Abstract
Despite the availability of over 20 antiepileptic drugs, about 30% of epileptic patients do not achieve seizure control. Thus, identification of additional molecules targeting novel molecular mechanisms is a primary effort in today’s antiepileptic drug research. This paper reviews the pharmacological development of retigabine, an antiepileptic drug with a novel mechanism of action, namely the activation of voltage-gated potassium channels of the Kv7 subfamily. These channels, which act as widespread regulators of intrinsic neuronal excitability and of neurotransmitter-induced network excitability changes, are currently viewed among the most promising targets for anticonvulsant pharmacotherapy. In particular, the present work reviews the pathophysiological role of Kv7 channels in neuronal function, the molecular mechanisms involved in the Kv7 channel-opening action of retigabine, the activity of retigabine in preclinical in vitro and in vivo studies predictive of anticonvulsant activities, and the clinical status of development for this drug as an add-on treatment for pharmacoresistant epilepsy. Particular efforts are devoted to highlighting the potential advantages and disadvantages of retigabine when compared with currently available compounds, in order to provide a comprehensive assessment of its role in therapy for treatment-resistant epilepsies.
Introduction
Epilepsy is a disorder characterized by seizures due to recurrent, spontaneous episodes of aberrant synchronization in neuronal networks.Citation1 It is the most common human neurologic disorder, with about 50 million people affected worldwide, an estimated prevalence of about 0.4%–1.0%, and an estimated incidence of 20–70 new cases/10.000 individuals.Citation2,Citation3
Aberrant neuronal synchronization in epilepsy can remain focal, spread to other sites, or engage all cortical regions simultaneously. According to the most recent International League Against Epilepsy (ILAE) classification, epileptic seizures can be classified as generalized or partial.Citation4 Generalized epileptic seizures engage bilaterally distributed networks which can include cortical and subcortical structures, but do not necessarily include the entire cortex and can be asymmetric. On the other hand, focal epileptic seizures originate within networks limited to one hemisphere, may be discretely localized or more widely distributed, and may originate in subcortical structures. The latest classification of the ILAE provides new concepts for the current understanding of epilepsies focusing on basic neurobiologic mechanisms rather than the clinical manifestations of the disease.
Except for a minority of cases which can be approached surgically, epilepsy treatment relies on the use of antiepileptic drugs to achieve symptomatic seizure control. Over 20 antiepileptic drugs, each with exquisite pharmacokinetics, spectrum of antiepileptic activity, and toxicity are currently available for this purpose. The mechanisms of action of most currently available antiepileptic drugs is restricted to voltage-gated sodium channels (phenytoin, carbamazepine), components of the γ-aminobutyric acid (GABA) system, including: GABAA receptors (phenobarbital, benzodiazepines), the GAT-1 GABA transporter (tiagabine), and GABA transaminase (vigabatrin); voltage-gated calcium channels (valproate, ethosuximide); and synaptic proteins involved in neurotransmitter release, such as SV2A (levetiracetam, brivaracetam).
Despite such a wide therapeutic armamentarium, it is currently estimated that about 30% of epileptic patients do not receive satisfactory treatment; this figure is even higher if one considers partial epilepsies in which developmental alterations leading to identifiable changes in brain structure are primary underlying causes. Thus, to improve some of the well known limitations of current anticonvulsant treatment, one of the most ambitious goals in today’s antiepileptic research is the identification of additional molecules targeting novel molecular mechanisms involved in neuronal excitability control. The present paper reviews the preclinical and clinical development of retigabine, a novel antiepileptic drug targeting voltage-gated potassium (K+) channels of the Kv7 subfamily. These channels, by acting as widespread regulators of intrinsic neuronal excitability and of neurotransmitter-induced network excitability changes, are currently viewed among the most promising “druggable” targets for anticonvulsant pharmacotherapy.
Neuronal potassium channels of the Kv7 subfamily
Potassium channels with distinct subcellular localization, biophysical properties, modulation, and pharmacologic profile are primary regulators of intrinsic electrical properties of neurons and their responsiveness to synaptic inputs.Citation5 An increase in membrane conductance to K+ ions causes neuronal hyperpolarization and, in most cases, reduces firing frequency, exerting a strong inhibitory function on neuronal excitability.
Among voltage-gated K+ currents, the M-current (IKM) is a primary transducer of changes in the extracellular chemical composition into modification of the intrinsic neuronal properties. IKM was first identified in amphibian peripheral neuronsCitation6 but later found to be widely distributed also in the mammalian peripheral and central nervous system.Citation7 IKM is a low-threshold, slowly activating and deactivating, and noninactivating voltage-dependent K+ current which limits repetitive firing and causes spike frequency adaptation.Citation8 Activation of several receptors linked to pertussis toxin-in-sensitive G proteins of the Gq/11 family, including M1, M3, and M5 subtypes of muscarinic receptors (hence its definition),Citation9 can control neuronal excitability by suppressing IKM.Citation10,Citation11
The identification of the molecular basis of IKM was achieved in the late 1990s upon the discovery that benign familial neonatal seizures, a rare autosomal dominant idiopathic epilepsy of the newborn characterized by the occurrence of focal or generalized seizures starting around day 3 of postnatal life and spontaneously disappearing after few weeks or months, was associated with mutations in Kv7.2Citation12,Citation13 or, more rarely, Kv7.3,Citation14 both genes encoding for K+ channel subunits.
Kv7.2 and Kv7.3 belong to the Kv7 (KCNQ) subfamily of K+ channel genes. This subfamily is composed of five members, also including Kv7.1 which is expressed in several non-neuronal tissues including the heart, where it underlies the IKs current involved in the late repolarization phase of the cardiac action potential,Citation15 and in epithelial cells of several organs; Kv7.4, which is expressed in primary sensory cells in the inner ear and in neurons of the central auditory pathway,Citation16,Citation17 as well as in skeletal muscle,Citation18 and visceral and vascular smooth muscle;Citation19 and Kv7.5, whose transcripts have been detected in the brain, as well as in skeletal and smooth muscle cells.Citation20 Because of their expression in the nervous system, Kv7.2–Kv7.5 subunits are commonly referred to as “neural” Kv7 subunits.Citation21
Most of the biophysical and pharmacologic properties of IKM in neurons are recapitulated upon heteromeric expression of Kv7.2 and Kv7.3 subunits.Citation22,Citation23 Functional studies in heterologous expression systems revealed that mutations in Kv7.2 and Kv7.3 genes causing benign familial neonatal seizures decrease the ability of IKM to suppress neuronal excitability.Citation24
Genetically-modified animal models further confirmed the specific involvement of Kv7.2 and Kv7.3 in brain excitability control. Heterozygous mice carrying a targeted deletion of the Kv7.2 gene displayed an increased sensitivity to proconvulsant stimuli.Citation25 In addition, transgenic mice conditionally expressing mutant Kv7.2 subunits causing dominant-negative suppression of IKM function showed signs of increased neuronal excitability in hippocampal CA1 pyramidal neurons, spontaneous seizures, behavioral hyperactivity, and deficits in hippocampus-dependent spatial memory.Citation26 A third model of Kv7.2 deficiency is represented by stz1 mice carrying an ethylnitrosurea-induced deletion of a large genomic region encompassing Kv7.2; heterozygous stz1 mice, similar to Kv7.2 knockout mice, have a reduced electroconvulsive threshold and an increased sensitivity to the convulsant pentylenetetrazole, together with structural abnormalities in the hippocampal formation.Citation27 More recently, mouse models carrying missense mutations that underlie human benign familial neonatal seizures into the orthologous murine Kv7.2 and Kv7.3 genes have been established. While heterozygous mice exhibited reduced thresholds to electrically induced seizures compared with wild-type littermate mice, homozygous mutant mice showed early-onset spontaneous generalized tonic-clonic seizures that triggered neuronal plasticity without hippocampal mossy fiber sprouting.Citation28
Kv7 channel openers: retigabine and novel mechanism of action of anticonvulsants
Potential role of IKM openers
Given the previously mentioned functional and genetic evidence, activation of neuronal IKM is currently considered as a primary strategy for pharmacologic intervention in epilepsy and other human diseases, such as migraine and chronic pain, all conditions in which neuronal hyperexcitability appears to play a fundamental pathogenetic role. In all these diseases, hyperpolarization of the resting membrane potential and decreased action potential generation, prompted by the activation of neuronal K+ currents in excitatory neurons, may result in an effective therapeutic strategy. As a matter of fact, compounds with IKM opening abilities have undergone extensive preclinical and clinical investigation as potential antiepileptics and analgesics, even before their molecular mechanism of action was discovered.
History of retigabine
Retigabine (D-23129, chemical name N-(2-amino-4- (4-fluorobenzylamino)-phenyl)-carbamic acid ethyl ester) is the prototype IKM opener. Retigabine is a structural analog of flupirtine (D-9998), a triaminopyridine successfully used in clinical practice in some European countries since 1984 as a centrally acting nonopioid analgesic, also provided with muscle relaxant and neuroprotective actions,Citation29,Citation30 but devoid of appreciable anti-inflammatory and antipyretic activity.Citation31
In the Antiepileptic Drug Development program supported by the US National Institutes of Health using in vivo animal screening strategies, flupirtine was shown in the late 1980s to have anticonvulsant activity, although at doses higher than those producing analgesia. Structure-activity optimization of flupirtine yielded retigabine, which showed more effective anticonvulsant activity when compared with flupirtine, being active in nearly every animal seizure model, with an overall profile different from any other known drug.Citation32 The broad profile of efficacy in animal models already suggested that the mechanism(s) involved in the anticonvulsant actions of retigabine were distinct from those previously known. In the late 1990s, retigabine was shown to activate voltage-dependent neuronal K+ currents at concentrations at least 10–30 times lower than those producing a potentiation of GABAA-mediated currents and a weak blocking action on ligand- (kainate and NMDA receptors) and voltage-gated (sodium and calcium) channels.Citation33,Citation34 However, these studies failed to identify the precise nature of the K+ current targeted by retigabine; this was only possible when drug effects were evaluated on native IKM in neuronal cells and on cloned Kv7.2 and Kv7.3 channels in heterologous expression systems; these experiments, clearly demonstrated that IKM formed by Kv7.2/3 subunits was the major molecular target for retigabine.Citation35–Citation37 In addition to IKM opening, both retigabine and flupirtine have been shown to possess antioxidant properties in vitro,Citation29,Citation38 a pharmacological property which might explain at least some of the neuroprotective effects shown by both compounds.
Mechanism of action of retigabine
The biophysical mechanism responsible for retigabine-induced IKM potentiation is rather complex, and involves a combination of two factors, ie, a hyperpolarizing shift in the voltage-dependence of the channel activation process and an increase in the maximal opening probability of these channels. Both these effects can contribute by a variable amount in channels formed by different combinations of Kv7 subunits. In fact, the hyperpolarizing shift is maximal for Kv7.3 (−43 mV), intermediate for Kv7.2 (−24 mV), smaller for Kv7.4 (−14 mV), and absent for Kv7.5 homomeric channels.Citation39,Citation40 In Kv7.5 channels, instead, retigabine markedly increases the current irrespective of the membrane potential; by contrast, in Kv7.2 and Kv7.2/3 combinations, the maximal amount of current elicited by strong depolarization is not affected, whereas homomeric Kv7.4 channels show both the shift in voltage-dependent opening probability and an increase in maximal conductance.Citation39 These changes are accompanied by a variable degree of drug-induced delay in channel closure (deactivation), suggesting that the primary molecular consequence of the interaction of retigabine with the channel protein is a stabilization of the channel pore in the open conformation.
Retigabine-induced enhancement of IKM hyperpolarizes the neuronal resting membrane potential, leading to an inhibition of spontaneous or synaptically-triggered neuronal activity. Intriguingly, some mutations causing benign familial neonatal seizures in Kv7.2 prompt functional changes in IKM biophysical properties which are opposite to those caused by retigabine.Citation41 As previously mentioned, retigabine affects IKM underlined by all different Kv7 subunits. However, the potency of retigabine in causing activation of the channels formed by distinct Kv7 subunits appears slightly different, with, in most cases, Kv7.2 and Kv7.3 showing higher (EC50 < 1 μM) and Kv7.4 and Kv7.5 lower (EC50 > 1 μM) sensitivity to the drug. Nevertheless, because these differences are quantitatively rather small, retigabine is currently regarded as a “nonselective” neural Kv7 channel opener. Noticeably, none of these actions are prompted by retigabine in cardiac Kv7.1 channels, an important observation to interpret the lack of cardiac toxicity shown by this compound.
Other IKM activators
The described results firmly establish IKM as a crucial target for pharmacological intervention in hyperexcitability diseases, and have led to an explosion of interest in the identification of congeners additional to flupirtine and retigabine acting as IKM activators. Among these, phenamates, such as meclofenamic acid and diclofenac, well known blockers of the cyclooxygenase enzymes, have been described to act as Kv7.2/3 K+ channel openers and to show robust antiepileptic properties in vivo.Citation42 Although these pharmacological actions require rather high drug concentrations, these compounds have been suggested to represent novel drug templates for the treatment of neuronal hyperexcitability diseases, including epilepsy, migraine, or neuropathic pain. Indeed, more recent studies demonstrated that diclofenac derivatives with poor (NH6)Citation43 or absent (NH29)Citation44 cyclooxygenase-blocking activity reduced neuronal excitability and/or showed anticonvulsant effects in murine models of seizures.
The interest in IKM (and in particular in Kv7.2/3 subunits) as a possible target in the treatment of epilepsy and other hyperexcitability diseases has also led to the development of ICA-27243, a compound belonging to the chemical class of substituted benzamides. In contrast with retigabine, which does not discriminate between channels formed by different Kv7 subunits, ICA-27243 showed high selectivity for current elicited by Kv7.2/Kv7.3 heterotetramers, with an EC50 of 0.4 μM, while its potency on Kv7.4 or Kv7.3/Kv7.5 channels was 20-fold and >100-fold lower, respectively.Citation45 Moreover, ICA-27243 exhibited anticonvulsant properties in preclinical models of seizures.Citation46 The search for new molecules with agonistic activity on Kv7 channels prompted Xiong et al to demonstrate that zinc pyrithione is an activator of heterologous expressed and native IKM.Citation47 Zinc pyrithione potentiated K+ currents elicited both by cardiac Kv7.1 and neural Kv7.2-5 subunits, except those sustained by Kv7.3 channels; in addition, its agonistic action was noncompetitive with that exerted by retigabine, giving rise to combinatorial effects when applied together.Citation48 Other molecules able to enhance Kv7-mediated current include BMS-204352, originally developed as an opener of Ca2+- and voltage-gated K+ channels, and the acrylamide compound, (S)-1. Although both these molecules can open channels formed by all neural Kv7 subunits, they display a noticeably stronger action on channels formed by Kv7.4 subunits (BMS- 204352)Citation49,Citation50 or on Kv7.4 and Kv7.5 subunits [(S)-1].Citation51
The binding site for retigabine on neuronal Kv7 channels
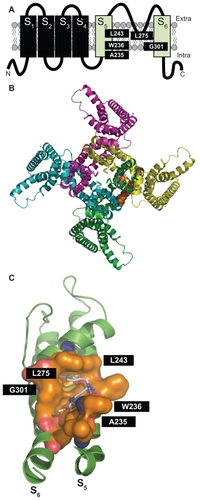
Results from mutagenesis and modeling experiments have suggested that, in channels formed by neural Kv7 subunits, retigabine binds to a hydrophobic pocket located between the cytoplasmic parts of the S5 and the S6 transmembrane domains in the open channel configuration. Within this cavity, a tryptophan residue in the intracellular end of the S5 helix (W236 in the Kv7.2 sequence) seems to play a crucial role.Citation52,Citation53 Replacement of this residue with a smaller and less hydrophobic amino acid (leucine) largely prevents the ability of retigabine to activate IKM. Additional amino acids (in Kv7.2 sequence: Leu243 in S5, Leu275 within the inner pore loop, Gly301 in the S6 segment, and Leu299 in S6 of the neighboring subunit) seem also to be involved in the formation of the complete retigabine binding siteCitation54 (see ). Except for Leu275, these amino acids are conserved among all neural Kv7 subunits but are missing in the Kv7.1 subunit primary sequence, an observation that may explain the lack of sensitivity to retigabine shown by channels formed by Kv7.1 subunits. The tryptophan in the S5 segment seems also to be important for the binding of other IKM activators, such as S-(1) and BMS-204352, both molecules failing to show additive effects with retigabine.Citation51 Another Kv7 activator, zinc pyrithione, also seems to bind to the pore region, although the molecular determinants for such an interaction appear to be only partially overlapping with those of retigabine, a result consistent with the additive effects on IKM exerted by these two compounds.Citation48 By contrast, phenamates (meclofenamic acid and diclofenac, as well as their derivatives) were still able to potentiate the K+ currents elicited by the Kv7.2 W236L mutant, suggesting their preferential binding in a different domain of the channel. In fact, NH29 appears to bind in close proximity to the external surface of the voltage-sensing domain, at the interface of helices S1, S2, and S4, with a mechanism of action similar to that of gating-modifier toxins.Citation55 Similar to NH29, the selective Kv7.2/Kv7.3 opener ICA-27243 was also found to bind to the voltage-sensing domain; the primary sequence of the region involved in ICA-27243 binding appears extremely variable among Kv7 subfamily members, allowing the drug to selectively affect channels formed by specific Kv7 subunits.Citation56
Figure 1 Panel A shows the schematic topology of a single Kv7.2 subunit, with each of the six transmembrane regions indicated. The green cylinders correspond to the S5 and S6 transmembrane regions. The amino acids involved in retigabine binding are indicated. Panel B shows a top view of the overall structure of the channel formed by four identical Kv7.2 subunits, with one retigabine molecule bound. Panel C shows an enlarged view of the retigabine binding site with the retigabine molecule docked into the channel cavity as obtained with ArgusLab 4.0.1 (Planaria Software LLC, Seattle, WA; available at http://www.arguslab.com). The residues involved in retigabine binding are indicated. The dashed yellow lines indicate the polar contacts between retigabine and the residues A235 and W236. Three-dimensional models of Kv7.2 subunits were generated by homology modelling using known structures of potassium channel subunits available in the Protein Data Bank), using SWISSMODEL, a program that performs automated sequence-structure comparisons, as previously described.Citation92 The model generated was analyzed using both the DeepView module of Swiss-PDBViewer (version 3.7, available at http://www.expasy.ch/spdbv/) and PyMOL (available at http://pymol.sourceforge.net/). In the present study, the homology model was built using the template structure (2R9RH) for a chimeric channel in which the voltage-sensor paddle (corresponding to the S3b–S4 region) of Kv2.1 was transferred into the Kv1.2 subunit.Citation93
In conclusion, the identification of the different regions on the channel molecule involved in binding distinct molecular entities might represent the starting point to design molecules selectively targeting strategic functional domains of Kv7 subunits, and may possibly allow for drug specificity at each Kv7 channel subtype. Whether this will translate into better efficacy and side effect profiles, and, therefore, into significant advantages for the treatment of hyperexcitability disorders with Kv7 channel openers, is still a matter of investigation.
Retigabine in preclinical models predictive of anticonvulsant activity
The therapeutic potential of retigabine as an antiepileptic drug has been assessed in several preclinical studies using various models. Retigabine was shown to be effective in reducing convulsions induced both by maximal electroshock (similarly to carbamazepine, phenytoin, and diazepam) and by pentylenetetrazole, picrotoxin, and NMDA (similarly to ethosuximide and valproate), two models predictive for drug efficacy in humans against generalized tonic–clonic, and generalized absence and myoclonic epilepsies, respectively.Citation57 Moreover, retigabine has been also tested in genetic models of epilepsy, such as DBA/2 miceCitation57 or epilepsy-prone rats (GEPR-3 and GEPR-9),Citation58 ie, models possessing some predictability for generalized tonic–clonic seizures (DBA/2 mouse and GEPR-9 rat) and absence seizures (GEPR-3 rat). In these three rodent models, retigabine was able to reduce seizures induced by loud noise stimulation. In addition, in DBA/2 mice, it showed an additive effect when administered in combination with classical anticonvulsants, most notably diazepam, phenobarbital, phenytoin, and valproate.Citation59 The administration of low doses of retigabine (0.01 mg/kg intraperitoneally or orally) also increased the threshold for induction of afterdischarges in the amygdala-kindling model of complex partial seizures; at higher doses (5 mg/kg intraperitoneally or 15 mg/kg orally) it reduced seizure severity and duration, total duration of behavioral changes, and afterdischarge duration. The potency of retigabine was much higher when compared with that of valproate; moreover, the doses required to increase the seizure threshold in amygdala-kindled rats are substantially lower than those needed to increase the threshold in the maximal electroshock test (an effect opposite to that of carbamazepine, phenytoin, and valproate), suggesting that retigabine is particularly effective against partial seizures.Citation60 Seizure models used in these studies lack specific predictivity against pharmacoresistant epilepsy. The fact that retigabine recognizes a molecular target distinct from currently available antiepileptic drugs makes this molecule particularly attractive for potential efficacy in drug-resistant epilepsies. To address this possibility, retigabine has been tested in vivo in lamotrigine-resistant kindled ratsCitation61 and in the 6 Hz psychomotor mouse model,Citation62 two preclinical models of epilepsy resistant to common antiepileptic drugs including lamotrigine, phenytoin, carbamazepine, topiramate, tiagabine, and felbamate. In these models, retigabine dose-dependently inhibited seizures and reduced afterdischarge duration induced by kindling stimulation and by 32 mA or 44 mA corneal stimulation, respectively.Citation63 In addition, retigabine was shown to reduce spontaneous bursting in hippocampal-entorhinal cortex slices from kainate-treated rats resistant to therapeutic concentrations of phenytoin, carbamazepine, and valproate,Citation64 and to suppress spontaneous spike waves and low magnesium induced-epileptiform field potential in neocortical slices prepared from human patients with pharmacoresistant epilepsy undergoing surgical treatment.Citation65 In conclusion, retigabine showed activity in a broad array of in vitro and in vivo epilepsy models, including some in which classical antiepileptic drugs failed to be effective.
Pharmacokinetics of retigabine in humans
Studies carried out to investigate retigabine pharmacokinetics in both healthy volunteers and patients mostly gave concordant results. The bioavailability of orally administered retigabine is 60%, with a very low first-pass metabolism. Absorption is rapid, and plasma peak concentration is achieved within 1.5 hours. Food does not significantly interfere with absorption of retigabine, causing only slight delays in the time required to reach plasma peak concentration (up to two hours). Retigabine plasma protein binding is about 80%, and the drug is widely distributed, with a volume of distribution of approximately 6.2 L/kg. Retigabine is metabolized only by Phase II reactions, primarily by acetylation (to a monoacetylated metabolite known as AWD21-360, which has similar half- life but lower anticonvulsant potency when compared with retigabine) and glucuronidation (via UGT1A1, A3, A4, and A9). Glucuronidation (which also occurs for AWD21- 360) leads to the formation mainly of N2- and of a minor amount of N4-glucuronide, both possessing no antiepileptic activity. Of note, Phase I clinical studies pointed to a constant ratio between retigabine and the N2-glucuronide metabolite, suggesting that the two compounds are coupled via enterohepatic circulation and glucuronidation/deglucuronidation reactions occurring in the liver and gut, respectively.Citation66 Retigabine does not induce or inhibit its own metabolism, and is eliminated primarily by the kidneys, with a mean terminal half-life of 8.0 hours and an apparent oral clearance of 0.70 L/h/kg. The pharmacokinetics of retigabine are linearly proportional to the dose. Steady-state pharmacokinetics were in agreement with single-dose pharmacokinetics, and the accumulation ratio was about 1.5.Citation67 Only a few differences regarding race, gender, and age have been observed in pharmacokinetic parameters. Ferron et al reported lower clearance and volume of distribution for retigabine in black subjects (25% and 30% lower, respectively), possibly due to differences in N-glucuronidation. In another study, retigabine was found to reach higher maximum concentration (56%) and exposure (20%) in young women when compared with young men, but showed similar clearance; this variability likely reflected differences in body weight. In elderly subjects, retigabine elimination was slower (30% lower apparent clearance normalized for weight), resulting in higher exposure (42%) and longer half-life (30%), although maximum concentration was similar to that in adults. These differences have been attributed to the decline of renal function with age.Citation68 No significant alterations in tolerability and safety were found in subjects with polymorphisms in the UGT1A1 (ie, Gilbert’s syndrome) and/or in N-acetyltransferase, NAT2 (ie, fast or slow acetylator) after a single and multiple (twice daily) 200 mg dose of oral retigabine administered over five days, although the total exposure to the monoacetylated metabolite, AWD21-360, was significantly related to the fast or slow acetylator status of subjects.Citation69 In vitro assays in human liver microsomal preparations showed that 6 μM retigabine (a concentration similar to the peak concentration in subjects receiving a therapeutic dose of the drug) has a moderate capacity to inhibit cytochrome (CYP)2A6 and low or no potential to inhibit CYP1A2, CYP2C8/9/19, CYP2D6, CYP2E1, CYP3A4/5, and CYP4A9/11. According to these studies, no clinically relevant inhibition of CYP-mediated drug metabolism is expected. The potential for drug-drug interaction due to competition for glucuronidation was investigated using valproic acid, lamotrigine, imipramine, and propofol, commonly used medications which are extensively glucuronidated. When assayed with human liver microsomes or UGT1 A9 enzyme, no interactions were found at clinically relevant concentrations.Citation70 Moreover, no clinically significant interactions between retigabine and phenobarbital were reported in clinical studies in healthy subjects.Citation71 By contrast, patients already treated with phenytoin and carbamazepine (two drugs known to induce UGT enzymes) showed an increased clearance of retigabine by 36% and 27%, respectively. Valproate and topiramate coadministration did not modify retigabine pharmacokinetics. Retigabine did not alter the pharmacokinetic profile of phenytoin, carbamazepine, valproate, phenobarbital, and topiramate.Citation72 Although in this latter study no significant interactions were observed in patients receiving retigabine in addition to lamotrigine, Hermann et al reported a modest reciprocal pharmacokinetic interaction between these two antiepileptic drugs; lamotrigine reduced the clearance of retigabine by 13%, while it increased the mean half-life and the area under the plasma concentration-time curve (AUC) by 7.5% and 15%, respectively. Retigabine also influenced lamotrigine pharmacokinetics, by increasing its clearance (by 22%) and reducing its mean half-life (by 15%) and AUC (by 18%).Citation73 While the reduced clearance of retigabine induced by lamotrigine is explained by the authors as competition for renal excretion, the effect of retigabine on lamotrigine pharmacokinetics remains unclear, because retigabine did not show enzyme induction in various other drug-drug interaction studies. Retigabine did not decrease contraceptive hormone exposure in women taking a low-dose oral contraceptive (ethinyl estradiol 0.035 mg + norethindrone 1 mg), suggesting no potential for reducing contraceptive efficacy. Moreover, the AUC values of retigabine measured in women receiving such combination therapy were not significantly different from those reported in other studies, suggesting that oral contraceptives did not alter retigabine pharmacokinetics.Citation72
Efficacy studies with retigabine
Retigabine efficacy in the treatment of partial onset seizures has been assessed by multicenter, randomized, double-blind, placebo-controlled Phase II and Phase III clinical trials ().
Table 1 Summary of Phase II and Phase III clinical trials with retigabine
Phase II studies
A Phase II studyCitation74,Citation75 was conducted to evaluate the 16-week (eight-week titration phase, eight-week maintenance phase) efficacy and safety of retigabine 600, 900, and 1200 mg/day (divided into three daily doses) versus placebo as an adjunctive therapy in partial onset seizures. Patients enrolled in this trial (n = 396) were men and women 16–70 years of age, with incomplete control of seizures despite therapy with 1–2 antiepileptic drugs or vagal nerve stimulation treatment (considered as an antiepileptic drug if stimulation parameters were kept constant during the study), which did not have to be discontinued throughout the study. Patients had to experience at least four partial onset seizures per month (with or without secondary generalization) in the eight-week baseline period, and no 30-day seizure-free period. The results of this study showed that retigabine caused a significant and dose-dependent reduction in median percent monthly seizure frequency from baseline by 23% (600 mg/day), by 29% (900 mg/day), and by 35% (1200 mg/day); in the placebo group the reduction was by 13%. The responders rates (patients with ≥50% seizure frequency reduction) were 23%, 32%, and 33% in the 600, 900, and 1200 mg/day groups, respectively, versus 16% in the placebo arm). It is interesting to note that the difference between the 600 mg/day arm and placebo group was not statistically significant. A total of 222 patients who completed the double-blind 16-week trial entered an open-label extension study, in which the retigabine dose was converted to 900 mg/day. Following this, each patient’s daily dose of retigabine or concomitant antiepileptic drugs could be either reduced or increased up to a maximum allowed dose of 1200 mg retigabine. The median decrease in monthly total partial seizure frequency from baseline was 48.3% and the responders rate was 46.4%.Citation72,Citation76
Phase III studies
Based on the positive results of the Phase II study, two multicenter, double-blind, placebo-controlled Phase III registration trials investigating the efficacy of retigabine as add-on therapy in the treatment of refractory partial onset epilepsy, called RESTORE 1 and RESTORE 2, have been performed and recently completed. Patients enrolled in these two studies showed similar clinical features to those of the Phase II clinical trial (18–75 years of age, partial onset epilepsy with or without secondary generalization, refractory to stable therapy with 1–3 approved antiepileptic drugs, ≥ four seizures per month and no 21-day seizure-free period during the eight-week baseline period), but differed in the target dosage and titration phase duration. In RESTORE 1, the target dose of retigabine was 1200 mg/day titrated over six weeks and with dosage adjustment allowed (to a minimum of 1050 mg/day); in RESTORE 2, retigabine 600 and 900 mg/day were administered, titrated over 2–4 weeks and with no change in the dosage allowed. The maintenance period was 12 weeks, with the possibility to continue with flexible dosing of the drug in an open-label extension of the studies. In both trials, retigabine significantly reduced the median percent in 28-day total partial seizure frequency from baseline (−44% in the RESTORE 1 1200 mg/day arm versus −17.5% in the placebo group; −39.9% in the RESTORE 2 900 mg/day and −27.9% in the RESTORE 2 600 mg/day arms versus −15.9% in the placebo group). The responder rate from baseline to maintenance phase were also significantly higher than placebo in all the arms considered (45% for 1200 mg/day versus 18% for placebo; 39.3% and 31.5% for 900 mg/day and for 600 mg/day, respectively, versus 17.3% for placebo).Citation72
Safety and tolerability
The potential toxicity of retigabine was investigated in many preclinical studies conducted in rat and mouse models of seizures.Citation57,Citation59,Citation60 In general, acute toxicity was limited to dose-related effects on the central nervous system, including hyperkinesia or hypokinesia, disturbed coordination, stilted gait, tremor, and convulsions.Citation72 Moreover, retigabine did not exert any toxic effect on cardiac function and on electrocardiographic parameters, but it was able to induce urinary retention, an action explained by the activation of Kv7 channels expressed in smooth muscle of the bladder combined with inhibitory effects on afferent nerve fiber activity.Citation77 No teratogenic and/or reproductive effects were observed in rats and rabbits.
In humans, the safety profile of retigabine has been investigated in large Phase II and III (RESTORE 1 and RESTORE 2) trials and their extension studies. In these studies, retigabine was used as additional therapy in patients already receiving 1–3 “classical” antiepileptic drugs. Thus, data concerning the efficacy and safety of retigabine in the form of monotherapy are lacking. As in animal models and similar to other antiepileptic drugs, retigabine showed minor and nonspecific adverse events involving the central nervous system, the most common of which were dizziness, somnolence, headache, and asthenia. These adverse events were generally dose-related, rarely serious, or a cause for discontinuation. Both in Phase II and III clinical trials, a large proportion of them occurred during the forced titration phase, and the proportion of patients withdrawing from the study was inversely related to the duration of the titration phase.Citation2 The most common reasons for discontinuation were dizziness, confusion, somnolence, and asthenia, with the highest proportion of withdrawals in patients receiving retigabine 1200 mg/kg.Citation72 In RESTORE 1, where retigabine was used at 1200 mg/kg, urinary or renal disorders were observed in 12% of patients. Among urinary disorders, urinary hesitancy was the most frequent adverse event. However, urinary adverse events did not correlate with objective assessments of urinary function, and most patients were able to continue on medication, while in all other patients (except one), this symptom disappeared after discontinuation. Although urinary adverse effects do not seem to be severe, retigabine should be used with caution in patients at risk for urinary dysfunction (benign prostatic hyperplasia, neurogenic bladder), in subjects with cognitive impairment, and in individuals already treated with drugs active on the bladder (anticholinergics).Citation78 As suggested by preclinical studies, retigabine did not exert any adverse effects on cardiac function, and did not prolong the QT interval. However, a study of cardiac conduction demonstrated that retigabine produced a slight (on average approximately 4 msec in the 24 hours postdosing) and transient QT-prolonging effect in healthy volunteers receiving 1200 mg/day. Although this extent of QT prolongation may have poor clinical relevance, retigabine should be administered with caution in patients at highest risk for arrhythmia.Citation78
Patient-focused perspectives
Due to the limited clinical experience with retigabine, data regarding quality of life, patient satisfaction, and adherence to therapy are rather scarce. However, the most frequent adverse events related to retigabine are similar to those caused by many other neuroactive drugs,Citation74 and they should not worsen quality of life more than other commonly used antiepileptic drugs. Moreover, the clinical relevance of such adverse events as a cause of discontinuation of treatment might be less severe than that predicted by controlled trials because, in these studies, contrary to common clinical practice, little or no adjustment of dose was allowed, thus increasing the rate of treatment-emergent adverse events and of subsequent interruption of therapy. The lack of clinically significant cardiac, hematologic, and hepatic toxicity (which can be associated, although not frequently, with treatment with other antiepileptic drugs, including phenytoin, carbamazepine, or valproate), the inability to induce or inhibit liver enzymes (that can reduce the efficacy of concomitant administered drug and, in particular, of contraceptive therapy), as well as the minimal clinical significance of drug-drug interactions, suggest that retigabine is a well tolerated new antiepileptic drug.
Conclusion
Nearly 30% of epileptic patients are resistant to pharmacological treatment, with persistence of frequent seizures despite assumption of one or more antiepileptic drugs. Uncontrolled seizures interfere with daily life, affecting lifespan (increased incidence of injuries and of sudden unexplained death),Citation79 and global quality of life (limitations on driving or job availability), causing a condition of “social disability”. Pharmacoresistant epilepsy has been associated with increased cognitive impairmentCitation80 and psychiatric disorders, such as depression.Citation81 Despite the availability of several antiepileptic drugs, the probability of obtaining seizure control diminishes progressively with successive antiepileptic drug regimens, and the success of therapy is only 4%–7% on the third drug used or on a combination of two antiepileptic drugs in an add-on regimen.Citation82 This reduced efficacy of commonly used medications may, in part, be a consequence of combining drugs with similar mechanisms of action. Moreover, antiepileptic drugs are frequently involved in wide and reciprocal drug-drug interactions, with increased frequency of adverse events and subsequent reduced tolerability for patients.Citation83 In this context, retigabine, with its different mechanism of action and its broad spectrum of activity demonstrated in numerous seizure and epilepsy models and suggested by Phase II and III clinical trials, could play an important role in the treatment of pharmacoresistant epilepsies. Moreover, retigabine and other neural Kv7 openers may be safe and useful tools for the treatment of pediatric epilepsy. Indeed, the role of IKM in reducing neuronal excitability is particularly relevant in early life; due to the excitatory effect of GABAergic neurotransmission in the immature brain, IKM represents the main inhibitory conductance in neonatal neurons.Citation84 The importance of IKM as a “brake” on neuronal excitability in newborns is also suggested by the natural clinical history of benign familial neonatal seizures, in which seizures spontaneously disappear after a few weeks or months, and by several in vitro studies showing that epileptiform activity caused by the pharmacological suppression of IKM decreased or disappeared with neuronal maturation.Citation85,Citation86 Moreover, it has been demonstrated that, in hippocampal neurons, IKM density increased after birth, causing the disappearance of intrinsic bursting and abnormal neuronal synchronization characteristic of the neonatal period.Citation87 Based on these data, IKM can represent a fascinating therapeutic target for the treatment of neonatal epilepsy, also in consideration of the fact that first-line drugs given to neonates (usually phenobarbital and phenytoin) are effective in less than 50% of casesCitation88 and that they have been demonstrated to cause widespread neuronal apoptosis when given to young rodents, thus raising concerns about their administration to children.Citation89 On these premises, flupirtine has been demonstrated to be more effective than phenobarbital and diazepam in arresting kainate- and flurothyl-induced seizures when administered in rats at postnatal day 10.Citation90 Moreover, in another study, retigabine has been shown to delay the development of focal seizures in postnatal day 14 rats, and to prevent the establishment of kindling-induced enhanced seizure susceptibility. Interestingly, in the latter study, the effect of retigabine appeared to be more pronounced in younger animals when compared with older ones.Citation91 Although these results argue in favor of the use of retigabine also in young children, further investigations are needed to demonstrate the efficacy and safety of this drug in the treatment of seizures occurring at early developmental stages, with special regard to the possible differences in pharmacokinetics (such as the decreased metabolism of retigabine due to the reduced glucuronidation capability of the newborn) which can enhance the frequency and severity of the side effects of retigabine in these patients.
Acknowledgments
Work on KCNQ/IKM channels in the author’s laboratories is currently supported by grants from the European Union (E-Rare 2007, EUROBFNS); Fondazione Telethon Italy (GGP07125); the Italian Ministry of University, Education, and Research (MIUR, PRIN 2007); and Regione Molise (Convenzione AIFA/Regione Molise).
Disclosure
The authors report no conflicts of interest in this work.
References
- NoebelsJLTargeting epilepsy genesNeuron19961622412448789939
- PloskerGLScottLJRetigabine: In partial seizuresCNS Drugs200620760160816800718
- ForsgrenLBeghiEOunASillanpääMThe epidemiology of epilepsy in Europe – a systematic reviewEur J Neurol200512424525315804240
- BergATBerkovicSFBrodieMJRevised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009Epilepsia201051467668520196795
- ShiehCCCoghlanMSullivanJPGopalakrishnanMPotassium channels: Molecular defects, diseases, and therapeutic opportunitiesPharmacol Rev200052455759411121510
- BrownDAAdamsPRMuscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neuroneNature198028357486736766965523
- HalliwellJVAdamsPRVoltage-clamp analysis of muscarinic excitation in hippocampal neuronsBrain Res1982250171926128061
- RogawskiMAKCNQ2/KCNQ3 K+ channels and the molecular pathogenesis of epilepsy: Implications for therapyTrends Neurosci200023939339810941184
- GuoJSchofieldGGActivation of muscarinic m5 receptors inhibits recombinant KCNQ2/KCNQ3 K+ channels expressed in HEK293T cellsEur J Pharmacol20034621–3253212591092
- MarrionNVControl of M-currentAnnu Rev Physiol1997594835049074774
- DelmasPBrownDAPathways modulating neural KCNQ/M (Kv7) potassium channelsNat Rev Neurosci200561185086216261179
- SinghNACharlierCStaufferDA novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newbornsNat Genet199818125299425895
- BiervertCSchroederBCKubischCA potassium channel mutation in neonatal human epilepsyScience199827953494034069430594
- CharlierCSinghNARyanSGA pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy familyNat Genet199818153559425900
- WangQCurranMESplawskiIPositional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmiasNat Genet199612117238528244
- KubischCSchroederBCFriedrichTKCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafnessCell199996343744610025409
- KharkovetsTHardelinJPSafieddineSKCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathwayProc Natl Acad Sci U S A20009784333433810760300
- IannottiFAPanzaEBarreseVViggianoDSoldovieriMVTaglialatelaMExpression, localization, and pharmacological role of Kv7 potassium channels in skeletal muscle proliferation, differentiation, and survival after myotoxic insultsJ Pharmacol Exp Ther2010332381182020040580
- GreenwoodIAOhyaSNew tricks for old dogs: KCNQ expression and role in smooth muscleBr J Pharmacol200915681196120319751313
- LercheCSchererCRSeebohmGMolecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversityJ Biol Chem200027529223952240010787416
- BrownDAPassmoreGMNeural KCNQ (Kv7) channelsBr J Pharmacol200915681185119519298256
- CooperECAldapeKDAboschAColocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsyProc Natl Acad Sci U S A20009794914491910781098
- WangHSPanSShiWKCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channelScience19982825395189018939836639
- BelliniGMiceliFSoldovieriMVMiraglia del GiudiceEPascottoATaglialatelaMBenign familial neonatal seizuresPagonRABirdTCDolanCRStephensKGeneReviews [Internet]Seattle, WAUniversity of Washington1993–2010
- WatanabeHNagataEKosakaiADisruption of the epilepsy KCNQ2 gene results in neural hyperexcitabilityJ Neurochem2000751283310854243
- PetersHCHuHPongsOStormJFIsbrandtDConditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behaviorNat Neurosci200581516015608631
- YangYBeyerBJOttoJFSpontaneous deletion of epilepsy gene orthologs in a mutant mouse with a low electroconvulsive thresholdHum Mol Genet200312997598412700166
- SinghNAOttoJFDahleEJMouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganizationJ Physiol2008586143405342318483067
- BosciaFAnnunziatoLTaglialatelaMRetigabine and flupirtine exert neuroprotective actions in organotypic hippocampal culturesNeuropharmacology200651228329416697426
- OttoMCepekLRatzkaPEfficacy of flupirtine on cognitive function in patients with CJD: A double blind studyNeurology200462571471815007119
- FriedelHAFittonAFlupirtine. A review of its pharmacological properties, and therapeutic efficacy in pain statesDrugs19934545485697684675
- PorterRJNohriaVRundfeldtCRetigabineNeurotherapeutics20074114915417199031
- RundfeldtCThe new anticonvulsant retigabine (D-23129) acts as an opener of K+ channels in neuronal cellsEur J Pharmacol19973362–32432499384239
- RundfeldtCCharacterization of the K+ channel opening effect of the anticonvulsant retigabine in PC12 cellsEpilepsy Res19993529910710372563
- RundfeldtCNetzerRThe novel anticonvulsant retigabine activates M-currents in Chinese hamster ovary-cells tranfected with human KCNQ2/3 subunitsNeurosci Lett20002821–2737610713399
- MainMJCryanJEDupereJRCoxBClareJJBurbidgeSAModulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabineMol Pharmacol200058225326210908292
- WickendenADYuWZouAJeglaTWagonerPKRetigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channelsMol Pharmacol200058359160010953053
- GassenMPergandeGYoudimMBAntioxidant properties of the triaminopyridine, flupirtineBiochem Pharmacol19985610132313299825731
- TatulianLDelmasPAbogadieFCBrownDAActivation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabineJ Neurosci200121155535554511466425
- DupuisDSSchrøderRLJespersenTActivation of KCNQ5 channels stably expressed in HEK293 cells by BMS-204352Eur J Pharmacol2002437312913711890900
- SoldovieriMVMiceliFBelliniGCoppolaGPascottoATaglialatelaMCorrelating the clinical and genetic features of benign familial neonatal seizures (BFNS) with the functional consequences of underlying mutationsChannels20071422823318698150
- PeretzADeganiNNachmanRMeclofenamic acid and diclofenac, novel templates of KCNQ2/Q3 potassium channel openers, depress cortical neuron activity and exhibit anticonvulsant propertiesMol Pharmacol20056741053106615598972
- PeretzASheininAYueCPre- and postsynaptic activation of M-channels by a novel opener dampens neuronal firing and transmitter releaseJ Neurophysiol200797128329517050829
- PeretzADegani-KatzavNTalmonMA tale of switched functions: From cyclooxygenase inhibition to M-channel modulation in new diphenylamine derivativesPLoS One2007212e133218159230
- WickendenADKrajewskiJLLondonBN-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide (ICA-27243): A novel, selective KCNQ2/Q3 potassium channel activatorMol Pharmacol200873397798618089837
- RoeloffsRWickendenADCreanCIn vivo profile of ICA- 27243 [N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide], a potent and selective KCNQ2/Q3 (Kv7.2/Kv7.3) activator in rodent anticonvulsant modelsJ Pharmacol Exp Ther2000326381882818577704
- XiongQSunHLiMZinc pyrithione-mediated activation of voltage-gated KCNQ potassium channels rescues epileptogenic mutantsNat Chem Biol20073528729617435769
- XiongQSunHZhangYNanFLiMCombinatorial augmentation of voltage-gated KCNQ potassium channels by chemical openersProc Natl Acad Sci U S A200810583128313318272489
- SchrøderRLJespersenTChristophersenPStrøbaekDJensenBSOlesenSPKCNQ4 channel activation by BMS-204352 and retigabineNeuropharmacology200140788889811378159
- KorsgaardMPHartzBPBrownWDAhringPKStrobaekDMirzaNRAnxiolytic effects of maxipost (BMS-204352) and retigabine via activation of neuronal Kv7 channelsJ Pharmacol Exp Ther2005314128229215814569
- BentzenBHSchmittNCalloeKDalby BrownWGrunnetMOlesenSPThe acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channelsNeuropharmacology20065161068107716904708
- WuttkeTVSeebohmGBailSMaljevicSLercheHThe new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gateMol Pharmacol20056741009101715662042
- SchenzerAFriedrichTPuschMMolecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabineJ Neurosci200525205051506015901787
- LangeWGeissendörferJSchenzerARefinement of the binding site and mode of action of the anticonvulsant retigabine on KCNQ K+ channelsMol Pharmacol200975227228019015229
- PeretzAPellLGofmanYTargeting the voltage sensor of Kv7.2 voltage-gated K+ channels with a new gating-modifierProc Natl Acad Sci U S A201010735156371564220713704
- PadillaKWickendenADGerlachACMcCormackKThe KCNQ2/3 selective channel opener ICA-27243 binds to a novel voltage-sensor domain siteNeurosci Lett2009465213814219733209
- RostockAToberCRundfeldtCD-23129: A new anticonvulsant with a broad spectrum activity in animal models of epileptic seizuresEpilepsy Res19962332112238739124
- DaileyJWCheongJHKoKHAdams-CurtisLEJobePCAnticonvulsant properties of D-20443 in genetically epilepsy-prone rats: Prediction of clinical responseNeurosci Lett1995195277807478272
- de SarroGdi PaolaEDConteGPasculliMPde SarroAInfluence of retigabine on the anticonvulsant activity of some antiepileptic drugs against audiogenic seizures in DBA/2 miceNaunyn Schmiedebergs Arch Pharmacol2001363333033611284448
- ToberCRostockARundfeldtCBartschRD-23129: A potent anticonvulsant in the amygdala kindling model of complex partial seizuresEur J Pharmacol199630331631698813562
- PostmaTKruppELiXLPostRMWeissSRLamotrigine treatment during amygdala-kindled seizure development fails to inhibit seizures and diminishes subsequent anticonvulsant efficacyEpilepsia200041121514152111114208
- BartonMEKleinBDWolfHHWhiteHSPharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsyEpilepsy Res200147321722711738929
- SrivastavaAKWhiteHSRetigabine decreases behavioral and electrographic seizures in the lamotrigine-resistant amygdala kindled rat model of pharmacoresistant epilepsyEpilepsia200546 Suppl 8S217
- SmithMDAdamsACSaundersGWWhiteHSWilcoxKSPhenytoin- and carbamazepine-resistant spontaneous bursting in rat entorhinal cortex is blocked by retigabine in vitroEpilepsy Res2007742–39710617395429
- StraubHKöhlingRHöhlingJEffects of retigabine on rhythmic synchronous activity of human neocortical slicesEpilepsy Res2001442–315516511325571
- HillerANguyenNStrassburgCPRetigabine N-glucuronidation and its potential role in enterohepatic circulationDrug Metab Dispos199927560561210220490
- FerronGMPaulJFruncilloRMultiple-dose, linear, dose-proportional pharmacokinetics of retigabine in healthy volunteersJ Clin Pharmacol200242217518211831540
- HermannRFerronGMErbKEffects of age and sex on the disposition of retigabineClin Pharmacol Ther2003731617012545144
- HermannRBorlakJMunzelUThe role of Gilbert’s syndrome and frequent NAT2 slow acetylation polymorphisms in the pharmacokinetics of retigabinePharmacogenomics J20066321121916402080
- BorlakJGasparicALocherMSchupkeHHermannRN-Glucuronidation of the antiepileptic drug retigabine: Results from studies with human volunteers, heterologously expressed human UGTs, human liver, kidney, and liver microsomal membranes of Crigler-Najjar type IIMetabolism200655671172116713428
- FerronGMPatatAParksVRolanPTroySMLack of pharmacokinetic interaction between retigabine and phenobarbitone at steady-state in healthy subjectsBr J Clin Pharmacol2003561394512848774
- BialerMJohannessenSILevyRHPeruccaETomsonTWhiteHSProgress report on new antiepileptic drugs: A summary of the Ninth EILAT Conference (EILAT IX)Epilepsy Res200983114319008076
- HermannRKnebelNGNiebchGRichardsLBorlakJLocherMPharmacokinetic interaction between retigabine and lamotrigine in healthy subjectsEur J Clin Pharmacol2003581279580212698305
- PorterRJPartiotASachdeoRNohriaVAlvesWMfor 205 Study GroupRandomized, multicenter, dose-ranging trial of retigabine for partial-onset seizuresNeurology200768151197120417420403
- BialerMJohannessenSIKupferbergHJLevyRHPeruccaETomsonTProgress report on new antiepileptic drugs: A summary of the Seventh EILAT Conference (EILAT VII)Epilepsy Res2004611–314815570674
- PorterRJNohriaVPechsteinBEfficacy, safety, and tolerability of retigabine as adjunctive therapy in patients with refractory partial-onset seizures in an open-label studyEpilepsia200445 Suppl 7S311
- RodeFSvaløJSheykhzadeMRønnLCFunctional effects of the KCNQ modulators retigabine and XE991 in the rat urinary bladderEur J Pharmacol20106381–312112720385123
- Advisory CommitteePotiga™ (ezogabine) tablets as adjunctive therapy for the treatment of partial onset seizuresPeripheral and central nervous system drugsValeant Pharmaceuticals2010Aug 11 Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM222587.pdfAccessed 2010 Nov 8
- PatiSAlexopoulosAVPharmacoresistant epilepsy: From pathogenesis to current and emerging therapiesCleve Clin J Med201077745746720601619
- HermannBSeidenbergMEpilepsy and cognitionEpilepsy Curr2007711617304341
- KannerAMBalabanovADepression and epilepsy: How closely related are they?Neurology2002588 Suppl 5S2S39
- KwanPSperlingMRRefractory seizures: Try additional antiepileptic drugs (after two have failed) or go directly to early surgery evaluation?Epilepsia200950 Suppl 8S57S62
- BrodieMJDo we need any more new antiepileptic drugs?Epilepsy Res2001451–33611461782
- OkadaMZhuGHiroseSAge-dependent modulation of hippocampal excitability by KCNQ-channelsEpilepsy Res2003531–2819412576170
- QiuCJohnsonBNTallentMKK+ M-current regulates the transition to seizures in immature and adult hippocampusEpilepsia200748112047205817651418
- PeñaFAlavez-PérezNEpileptiform activity induced by pharmacologic reduction of M-current in the developing hippocampus in vitroEpilepsia20064714754
- SafiulinaVFZacchiPTaglialatelaMYaariYCherubiniELow expression of Kv7/M channels facilitates intrinsic and network bursting in the developing rat hippocampusJ Physiol2008586225437545318801845
- PainterMJSherMSSteinADPhenobarbital compared with phenytoin for the treatment of neonatal seizuresN Engl J Med1999341748548910441604
- OlneyJWYoungCWozniakDFJevtovic-TodorovicVIkonomidouCDo pediatric drugs cause developing neurons to commit suicide?Trends Pharmacol Sci200425313513915019268
- RaolYHLapidesDAKeatingJGBrooks-KayalARCooperECA KCNQ channel opener for experimental neonatal seizures and status epilepticusAnn Neurol200965332633619334075
- MazaratiAWuJShinDKwonYSSankarRAntiepileptogenic and antiictogenic effects of retigabine under conditions of rapid kindling: An ontogenic studyEpilepsia200849101777178618503560
- MiceliFSoldovieriMVLugliLNeutralization of a unique, negatively-charged residue in the voltage sensor of Kv7.2 subunits in a sporadic case of benign familial neonatal seizuresNeurobiol Dis200934350151019344764
- LongSBTaoXCampbellEBMacKinnonRAtomic structure of a voltage-dependent K+ channel in a lipid membrane-like environmentNature2007450716837638218004376