosis [13]. Oxidative stress may also be responsible for the disruption of VEGF signaling [14], leading to alveolar endothelial cell apoptosis, and, through a lack of physical support, promoting alveolar epithelial cell apoptosis [15].
Based on this model of hypothesis, tools such as DNA and protein microarrays and softwares for cellular pathway interactions analysis will help to see in a more global way the pathways involved in the disease development while new primary cell isolation and culture will give better cellular models. All this will increase the comprehension of the different mechanisms involved in emphysema pathogenesis and ultimately lead the elaboration of treatments that will stop the development or the progression of the disease.
Disclosure
The authors report no conflicts of interest in this work. MCM is the recipient of a studentship from the Fonds de Recherche en Santé du Québec.
References
- MurrayCJLopezADAlternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease StudyLancet1997349149815049167458
- HoggJCPathophysiology of airflow limitation in chronic obstructive pulmonary diseaseLancet200436470972115325838
- MulgrewATTaggartCCMcElvaneyNGAlpha-1-antitrypsin deficiency: current conceptsLung200718519120117562108
- QuintJKWedzichaJAThe neutrophil in chronic obstructive pulmonary diseaseJ Allergy Clin Immunol20071191065107117270263
- BarnesPJAlveolar macrophages in chronic obstructive pulmonary disease (COPD)Cell Mol Biol (Noisy-le-grand)200450OL627OL63715579256
- CosioMGMajoJCosioMGInflammation of the airways and lung parenchyma in COPD: role of T cellsChest2002121160S165S12010846
- StrasserAO’ConnorLDixitVMApoptosis signalingAnnu Rev Biochem20006921724510966458
- RoosWPKainaBDNA damage-induced cell death by apoptosisTrends Mol Med20061244045016899408
- ElmoreSApoptosis: a review of programmed cell deathToxicol Pathol20073549551617562483
- ImaiKMercerBASchulmanLLCorrelation of lung surface area to apoptosis and proliferation in human emphysemaEur Respir J20052525025815684288
- YokohoriNAoshibaKNagaiAIncreased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysemaChest200412562663214769747
- YangJJKettritzRFalkRJApoptosis of endothelial cells induced by the neutrophil serine proteases proteinase 3 and elastaseAm J Pathol1996149161716268909251
- GinzbergHHShannonPTSuzukiTLeukocyte elastase induces epithelial apoptosis: role of mitochondial permeability changes and AktAm J Physiol Gastrointest Liver Physiol2004287G286G29815194561
- SuzukiTMoraesTJVachonEProteinase-activated receptor-1 mediates elastase-induced apoptosis of human lung epithelial cellsAm J Respir Cell Mol Biol20053323124715891109
- PetracheIFijalkowskaIMedlerTRalpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosisAm J Pathol20061691155116617003475
- AldonyteRHutchinsonETJinBEndothelial alpha-1-antitrypsin attenuates cigarette smoke induced apoptosis in vitroCOPD2008515316218568839
- ChircoRLiuXWJungKKNovel functions of TIMPs in cell signalingCancer Metastasis Rev2006259911316680576
- ErikssonSPulmonary emphysema and alpha1-antitrypsin deficiencyActa Med Scand196417519720514124635
- GrossPPfitzerEATolkerEExperimental emphysema: its production with papain in normal and silicotic ratsArch Environ Health196511505814312390
- YoshiokaABetsuyakuTNishimuraMExcessive neutrophil elastase in bronchoalveolar lavage fluid in subclinical emphysemaAm J Respir Crit Care Med1995152212721328520785
- TakeyabuKBetsuyakuTNishimuraMCysteine proteinases and cystatin C in bronchoalveolar lavage fluid from subjects with subclinical emphysemaEur Respir J199812103310399863993
- Segura-ValdezLPardoAGaxiolaMUpregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPDChest200011768469410712992
- ImaiKDalalSSChenESHuman collagenase (matrix metalloproteinase-1) expression in the lungs of patients with emphysemaAm J Respir Crit Care Med200116378679111254539
- OhnishiKTakagiMKurokawaYMatrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysemaLab Invest199878107710879759652
- BabusyteAStravinskaiteKJerochJPatterns of airway inflammation and MMP-12 expression in smokers and ex-smokers with COPDRespir Res200788118001475
- StonePJLuceyECCaloreJDDefenses of the hamster lung against human neutrophil and porcine pancreatic elastaseRespiration1988541152469116
- LuceyECKeaneJKuangPPSeverity of elastase-induced emphysema is decreased in tumor necrosis factor-alpha and interleukin-1beta receptor-deficient miceLab Invest200282798511796828
- ZhengTKangMJCrothersKRole of cathepsin S-dependent epithelial cell apoptosis in IFN-gamma-induced alveolar remodeling and pulmonary emphysemaJ Immunol20051748106811515944319
- FerraraNGerberHPLeCouterJThe biology of VEGF and its receptorsNat Med2003966967612778165
- GerberHPDixitVFerraraNVascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cellsJ Biol Chem199827313313133169582377
- GuptaKKshirsagarSLiWVEGF prevents apoptosis of human microvascular endothelial cells via opposing effects on MAPK/ERK and SAPK/JNK signalingExp Cell Res199924749550410066377
- GerberHPMcMurtreyAKowalskiJVascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activationJ Biol Chem199827330336303439804796
- CardoneMHRoyNStennickeHRRegulation of cell death protease caspase-9 by phosphorylationScience1998282131813219812896
- del PesoLGonzalez-GarciaMPageCInterleukin-3-induced phosphorylation of BAD through the protein kinase AktScience19972786876899381178
- TranJRakJSheehanCMarked induction of the IAP family antiapoptotic proteins survivin and XIAP by VEGF in vascular endothelial cellsBiochem Biophys Res Commun199926478178810544009
- KanazawaHRole of vascular endothelial growth factor in the pathogenesis of chronic obstructive pulmonary diseaseMed Sci Monit200713RA18919517968307
- VoelkelNFVandivierRWTuderRMVascular endothelial growth factor in the lungAm J Physiol Lung Cell Mol Physiol2006290L209L22116403941
- MarwickJAStevensonCSGiddingsJCigarette smoke disrupts VEGF165-VEGFR-2 receptor signaling complex in rat lungs and patients with COPD: morphological impact of VEGFR-2 inhibitionAm J Physiol Lung Cell Mol Physiol2006290L89790816361360
- KasaharaYTuderRMCoolCDEndothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysemaAm J Respir Crit Care Med200116373774411254533
- KasaharaYTuderRMTaraseviciene-StewartLInhibition of VEGF receptors causes lung cell apoptosis and emphysemaJ Clin Invest20001061311131911104784
- TuderRMZhenLChoCYOxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockadeAm J Respir Cell Mol Biol200329889712600822
- DemuraYTaraseviciene-StewartLScerbaviciusRN-acetylcysteine treatment protects against VEGF-receptor blockade-related emphysemaCOPD20041253216997736
- Committee ahSMechanisms and limits of induced postnatal lung growthAm J Respir Crit Care Med200417031934315280177
- AcarreguiMJPenistenSTGossKLVascular endothelial growth factor gene expression in human fetal lung in vitroAm J Respir Cell Mol Biol19992014239870913
- ShifrenJLDoldiNFerraraNIn the human fetus, vascular endothelial growth factor is expressed in epithelial cells and myocytes, but not vascular endothelium: implications for mode of actionJ Clin Endocrinol Metab1994793163228027247
- NgYSRohanRSundayMEDifferential expression of VEGF isoforms in mouse during development and in the adultDev Dyn200122011212111169844
- RyterSWKimHPHoetzelAMechanisms of cell death in oxidative stressAntioxid Redox Signal20079498917115887
- JingYDaiJChalmers-RedmanRMArsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathwayBlood1999942102211110477740
- SerranoJPalmeiraCMKuehlDWCardioselective and cumulative oxidation of mitochondrial DNA following subchronic doxorubicin administrationBiochim Biophys Acta1999141120120510216166
- HugHStrandSGrambihlerAReactive oxygen intermediates are involved in the induction of CD95 ligand mRNA expression by cyto-static drugs in hepatoma cellsJ Biol Chem199727228191281939353266
- SuzukiSHiguchiMProskeRJImplication of mitochon-dria-derived reactive oxygen species, cytochrome C and caspase-3 in N-(4-hydroxyphenyl)retinamide-induced apoptosis in cervical carcinoma cellsOncogene1999186380638710597238
- CarvalhoHEvelsonPSigaudSMitogen-activated protein kinases modulate H(2)O(2)-induced apoptosis in primary rat alveolar epithelial cellsJ Cell Biochem20049250251315156562
- RahmanIBiswasSKKodeAOxidant and antioxidant balance in the airways and airway diseasesEur J Pharmacol200653322223916500642
- PryorWACigarette smoke radicals and the role of free radicals in chemical carcinogenicityEnviron Health Perspect1997105Suppl 48758829255574
- WillemseBWPostmaDSTimensWThe impact of smoking cessation on respiratory symptoms, lung function, airway hyperrespon-siveness and inflammationEur Respir J20042346447615065840
- RussellRECulpittSVDeMatosCRelease and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary diseaseAm J Respir Cell Mol Biol20022660260911970913
- RussellREThorleyACulpittSVAlveolar macrophage-mediated elastolysis: roles of matrix metalloproteinases, cysteine, and serine proteasesAm J Physiol Lung Cell Mol Physiol2002283L8677312225964
- LacosteJYBousquetJChanezPEosinophilic and neutrophilic inflammation in asthma, chronic bronchitis, and chronic obstructive pulmonary diseaseJ Allergy Clin Immunol1993925375488409114
- KeatingsVMBarnesPJGranulocyte activation markers in induced sputum: comparison between chronic obstructive pulmonary disease, asthma, and normal subjectsAm J Respir Crit Care Med19971554494539032177
- GwinnMRVallyathanVRespiratory burst: role in signal transduction in alveolar macrophagesJ Toxicol Environ Health B Crit Rev20069273916393868
- DahlgrenCKarlssonARespiratory burst in human neutrophilsJ Immunol Methods199923231410618505
- RahmanIvan SchadewijkAACrowtherAJ4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200216649049512186826
- MalhotraDThimmulappaRNavas-AcienADecline in NRF2 Regulated Antioxidants in COPD Lungs due to Loss of its Positive Regulator DJ-1Am J Respir Crit Care Med2008
- FalschlehnerCEmmerichCHGerlachBTRAIL signalling: decisions between life and deathInt J Biochem Cell Biol2007391462147517403612
- BonavidaBNgCPJazirehiASchillerGMizutaniYSelectivity of TRAIL-mediated apoptosis of cancer cells and synergy with drugs: the trail to non-toxic cancer therapeuticsInt J Oncol19991579380210493964
- ZhangLFangBMechanisms of resistance to TRAIL-induced apoptosis in cancerCancer Gene Ther20051222823715550937
- HuHJiangCSchusterTInorganic selenium sensitizes prostate cancer cells to TRAIL-induced apoptosis through superoxide/p53/Bax-mediated activation of mitochondrial pathwayMol Cancer Ther200651873188216891474
- FanQLZouWYSongLHSynergistic antitumor activity of TRAIL combined with chemotherapeutic agents in A549 cell lines in vitro and in vivoCancer Chemother Pharmacol20055518919615290100
- FreseSBrunnerTGuggerMEnhancement of Apo2L/TRAIL (tumor necrosis factor-related apoptosis-inducing ligand)-induced apoptosis in non-small cell lung cancer cell lines by chemotherapeutic agents without correlation to the expression level of cellular protease caspase-8 inhibitory proteinJ Thorac Cardiovasc Surg200212316817411782771
- UbertiDFerrari-ToninelliGBoniniSABlockade of the tumor necrosis factor-related apoptosis inducing ligand death receptor DR5 prevents beta-amyloid neurotoxicityNeuropsychopharmacology20073287288016936710
- RobertsonNMZangrilliJGSteplewskiADifferential expression of TRAIL and TRAIL receptors in allergic asthmatics following segmental antigen challenge: evidence for a role of TRAIL in eosinophil survivalJ Immunol20021695986599612421985
- RobertsonNMRosemillerMLindemeyerRGTRAIL in the airwaysVitam Horm20046714916715110176
- MirandolaPPontiCGobbiGActivated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicityBlood20041042418242415205263
- MorissetteMCVachon-BeaudoinGParentJIncreased p53 level, Bax/Bcl-x(L) ratio, and TRAIL receptor expression in human emphysemaAm J Respir Crit Care Med200817824024718511705
- AndersenMHSchramaDThor StratenPCytotoxic T cellsJ Invest Dermatol2006126324116417215
- FaragSSCaligiuriMAHuman natural killer cell development and biologyBlood Rev20062012313716364519
- AtassiMZCasaliPMolecular mechanisms of autoimmunityAutoimmunity20084112313218324481
- GaiplUSKuhnASheriffAClearance of apoptotic cells in human SLECurr Dir Autoimmun2006917318716394661
- ScofieldRHAutoantibodies as predictors of diseaseLancet20043631544154615135604
- GaoHNeffTWardPARegulation of lung inflammation in the model of IgG immune-complex injuryAnnu Rev Pathol2006121524218039114
- SmythMJCretneyEKellyJMActivation of NK cell cytotoxicityMol Immunol20054250151015607806
- ElsonCJBarkerRNHelper T cells in antibody-mediated, organ-specific autoimmunityCurr Opin Immunol20001266466911102770
- WalterUSantamariaPCD8+ T cells in autoimmunityCurr Opin Immunol20051762463116226438
- Feghali-BostwickCAGadgilASOtterbeinLEAutoantibodies in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200817715616317975205
- Taraseviciene-StewartLScerbaviciusRChoeKHAn animal model of autoimmune emphysemaAm J Respir Crit Care Med200517173474215563631
- LeeSHGoswamiSGrudoAAntielastin autoimmunity in tobacco smoking-induced emphysemaNat Med20071356756917450149
- SullivanAKSimonianPLFaltaMTOligoclonal CD4+ T cells in the lungs of patients with severe emphysemaAm J Respir Crit Care Med200517259059615937291
- SullivanAKSimonianPLFaltaMTActivated oligoclonal CD4+ T cells in the lungs of patients with severe emphysemaProc Am Thorac Soc2006348616921120
- KornSWiewrodtRWalzYCCharacterization of the interstitial lung and peripheral blood T cell receptor repertoire in cigarette smokersAm J Respir Cell Mol Biol20053214214815539458
- ChrysofakisGTzanakisNKyriakoyDPerforin expression and cytotoxic activity of sputum CD8+ lymphocytes in patients with COPDChest2004125717614718423
- HodgeSHodgeGNairnJIncreased airway granzyme b and perforin in current and ex-smoking COPD subjectsCOPD2006317918717361498
- MorissetteMCParentJMilotJPerforin, granzyme B, and FasL expression by peripheral blood T lymphocytes in emphysemaRespir Res200786217822550
- LiuCYLinHCYuCTThe concentration-dependent chemokine release and pro-apoptotic effects of neutrophil-derived alpha-defensin-1 on human bronchial and alveolar epithelial cellsLife Sci20078074975817141275
- AarbiouJTjabringaGSVerhooselRMMechanisms of cell death induced by the neutrophil antimicrobial peptides alpha-defensins and LL-37Inflamm Res20065511912716673155
- Taraseviciene-StewartLVoelkelNFMolecular pathogenesis of emphysemaJ Clin Invest200811839440218246188
- VandivierRWHensonPMDouglasISBurying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung diseaseChest20061291673168216778289
- CalabreseFGiacomettiCBegheBMarked alveolar apoptosis/proliferation imbalance in end-stage emphysemaRespir Res200561415705190
- AoshibaKYokohoriNNagaiAAlveolar wall apoptosis causes lung destruction and emphysematous changesAm J Respir Cell Mol Biol20032855556212707011
- BartalesiBCavarraEFineschiSDifferent lung responses to cigarette smoke in two strains of mice sensitive to oxidantsEur Respir J200525152215640318
- KuoWHChenJHLinHHInduction of apoptosis in the lung tissue from rats exposed to cigarette smoke involves p38/JNK MAPK pathwayChem Biol Interact2005155314215970277
- PetracheINatarajanVZhenLCeramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in miceNat Med20051149149815852018
- BrassDMHollingsworthJWCinqueMChronic LPS Inhalation Causes Emphysema-like Changes in Mouse Lung that is Associated with ApoptosisAm J Respir Cell Mol Biol2008