Abstract
Although there have been significant advances in the therapy of heart failure in recent decades, such as the introduction of β-blockers and antagonists of the renin–angiotensin–aldosterone system, this devastating disease still carries tremendous morbidity and mortality in the western world. G protein-coupled receptors, such as β-adrenergic and angiotensin II receptors, located in the membranes of all three major cardiac cell types, ie, myocytes, fibroblasts, and endothelial cells, play crucial roles in regulation of cardiac function in health and disease. Their importance is reflected by the fact that, collectively, they represent the direct targets of over one-third of the currently approved cardiovascular drugs used in clinical practice. Over the past few decades, advances in elucidation of the signaling pathways they elicit, specifically in the heart, have led to identification of an increasing number of new molecular targets for heart failure therapy. Here, we review these possible targets for heart failure therapy that have emerged from studies of cardiac G protein-coupled receptor signaling in health and disease, with a particular focus on the main cardiac G protein-coupled receptor types, ie, the β-adrenergic and the angiotensin II type 1 receptors. We also highlight key issues that need to be addressed to improve the chances of success of novel therapies directed against these targets.
Introduction
Heart failure (HF) is a complex pathophysiological syndrome that arises from a primary defect in the ability of the heart to fill and/or eject blood sufficiently. The clinical manifestations of HF result from the primary myocardial insult (most commonly coronary artery disease, hypertension, or genetic factors) and the attendant sequelae. In general, the primary insult brings about an increase in myocardial wall stress that induces an orchestrated cascade of remodeling stimuli within the heart, as well as neurohormonal, vascular, renal, and skeletal muscle alterations. Within this conceptual framework, chronic HF is generally a progressive disorder that results from continued left ventricular (LV) remodeling and a progressive loss of function. It should be noted that abnormalities of systolic and/or diastolic function can result in similar symptoms and they might share some common underlying mechanisms. It is estimated that symptomatic HF currently affects 0.4%–2% of the general population in the western world.Citation1–Citation3 Importantly, however, the incidence of symptomatic HF rises substantially with increasing age; estimates of symptomatic HF prevalence for individuals over 65 years of age range between 6% and 10%.Citation1,Citation4,Citation5 Up to 50% of patients diagnosed with HF will die within 4 years, and for patients with end-stage HF, the 1 year survival rate is 50% – worse than most advanced malignancies.Citation6,Citation7
The most important neurohormonal receptors that regulate cardiac function and physiology belong to the superfamily of G protein-coupled receptors (GPCRs) (or seven transmembrane-spanning receptors [7TMRs]).Citation8 For instance, cardiac function (contractility) is tightly controlled by the activity of β-adrenergic receptors (β1- and β2ARs) located in the membranes of cardiac myocytes.Citation9 Cardiac structure and morphology are regulated by angiotensin II (AngII) type 1 receptors (AT1Rs) present (mainly) in cardiac fibroblast and endothelial cell membranes, but also, to a lesser extent, in cardiomyocyte membranes.Citation8 Heart rate (HR) is modulated by the balance between the activities of β-adrenergic and muscarinic cholinergic (mAChR) receptors located in various anatomical segments of the cardiac electrical conduction system.Citation8,Citation9 Furthermore, even the neurohormonal control of the circulatory system, whether it be catecholamine and corticosteroid release by the adrenal glands or activation of the renin–angiotensin–aldosterone system (RAAS) by the juxtaglomerular apparatus of the kidneys or release of neurotransmitters by central and peripheral neurons innervating cardiovascular organs, is under tight regulation by various GPCRs (eg, α2ARs) as well.Citation8,Citation9 Thus, given that signaling from all these cardiac GPCRs constitutes an integral part of regulation of cardiac function, it comes as no surprise that drugs directly targeting (ie, binding) these receptors represent over one-third of currently used cardiovascular drugs in clinical practice, and the vast majority of currently approved HF drugs target GPCR function and signaling in one way or another.Citation8,Citation9 However, there is still an enormous potential for development of novel HF therapies targeting these receptors, either directly (ie, the GPCR per se) or some other signaling molecule down the pathway the receptor activates. The cloning and molecular and structural characterizations of GPCRs, highlighted by last year’s Nobel prize in chemistry award to the two pioneers of the field, Bob Lefkowitz and Brian Kobilka,Citation10 has spurred many significant advances in delineation and understanding of cardiac GPCR signaling in health and disease over the past couple of decades. The present review will discuss, receptor and signaling molecule type-by-type, all the important findings in the field of cardiac GPCR signaling that can be harnessed for development of novel HF therapeutics, and will also highlight the salient issues that complicate exploitation of these GPCR signaling targets for future HF clinical therapies.
βAR signaling targets for HF therapy
The sympathetic nervous system (SNS) neurotransmitters norepinephrine (NE) and epinephrine (Epi) mediate their effects in cells and tissues by binding to specific cell surface adrenoceptors (ARs), three α1 ARs, three α2 ARs, and three βARs (β1, β2, β3).Citation9 All ARs primarily signal through heterotrimeric G proteins. The human heart contains all three βAR subtypes.Citation9 β1AR is the predominant subtype in the (normal, healthy) myocardium, representing 75%–80% of total βAR density; followed by β2AR, which accounts for about 15%–18% of total cardiomyocyte βARs; and the remaining 2%–3% is β3ARs (under normal conditions).Citation9,Citation11 The principal role of βARs in the heart is the regulation of cardiac rate and contractility in response to NE and Epi. Stimulation of β1ARs (mainly) and of β2ARs (to a lesser extent) increases cardiac contractility (positive inotropic effect), frequency (positive chronotropic effect), and rate of relaxation (lusitropic effect), and accelerates impulse conduction through the atrioventricular node (positive dromotropic effect) and pacemaker activity from the sinoatrial node.Citation9 β3ARs are predominantly inactive during normal physiologic conditions;Citation12 however, their stimulation seems to produce a negative inotropic effect opposite to that induced by β1ARs and β2ARs, involving the nitric oxide synthase (NOS) pathway,Citation13 thus acting as a “fuse” against cardiac adrenergic overstimulation.Citation14 The most powerful physiologic mechanism to increase cardiac performance is activation of cardiomyocyte β1ARs and β2ARs, which, in turn, activates Gs proteins (stimulatory G proteins). Gs protein signaling stimulates the effector adenylate cyclase (AC), which converts adenosine triphosphate (ATP) to the second messenger adenosine 3′,5′-monophosphate or cyclic AMP (cAMP), which in turn binds to and activates the cAMP-dependent protein kinase (protein kinase A [PKA]).Citation9 PKA is the major effector of cAMP and by phosphorylating a variety of substrates, it ultimately results in a significant raise in free intracellular Ca2+ concentration, which is the master regulator of cardiac muscle contraction ().Citation15 Of note, PKA can phosphorylate the βARs themselves (and other GPCRs) in the heart, causing G protein uncoupling and functional desensitization of the receptor (heterologous or agonist-independent desensitization).Citation16 Given that cAMP and PKA augment cardiac contractility, drugs that enhance signaling through these molecules (such as inhibitors of cAMP-specific phosphodiesterase, an enzyme that degrades cAMP and reduces PKA activation) have been developed for HF ().Citation17 Although they might be useful for acute decompensated HF, when acute increases in contractility are needed to sustain life, these drugs increase mortality in human HF in the long run (possibly because they increase cardiac workload and oxygen demand) and are nowadays contraindicated in chronic HFCitation17 This is entirely consistent with the notion that chronic cardiac β1AR activation is detrimental and pro-apoptotic in the heart (see below).
Figure 1 G protein-coupled receptors and their signaling pathways involved in heart failure pathophysiology.
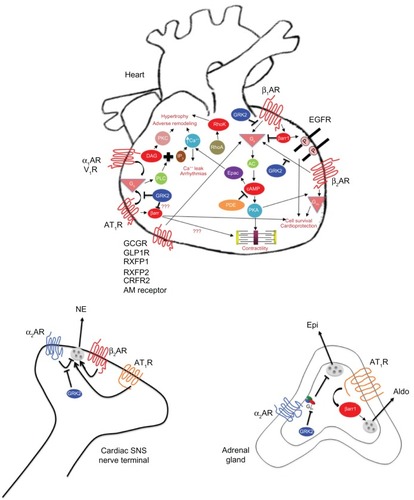
Importantly, the β2AR also mediates the effects of catecholamines in the heart, but in a qualitatively different manner from β1AR, as it can also couple to the AC inhibitory G protein (Gi). In fact, this switching of β2AR signaling from Gs to Gi proteins is postulated to be induced by the phosphorylation of the β2AR by PKA ().Citation16 It is now generally accepted that in the heart, β2AR signals and functions in a substantially different way than β1AR:Citation18–Citation20 whereas β1AR activation enhances cardiomyocyte apoptosis, β2AR exerts anti-apoptotic effects in the heart.Citation18–Citation21 This essential difference between the two receptor subtypes is ascribed to the signal of β2AR through Gi/o proteins.Citation19 This is the rationale behind the notion that promotion of Gi/o protein signaling might serve as a therapeutic strategy in HF in an effort to augment cardiac survival (anti-apoptotic, cardioprotective) ( and ). Nevertheless, studies using transgenic mice, β2AR-selective stimulation, and adenoviral-mediated β2AR overexpression have clearly demonstrated the protective effects of β2AR signaling in the myocardium, including improved cardiac function and decreased apoptosis. Conversely, hyperstimulation or overexpression of β1AR has detrimental effects in the heart.Citation21,Citation22
Table 1 Potential G protein-coupled receptor signaling targets for drug development in heart failure
Of note, the differences between the two predominant cardiac βARs, ie, β1AR and β2AR, in terms of their signaling properties, might take quite a different shape and have a much bigger bearing on pathophysiologic implications in the setting of human HF: for instance, and as discussed in more detail in subsequent sections, β1AR is selectively downregulated (ie, functional receptor number reduced) in human HF, thus shifting the above-mentioned stoichiometry of β1AR:β2AR toward 50:50 in the failing heart from ∼75%:∼20% in the normal, healthy heart.Citation23,Citation24 However, β2AR is also nonfunctional and does not signal properly in the failing heart.Citation23,Citation24 In addition, emerging evidence suggests that β2AR signaling in the failing heart is quite different from that in the normal heart, ie, is more diffuse and noncompartmentalized and resembles more the pro-apoptotic “diffuse” cAMP signaling pattern of the β1AR.Citation25 Therefore, this stoichiometric shift in favor of the supposedly “good” β2AR in HF appears unable to help the heart improve its structure and function.
Chronic β-blocker (ie, βAR antagonist) therapy reverses LV remodeling in H F, reduces risk of hospitalization, improves survival, reduces risk of arrhythmias (sudden cardiac death), improves coronary blood flow to the heart (relieves angina), and protects the heart against cardiotoxic overstimulation by the catecholamines (NE and Epi).Citation9 All of these effects result in a decrease in the oxygen/energy and metabolic demands of the heart (cardiac workload is decreased) and in an increase in its oxygen/energy supply, thereby improving, in the long run, LV function and performance. Various molecular mechanisms underlying these effects have been postulated: 1) direct antagonism of catecholaminergic cardiotoxic effects; 2) cardiac βAR upregulation and restoration of their signaling and function (ie, increase in adrenergic and inotropic reserves of the heart), which is needed in situations where the heart needs to work and sustain systemic circulation (eg, in acute stress or in acute HF episodes);Citation26 3) suppression of the elevated cardiotoxic, adverse remodeling–promoting, and pro-apoptotic neurohormonal systems (RAAS, endothelin); 4) coronary blood flow enhancement (as a result of diastolic prolongation); and 5) restoration of the reflex controls on the heart and the circulation.Citation27 Given that β2AR signaling, unlike β1AR signaling, might be cardioprotective in HF, novel β1AR-selective β-blockers (ie, like metoprolol) or combinations of a β-blocker with a β2AR-selective agonist (eg, clenbuterol) might be preferable for HF therapy (),Citation28 although carvedilol, a very successful and efficacious β-blocker in HF therapy, lacks βAR subtype selectivityCitation29
α1AR signaling targets for HF therapy
The human heart also expresses α1 ARs, albeit at much lower levels than βARs (∼20% of total βARs).Citation30 Their role in cardiac physiology is still a matter of debate, contrary to their well established effects in regulation of blood flow by inducing constriction in the smooth muscle wall of major arteries (eg, aorta, pulmonary arteries, mesenteric vessels, coronary arteries, etc).Citation31 The α1 ARs couple to the Gq/11 family of heterotrimeric G proteins, thereby activating phospholipase C (PLC)-β. PLCβ generates the second messengers, inositol [1,4,5]-trisphosphate (IP3) and 2-diacylglycerol (DAG) from the cell membrane component phospholipid phosphatidylinositol (4,5)-bisphosphate (PIP2). IP3 binds specific receptors in the sarcoplasmic reticulum (SR) membrane, which cause release of Ca2+ from intracellular stores, whereas DAG activates protein kinase C (PKC) ().Citation9 The end result is raised intracellular [Ca2+], which leads to contraction in vascular smooth muscle (vasoconstriction) and to activation of hypertrophic programs in the heart ().Citation15 α1ARs in HF may function in a compensatory fashion to maintain cardiac inotropy, but their involvement in cardiac pathophysiology appears limited to situations of cardiac hypertrophy that ultimately lead to HFCitation32 For instance, in the presence of pressure overload cardiac α1AARs get activated and promote cardiomyocyte survival (ie, block apoptosis), protecting against adverse remodeling and decompensation to HFCitation33,Citation34 Thus, cardiac α1AR antagonism, as well as inhibition of some cardiac α1AR signaling components, eg, Gq proteins or PKC (PKCβ2, for instance),Citation35,Citation36 have been pursued as HF therapeutic modalities ( and ), but the jury is still out with regards to their effectiveness.
α2AR signaling targets for HF therapy
Centrally located α2 ARs reduce SNS outflow (presynaptic inhibitory autoreceptors) and thus lower systemic blood pressure.Citation37,Citation38 The release of NE from cardiac sympathetic nerve terminals is controlled by both presynaptic α2A- and α2CARs,Citation39,Citation40 and secretion of Epi (mainly) and NE from the adrenal medulla is also controlled (ie, inhibited) by α2CARs present in chromaffin cell membranes ().Citation41,Citation42 Genetic deletion of both α2A- and α2CAR subtypes leads to cardiac hypertrophy and HF due to chronically enhanced cardiac NE release, as well as enhanced NE and Epi secretion from the adrenal medulla.Citation40–Citation42 In addition, the human α2CAR Del322–325 genetic variant that displays impaired signaling and sympatho-inhibitory function is associated with increased risk of HF in homozygous African-American carriers, especially when co-carried with the hyperfunctional cardiac β1AR Arg389 genetic variant, with the most probable mechanism being attenuated auto-inhibitory feedback of, and thus enhanced NE release from, the cardiac sympathetic nerves.Citation43 In fact, even in healthy humans, the α2CAR Del322–325 variant is associated with increased sympathetic nervous and adrenomedullary hormonal activities, during both supine rest and pharmacologically evoked catecholamine release.Citation44 Thus, presynaptic inhibitory α2-adrenergic autoreceptors crucially regulate SNS cardiac nerve activity and NE release into the heart, and any dysfunction of these receptors, either due to genetic polymorphisms or enhanced desensitization/downregulation (see GRK targets for HF therapy), translate into increased morbidity and mortality in chronic HF (). Perhaps the crucial role of presynaptic α2ARs in regulating NE release from cardiac SNS nerves stems from the fact that they are the only presynaptic ARs that can inhibit NE release;Citation9 presynaptic βARs (of the β2AR subtype, mainly) are facilitatory autoreceptors enhancing NE release at sympathetic nerve terminals,Citation9 a phenomenon whose inhibition may contribute to the therapeutic benefit of β-blockers in HF (see βAR signaling targets for HF therapy) ().
One of the hallmark abnormalities of chronic HF, contributing significantly to its morbidity and mortality, is chronically elevated SNS activity/outflow, as reflected by enhanced NE release in the heart (increased NE spillover) and enhanced adrenal catecholamine secretion leading to elevated circulating catecholamines.Citation45–Citation47 Initially an adaptive mechanism aiming to compensate decreased contractility following cardiac insult, it becomes progressively maladaptive, contributing to HF establishment and progression and to its morbidity and mortality.Citation48,Citation49 Based on this, sympatholysis (ie, reduction of SNS outflow and of circulating NE and Epi) is among the desirable goals of chronic HF therapy, and α2AR agonism has been employed in HF clinical trials as one such strategy ( and ). However, one such drug, moxonidine, failed to provide any survival or hemodynamic benefits in its clinical trial (Sustained Release Moxonidine for Congestive Heart Failure [MOXCON]),Citation50 an unexpected and unfortunate outcome attributed (somewhat paradoxically) to excessive, incompatible with life sympatho-inhibition caused by the drug (). Unfortunately, this misfortune essentially ended further interest by industry in developing sympatholytic strategies for chronic HF, although bucindolol, a β-blocker with strong sympatholytic properties, is currently being promoted for chronic HF treatment based (in part) also on its SNS activity-lowering effects.Citation51 Of note, chronic HF is also accompanied by enhanced α2AR desensitization and downregulation (see GRK targets for HF therapy and ),Citation52 which might be another reason that moxonidine failed in its clinical trials for HF therapy.
AT1R signaling targets for HF therapy
Landmark studies have shown that antagonism of RAAS provides HF patients with a substantial symptomatic and survival advantage.Citation53,Citation54 These observations are supported by studies unequivocally showing activation of the RAAS in both clinical and experimental HF.Citation55 Furthermore, the pivotal role of angiotensin II, acting through AT1Rs, in the process of cardiac adverse remodeling has been clearly documented in both clinical and experimental HF models.Citation56 Thus, both angiotensin-converting enzyme (ACE) inhibitors, which inhibit synthesis of angiotensin II, and AT1R antagonists (the so-called “angiotensin receptor blockers” [ARBs]) () ameliorate HF-associated LV adverse remodeling, such as fibrosis, hypertrophy, dilatation, myocardial stiffness, and oxidative stress, particularly after myocardial infarction, and RAAS antagonism is the cornerstone of all pharmacotherapeutic regimens currently employed in HF treatment.Citation57 Further adding to the importance of RAAS inhibition for HF therapy, antagonism of aldosterone, which is produced by the adrenal cortex (and possibly also in the heart per se) in response to angiotensin II activation of AT1Rs and is the last hormone activated in the RAAS hormonal axis, has recently emerged as a very effective therapeutic strategy for advanced-stage HF, promoting patient survival and ameliorating LV adverse remodeling ().Citation58 AT1R is a classic Gq/11-coupled receptor, ie, it signals through the very same pathway as do α1ARs (see α1AR signaling targets for HF therapy) (), although it can also couple to Gi/o and Gs proteins in certain cell types.Citation8 In addition, it also signals via β arrestins (βarrs), independently of G proteins (see βarr targets for HF therapy).Citation8 Finally, in the central nervous system (CNS), the AT1R can elevate SNS activity/outflow, which also contributes to the adverse hemodynamic and LV remodeling responses to myocardial infarction that angiotensin II activation of this receptor elicits.Citation9 Thus, part of the benefit of RAAS inhibitors (and of AT1R antagonists in particular) in HF might also derive from centrally mediated suppression of SNS activity ( and ).
Targets for HF therapy in signaling from other GPCRs
Endothelin receptors
Unlike blockade of the SNS and RAAS, which have been successful in HF therapy, strategies targeting the endothelin system have largely failed.Citation1 Endothelin is another cardiotoxic hormone, the plasma concentrations of which are considerably increased in patients with HF and correlate with disease severity.Citation59 Additionally, endothelin is the most potent endogenous vasoconstrictor substance produced in the body (much more potent than angiotensin II).Citation59 Therefore, development of endothelin receptor antagonists for HF therapy made perfect sense on paper, but, although the hemodynamic profile of various endothelin receptor inhibitors has been favorable and these drugs have proven to be of significant therapeutic value for pulmonary arterial hypertension (PAH) and might also be of value in coronary artery disease (CAD), they have not provided any substantial benefit for HF patients in terms of survival or disease progression.Citation59,Citation60 This might indicate that endothelin receptor signaling has only a minimal impact on cardiac function and certainly negligible compared with the impact of adrenergic or angiotensin II receptor signaling in the heart. The only HF indication for which endothelin receptor antagonists might hold promise right now appears to be right atrial/ventricular HF, given the benefits they provide in PAH ().
Adenosine receptors
Adenosine is a purine nucleoside that exerts a variety of physiological actions by binding to four adenosine cell surface GPCR subtypes, namely A1, A2a, A2b, and A3. The cardioprotective effects of adenosine have been extensively studied and are primarily mediated by activation of the A1-receptor (A1R) subtype in ischemic preconditioning.Citation61 However, activation of A1R, which primarily couples to (inhibitory or other) Gi/o proteins, also slows HR, which is therapeutically exploited in treatment of certain supraventricular arrhythmias, but, in the context of chronic HF, it might constitute an undesirable effect, as it can lead to bradycardias and atrioventricular blocks.Citation61 A partial A1R agonist, capadenoson,Citation62 was very recently shown to improve LV function and prevent progressive cardiac adverse remodeling in a canine chronic HF model.Citation63 Importantly, improvement of LV systolic function seemed to occur early after treatment initiation with capadenoson, and, since the compound is not a full agonist at the A1R, it appears devoid of the HR-lowering complications with which full A1R agonism is hampered.Citation63 Although the precise signaling mechanism(s) that mediate these beneficial effects of partial A1R agonism in chronic HF remain to be worked out, this study strongly indicates that A1R-selective (partial) agonists might have a place in the chronic HF drug armamentarium in the future ().
Adrenomedullin receptor
Adrenomedullin is a peptide hormone released from multiple tissue types, including the kidneys and the adrenal medulla, in response to pressure and volume overloads, and its plasma levels have been shown to be elevated in acute decompensated HF.Citation64 In chronic HF, its levels appear to be independently predictive of 2 year mortality, especially in non-ischemic and in New York Heart Association (NYHA) class II or lower HF.Citation64 Its receptor is also a GPCR, albeit an unusual one: the receptor protein is encoded by the calcitonin receptor-like receptor (CRLR) gene, but, on its own, it is not functional. The receptor protein has to structurally couple to one of the receptor activity-modifying proteins (RAMPs) in order to become functional and capable of signaling.Citation65 The adrenomedullin receptor thus consists of the CRLR bound to RAMP2 (or RAMP3) and primarily couples to the G protein Gs, ie, activates, similarly to the βARs, the classic AC–cAMP–PKA signaling pathway, which, in the cardiac myocyte, leads to positive inotropy (increased contractility) ().Citation65 Therefore, adrenomedullin and its analogs exert potent positive inotropic effects in the heart, which, coupled with their other beneficial effects on the circulation, such as hypotension and natriuresis,Citation66 make adrenomedullin receptor agonists (adrenomedullin non-peptide analogs, for instance) attractive possibilities for future HF drug development (). What’s more, these drugs might prove useful for both acute and chronic HF treatments.
Relaxin receptors
The relaxins are a multi-member peptide hormone family comprising several relaxin-like and insulin-like peptides.Citation67 They are primarily involved in functional regulation of the reproductive and neuroendocrine systems, but they are also present in the brain and in the cardiovascular system, where relaxin-1 and -2 exert potent vasoactive (vasodilatory) effects.Citation67 Their cellular effects are mediated by four different types of relaxin family peptide (RXFP) receptors, which are all GPCRs. RXFP1 and RXFP2 receptors primarily couple to the Gs protein–AC–cAMP–PKA signaling pathway, which leads to positive inotropy in the heart and vasodilatation in vascular smooth muscle ().Citation67 The RXFP1 receptor has also been shown to activate the phosphatidylinositol 3-kinase (PI3K) and extracellular signal-regulated kinase (ERK) signaling pathways.Citation67 Given that RXFP1 and RXFP2 receptors, which are both activated by relaxin-2, can lead to positive inotropy in the myocardium,Citation67 recombinant relaxin peptides and peptide analogs are currently pursued for HF drug development, and especially for acute decompensated HF (). One such agent, recombinant human relaxin-2 or serelaxin, is currently in clinical trials by Novartis for acute HF treatment, and the results have so far been more than encouraging.Citation68 Thus, development of relaxin peptide analogs and, specifically, of RXFP1 or RXFP2 receptor peptide or non-peptide agonists for acute HF is another area of HF drug development research that currently holds great promise, even though the chances of these agents also proving useful for chronic HF are low ().
Corticotrophin-releasing factor receptors
Corticotrophin-releasing factor (CRF) receptors (CRFRs) are GPCRs for the CRF family of peptide hormones, which includes urotensin-1, urocortins (Ucns), and CRF (or corticotrophin-releasing hormone [CRH]) itself, of course.Citation69 CRFR type 2 (CRFR2) is abundantly expressed in the human heart and primarily couples to Gs proteins, thereby activating the AC–cAMP–PKA (ie, the positive inotropic) signaling pathway of the cardiomyocytes ().Citation69 This means that CRFR2 agonism can have cardiostimulatory effects and, indeed, in sheep HF, CRFR2 activation by Ucn1 induces sustained reductions in cardiac preload and afterload, improvements in cardiac output, and inhibition of a variety of cardiotoxic neurohormonal systems (eg, RAAS, endothelin, vasopressin, etc).Citation70 Antagonism of this receptor produces the mirror opposite effects in the same animals.Citation70 On the other hand, in human HF, CRFR2 activation by Ucn2 or stresscopin (a CRF-like peptide that selectively activates CRFR2) induces increases in cardiac output and LV ejection fraction, along with a fall in systemic vascular resistance.Citation71,Citation72 Thus, similarly to the adrenomedullin and relaxin receptors above, Ucn-dependent CRFR2 activation produces positive inotropic, vasodilatory, and diuretic effects simultaneously, thereby making CRFR2 agonism another possible avenue for future HF drug development ().
Vasopressin receptors
Vasopressin is another very potent vasoactive peptide hormone that exerts its cellular actions via GPCRs (three different vasopressin receptor types: V1R, V2R, V3R).Citation73 The human cardiac myocyte expresses (albeit to a limited extent) V1R, which is a Gq/11 protein-coupled receptor, ie, activates the PLCβ–IP3–DAG–PKC signaling pathway, similarly to the α1ARs (see α1AR signaling targets for HF therapy).Citation73 This signaling pathway leads to vasoconstriction in vascular smooth muscle cells and to hypertrophy in the myocardium, again similarly to the α1ARs (). Thus, vasopressin receptor antagonism poses as a possible therapeutic strategy in HF to combat cardiac hypertrophy, high blood pressure and systemic vascular resistance, overactivation of other cardiotoxic neurohormonal systems (eg, RAAS, SNS), and, of course, to reduce volume overload of the heart, along with its accompanying water and electrolyte abnormalities, which stem from the excessive V2R-dependent anti-diuresis in the kidneys that causes hyponatremia.Citation74,Citation75 Nevertheless, the main indication for vasopressin receptor antagonist drugs currently is hyponatremia (along with some other renal indications), and only in very few countries (eg, Japan but not in the USA) are they currently approved for congestive HF ().Citation76 Pretty much like α1AR and endothelin receptor antagonism, cardiac V1R antagonism, as well as antagonism of vasopressin receptors in general, clearly warrants further investigation before it can be considered a legitimate therapeutic strategy for HF.
Glucagon peptide hormone receptors
Glucagon receptor (GCGR) and glucagon-like peptide (GLP)-1 receptor (GLP1R) are class B GPCRs mediating some of the cardiovascular effects of glucagon and GLP1, respectively, both members of the glucagon family of peptide hormones.Citation77 GCGR is a Gs protein-coupled receptor and is present in cardiac myocyte membranes, where it can activate the positive inotropic AC–cAMP–PKA signaling pathway, pretty much like the β1AR does ().Citation77 This constitutes the molecular basis for the clinical use of glucagon to acutely raise cardiac output in acute decompensated HF patients who are on β-blockers,Citation77 a situation in which use of adrenergic positive inotropes (eg, dobutamine, dopamine) would be ineffective (). Glucagon receptor agonism for chronic HF has not been studied but is probably not recommended, given that it is, in essence, a positive inotropic therapy that raises workload and oxygen and metabolic demands of the heart.
The GLP1R is also expressed in the heart, where it seems to mediate some of the beneficial cardiac effects of GLP1, such as inhibition of apoptosis, myocyte proliferation, and even positive inotropy.Citation78,Citation79 The signaling pathways initiated by GLP1R in the heart underlying these GLP1 effects are not entirely clear, but probably involve activation of the PI3K-Akt and the ERK1/2 signaling cascades (see Relaxin receptors), pathways known to result in cell survival and proliferation.Citation78,Citation79 Given these beneficial effects of GLP1R in the heart, GLP1 analogs have been tried as potential therapies in a number of experimental HF studies, including some with concurrent diabetes mellitus but also some independent of diabetic complications, and the results are overall promising.Citation78,Citation79 Thus, GLP1R agonism represents another potential avenue for future HF drug development, especially for those types of HF that are of diabetic etiology or are complicated by diabetes ().
G protein targets for HF therapy
With regards to heterotrimeric G proteins as potential targets for HF therapy, the reader is referred to the preceding sections of this article (specifically, the sections on ARs). In addition to the heterotrimeric G proteins, small (or Ras-like) G proteins also exist and provide an important link between the cell surface and intracellular signaling pathways.Citation80 Among these, the small G protein “Ras homolog gene family member A” (RhoA) activates a protein kinase, the Rho kinase, which can drive the cardiac hypertrophic process ().Citation80 Inhibition of Rho kinase with the drug fasudil has been shown to reverse LV remodeling in experimental myocardial infarction, thus posing Rho kinase (or perhaps even RhoA itself) as a novel molecular target for HF therapy ( and ).Citation81,Citation82
Moreover, the second messenger cAMP can activate, in addition to PKA (see βAR signaling targets for HF therapy), another effector in the cardiac myocyte, the “Exchange protein directly activated by cAMP” (Epac) ().Citation83 Epac was initially discovered as a cAMP-dependent and PKA-independent guanine nucleotide exchange factor (GEF) for the small G protein Rap1 (ie, Rap1 activator) but it is now known to be a multi-purpose adapter protein mediating a plethora of protein–protein interactions in the cell.Citation83 Epac2 was recently shown to mediate β1AR-dependent SR Ca2+ leak and arrhythmias in the heart, upon its activation by cAMP generated by cardiac β1AR stimulation.Citation84 This suggests that Epac inhibition might be of therapeutic value for cardiac arrhythmias and potentially also for HF, but further studies are needed to delineate the precise role(s) of Epac in the heart and in HF ( and ).
GRK targets for HF therapy
The majority of GPCRs are subject to agonist-promoted (homologous) desensitization and downregulation, a regulatory process that diminishes receptor response to continuous or repeated agonist stimulation.Citation85,Citation86 At the molecular level, this process is initiated by receptor phosphorylation by a family of kinases, termed GPCR kinases (GRKs), followed by binding of βarrs to the GRK-phosphorylated receptor. The βarrs then uncouple the receptor from its cognate G proteins, sterically hinder its further binding to them (functional desensitization), and subsequently target the receptor for internalization.Citation85,Citation86 Across all mammalian species, GRK2 and GRK5 are the most physiologically important members of the GRK family because they are expressed ubiquitously and regulate the vast majority of GPCRs. They are particularly abundant in neuronal tissues and in the heart.Citation87,Citation88 The elevated SNS outflow and NE and Epi levels in chronic HF lead to chronically elevated stimulation of the cardiac βAR system, which has detrimental repercussions for the failing heart. Extensive investigations over the past 3 decades have helped delineate the molecular alterations afflicting the cardiac βAR system that occur during HF, and it is now well known that, in chronic human HF, cardiomyocyte βAR signaling and function are significantly deranged and the adrenergic reserve of the heart is diminished.Citation9,Citation48 Cardiac βAR dysfunction in human HF is characterized at the molecular level by selective reduction of β1AR density at the plasma membrane (downregulation) and by uncoupling of the remaining membrane β1ARs and β2ARs from G proteins (functional desensitization).Citation23,Citation24 Importantly, myocardial levels and activities of GRK2 and GRK5 are elevated, both in humans and in animal models of HF.Citation89,Citation90 This GRK elevation possibly serves as a homeostatic protective mechanism aimed at defending the heart against excessive catecholaminergic toxicity. However, several studies soon refuted this assumption, demonstrating that GRK2 upregulation is detrimental for the heart and causes the functional uncoupling of βARs in vivo.Citation91 This finding prompted investigations of the role GRK2 plays in cardiac function, which revealed that cardiac GRK2 is an absolutely critical regulator of cardiac βAR-dependent contractility and function. Specifically, cardiomyocyte-restricted overexpression of GRK2 to the same level of upregulation found in human HF (ie, three- to four-fold) markedly attenuated βAR signaling and contractile reserve, showing that GRK2 is the main culprit for the functional desensitization of cardiac βARs in HF ().Citation92 The proof for this was provided by studies of the in vivo inhibition of cardiomyocyte GRK2, which were enabled by the development of the βARKct (beta adrenergic receptor kinase C-terminal fragment) mini-gene, which blocks cell membrane translocation and hence activation of GRK2, and its cardiomyocyte-specific expression in vivo in transgenic mice by virtue of the αMHC (myosin heavy chain) gene promoter.Citation90 Indeed, GRK2 inhibition in vivo in the heart with βARKct (or its partial genetic deletion) enhances cardiac contractility both at baseline and after adrenergic stimulation, reverses the contractile and βAR dysfunctions, and preserves or even augments cardiac function and survival in HF.Citation90 Of note, the antidepressant paroxetine was recently shown to inhibit GRK2, thus providing a lead for the development of GRK2-specific small molecule inhibitors.Citation93 In summary, elevated SNS activity in chronic HF causes enhanced GRK2-mediated cardiac β1- and β2AR desensitization and β1AR downregulation, which leads to the progressive loss of the adrenergic and inotropic reserves of the heart, the hallmark molecular abnormality of this disease ().Citation94
Additionally, GRK2 expression and activity are also increased in the adrenal gland during HF.Citation95 Specifically, our studies over the past few years have established that adrenal GRK2 upregulation is responsible for severe adrenal α2AR dysfunction in chronic HF, which causes a loss of the sympatho-inhibitory function of these receptors in the adrenal gland, and catecholamine secretion is thus chronically elevated ().Citation9,Citation11,Citation96 This emerging crucial role for adrenal GRK2 in HF is underlined by the fact that its specific inhibition, via adenoviral-mediated βARKct adrenal gene delivery (see above), leads to a significant reduction in circulating catecholamine levels, restoring not only adrenal, but also cardiac function in HF.Citation95 Additional evidence for the crucial role of adrenal GRK2-regulated α2ARs in regulating adrenal SNS tone in HF comes from the phenylethanolamine-N-methyl transferase (PNMT)-driven GRK2 knockout (KO) mice.Citation97 These mice, which do not express GRK2 in their adrenal medullae from birth, display decreased SNS outflow and circulating catecholamines in response to myocardial infarction, which translates into preserved cardiac function and morphology over the course of the ensuing HF.Citation97 Of note, elevated GRK2-dependent α2AR dysfunction during HF might also occur in other peripheral sympathetic nerve terminals of the heart () and of other organs, thus contributing to the increased NE release and spillover, as well as to the presynaptic α2AR dysfunction in SNS neurons observed in chronic HF (see α2AR signaling targets for HF therapy).Citation9,Citation11 Thus, GRK2 inhibition poses not only as a positive inotropic therapy in the heart per se, but also as a novel sympatholytic strategy in HF, blocking catecholamine release at the sources of these hormones (ie, adrenals and cardiac SNS terminals) and preventing their toxic effects on peripheral organs, like the heart. In addition, adrenal βARKct expression might have a synergistic action with β-blockers, as both of these therapeutic strategies target adrenergic hyperactivity in HF ( and ).
βarr targets for HF therapy
βarrs comprise two ubiquitously expressed isoforms, βarr1 and βarr2 (arrestin-2 and -3 respectively), both of which are abundantly expressed in cardiac muscle.Citation98 As co-factors of GRKs in βAR desensitization/downregulation, they contribute to the diminished inotropic and adrenergic reserves of the failing heart and their inhibition should theoretically be beneficial in acute HF, as it would enhance the Gs–AC–PKA axis of pro-contractile signaling of cardiac βARs (see βAR signaling targets for HF therapy), thereby increasing cardiac contractility.Citation98 However, βarrs do not merely terminate G protein-mediated signaling by GPCRs. It is now well established that they promote signaling in their own right, independently of G proteins, and a number of recent studies point to a beneficial role played by them in the heart, especially when they engage the cardiac β1AR.Citation99 More specifically, they have been reported to mediate epidermal growth factor receptor (EGFR) transactivation by the β1AR.Citation99 Consistent with this, a mutant β1AR lacking 14 GRK phosphorylation sites in its C-terminal tail that cannot undergo βarr-dependent desensitization, fails to transactivate the EGF receptors.Citation100 In response to chronic isoproterenol stimulation, transgenic mice expressing this β1AR mutant develop severe dilated cardiomyopathy with significantly increased LV dilatation, decreased fractional shortening, and increased myocardial apoptosis compared with wild-type β1AR-expressing transgenic mice.Citation100 In this model, inhibition of EGF receptors worsens the dilated cardiomyopathy, suggesting a protective rather than deleterious role for transactivated EGFRs in the heart and prompting the investigators to speculate that βarr-dependent EGFR transactivation exerts a cardioprotective effect and thus, βarr-mediated (in contrast to the classical G protein-dependent) β1AR signaling might be of therapeutic benefit in HF ( and ).Citation99,Citation100
Effects of cardiac βarr-dependent signaling can be quite different when the AT1R is bound by βarrs, An artificially constructed AT1AR mutant (AT1-i2m), which fails to activate G proteins but nonetheless interacts with βarrs, activates the mitogenic Src–Ras–ERK1/2 pathway in vitro.Citation101 In vivo, cardiomyocyte-specific overexpression of this receptor mutant leads to greater cardiomyocyte hypertrophy, bradycardia, and fetal cardiac gene expression than comparable overexpression of the wild-type receptor.Citation101 Conversely, overexpressed wild-type AT1AR produces greater cardiomyocyte apoptosis and interstitial fibrosis than the G protein-uncoupled mutant, suggesting that G protein-dependent and -independent AT 1AR signals mediate different aspects of the hypertrophic response.Citation101 Of course, these studies do not directly implicate βarr signaling; another series of studies using the AngII peptide analog SII ([SarCitation1-IleCitation4-IleCitation8]-AngII), which, when bound to the AT1AR, elicits βarr signaling but no Gq protein signaling,Citation98 provide direct evidence for potential roles of βarrs in cardiac AT1AR signaling. Several studies have shown that AT 1R βarr-dependent signaling in cardiac myocytes leads to cardiomyocyte proliferation without hypertrophy (which requires Gq/11 protein signaling) and can even result in positive inotropy and lusitropy.Citation102 These effects require GRK6 and βarr2, whereas GRK2 seems to oppose them, consistent with the specialized role of GRK isoforms described in a transfected system.Citation102 On the other hand, AT1R-bound βarrs do not produce inotropic or chronotropic effects in isolated Langendorff-perfused cardiac preparations despite activating ERK1/2.Citation103 Thus, it seems that, while cardiac AT1R promotes hypertrophy and cardiomyocyte proliferation via the classical Gq protein–PKC pathway, it can increase cardiac contractility and function via βarr2-dependent signaling (). Since βarr2 is bound to stop the G protein-mediated signaling of the receptor, and GRK2 also seems to oppose this pro-contractile signaling of βarr2, it follows that stimulation of βarr2 activity and/or GRK2/βarr1 inhibition at the cardiac AT1R might be of therapeutic value in HF and/or cardiac hypertrophy treatments ( and ).Citation8 In fact, since βarr1 also mediates AT1R-induced aldosterone production and secretion in the adrenal cortex (),Citation8,Citation104,Citation105 βarr1 inhibition in both the heart and adrenals might be of therapeutic value in chronic HF ().
Conclusions and future perspectives
The tremendous progress of molecular biology, physiology, and pharmacology over the past 2 decades or so, coupled with the advent of the first GPCR structures and of the so-called “rational” (ie, target- and structure-based) drug design have provided clinicians and pharmacologists with a tremendous expansion of the therapeutic arsenal for HF. Nevertheless, HF still remains the most devastating, in terms of morbidity, mortality, quality of life, and health care costs, cardiovascular disease and the number one killer in the western world.Citation1–Citation7 Thus, new and innovative drugs are desperately needed in order to, if not cure, at least improve quality of life of HF patients.Citation1 The field of GPCR signaling keeps providing exciting new possibilities and targets for HF drug development. For instance, targeting intracellular signaling components of the traditional cardiac GPCR drug targets, βARs and AT1Rs, such as heterotrimeric G proteins per se (Gi protein activation or Gq protein inhibition), small G proteins like RhoA, and Epac, might produce novel useful HF drugs. Another exciting avenue for future HF drug development is targeting and exploitation of new GPCRs, the important roles of which in cardiac physiology and pathophysiology keep getting uncovered, such as select adenosine receptor agonism or agonism of certain vasoactive peptide hormone receptors, eg, adrenomedullin, relaxins, or Ucns (CRFR2). In addition, further studies on signaling of cardiac α1ARs, endothelin, and vasopressin receptors, as well as on central and adrenal α2AR signaling, might also finally yield some valid therapeutic targets. Finally, targeting molecules that regulate cardiac GPCR signaling, such as cardiac GRKs and βarrs, is perhaps the most exciting area for future HF drug development. For instance, GRK2 inhibition, which can provide a positive inotropic and sympatholytic therapy at the same time, has the potential to revolutionize current chronic HF therapy. On the other hand, as the physiological relevance of cardiac βarr signaling becomes fully elucidated, selective targeting of βarr1 or βarr2 in the heart or development of “biased” GPCR ligands, which selectively recruit βarrs over G proteins (or vice versa) at cardiac GPCRs,Citation98 will also offer attractive options for future HF drug development. The research conducted to date already indicates that inhibition of βarr1 or stimulation of βarr2 in the heart per se might be beneficial for cardiac function and in HF. Future studies will help clarify the picture regarding the potential of cardiac GRK2 and βarr targeting for HF treatment. Nonetheless, cardiac GPCR signal transduction appears to be among the research fields that currently hold the largest potential and the biggest promise for the future of HF drug design and development.
Acknowledgments
AL is supported by a Scientist Development Grant from the American Heart Association (AHA No 09SDG2010138, National Center).
Disclosure
The authors report no conflicts of interest in this work.
References
- Kaye DM Krum H Drug discovery for heart failure: a new era or the end of the pipeline? Nat Rev Drug Disc 2007 6 2 127 139
- Swedberg K Cleland J Dargie H Task Force for the Diagnosis and Treatment of Chronic Heart Failure of the European Society of Cardiology Guidelines for the diagnosis and treatment of chronic heart failure: executive summary (update 2005): the Task Force for the Diagnosis and Treatment of Chronic Heart Failure of the European Society of Cardiology Eur Heart J 2005 26 11 1115 1140 15901669
- Abhayaratna W Smith WT Becker NG Marwick TH Jeffery IM McGill DA Prevalence of heart failure and systolic ventricular dysfunction in older Australians: the Canberra Heart Study Med J Aust 2006 184 4 151 154 16489896
- Levy D Kenchaiah S Larson MG Long-term trends in the incidence of and survival with heart failure N Engl J Med 2002 347 18 1397 1402 12409541
- Roger VL Weston SA Redfield MM Trends in heart failure incidence and survival in a community-based population JAMA 2004 292 3 344 350 15265849
- Mosterd A Hoes AW de Bruyne MC Prevalence of heart failure and left ventricular dysfunction in the general population; the Rotterdam Study Eur Heart J 1999 20 6 447 455 10213348
- Cleland JG Khand A Clark A The heart failure epidemic: exactly how big is it? Eur Heart J 2001 22 8 623 626 11286518
- Lymperopoulos A Bathgate A Arrestins in the cardiovascular system Prog Mol Biol Transl Sci 2013 118 297 334 23764059
- Lymperopoulos A Rengo G Koch WJ Adrenergic nervous system in heart failure: pathophysiology and therapy Circ Res 2013 113 739 753 23989716
- Lin HH G-protein-coupled receptors and their (Bio) chemical significance win 2012 Nobel Prize in chemistry Biomed J 2013 36 3 118 124 23806881
- Lymperopoulos A Rengo G Koch WJ GRK2 inhibition in heart failure: something old, something new Curr Pharm Des 2012 18 2 186 191 22229578
- Skeberdis VA Gendviliene V Zablockaite D Beta3-adrenergic receptor activation increases human atrial tissue contractility and stimulates the L-type Ca2+ current J Clin Invest 2008 118 9 3219 3227 18704193
- Gauthier C Leblais V Kobzik L The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle J Clin Invest 1998 102 7 1377 1384 9769330
- Rozec B Erfanian M Laurent K Trochu JN Gauthier C Nebivolol, a vasodilating selective beta(1)-blocker, is a beta(3)-adrenoceptor agonist in the nonfailing transplanted human heart J Am Coll Cardiol 2009 53 17 1532 1538 19389564
- Bers DM Calcium cycling and signaling in cardiac myocytes Annu Rev Physiol 2008 70 23 49 17988210
- Daaka Y Luttrell LM Lefkowitz RJ Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A Nature 1997 390 6655 88 91 9363896
- Movsesian MA Kukreja RC Phosphodiesterase inhibition in heart failure Handb Exp Pharmacol 2011 204 237 249 21695643
- Communal C Singh K Sawyer DB Colucci WS Opposing effects of beta(1)- and beta(2)-aadrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein Circulation 1999 100 22 2210 2212 10577992
- Chesley A Lundberg MS Asai T The beta(2)-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through G(i)-dependent coupling to phosphatidylinositol 3′-kinase Circ Res 2000 87 12 1172 1179 11110775
- Zhu WZ Zheng M Koch WJ Lefkowitz RJ Kobilka BK Xiao RP Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes Proc Natl Acad Sci U S A 2001 98 4 1607 1612 11171998
- Dorn GW2nd Tepe NM Lorenz JN Koch WJ Liggett SB Low-and high-level transgenic expression of beta2-adrenergic receptors differentially affect cardiac hypertrophy and function in Galphaq-overexpressing mice Proc Natl Acad Sci U S A 1999 96 11 6400 6405 10339599
- Liggett SB Tepe NM Lorenz JN Early and delayed consequences of beta(2)-adrenergic receptor overexpression in mouse hearts: critical role for expression level Circulation 2000 101 14 1707 1714 10758054
- Bristow MR Ginsburg R Umans V Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure Circ Res 1986 59 3 297 309 2876788
- Bristow MR Ginsburg R Minobe W Decreased catecholamine sensitivity and beta-adrenergic receptor density in failing human hearts N Engl J Med 1982 307 4 205 211 6283349
- Nikolaev VO Moshkov A Lyon AR Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation Science 2010 327 5973 1653 1657 20185685
- Iaccarino G Tomhave ED Lefkowitz RJ Koch WJ Reciprocal in vivo regulation of myocardial G protein-coupled receptor kinase expression by beta-adrenergic receptor stimulation and blockade Circulation 1998 98 17 1783 1789 9788834
- Adamson PB Gilbert EM Reducing the risk of sudden death in heart failure with beta-blockers J Card Fail 2006 12 9 734 746 17174236
- Birks EJ Tansley PD Hardy J Left ventricular assist device and drug therapy for the reversal of heart failure N Engl J Med 2006 355 18 1873 1884 17079761
- DiNicolantonio JJ Lavie CJ Fares H Menezes AR O’Keefe JH Meta-analysis of carvedilol versus beta 1 selective beta-blockers (atenolol, bisoprolol, metoprolol, and nebivolol) Am J Cardiol 2013 111 5 765 769 23290925
- Woodcock EA Du XJ Reichelt ME Graham RM Cardiac alpha 1-adrenergic drive in pathological remodelling Cardiovasc Res 2008 77 3 452 462 18032391
- Shannon R Chaudhry M Effect of alpha1-adrenergic receptors in cardiac pathophysiology Am Heart J 2006 152 5 842 850 17070143
- Knowlton KU Michel MC Itani M The alpha 1 A-adrenergic receptor subtype mediates biochemical, molecular, and morphologic features of cultured myocardial cell hypertrophy J Biol Chem 1993 268 21 15374 15380 8393439
- Du XJ Gao XM Kiriazis H Transgenic alpha1 A-adrenergic activation limits post-infarct ventricular remodeling and dysfunction and improves survival Cardiovasc Res 2006 71 4 735 743 16859660
- Huang Y Wright CD Merkwan CL An alpha1A-adrenergic extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes Circulation 2007 115 6 763 772 17283256
- Esposito G Prasad SV Rapacciuolo A Mao L Koch WJ Rockman HA Cardiac overexpression of a G(q) inhibitor blocks induction of extracellular signal-regulated kinase and c-Jun NH(2)-terminal kinase activity in in vivo pressure overload Circulation 2001 103 10 1453 1458 11245652
- Hwang H Robinson DA Stevenson TK PKCβII modulation of myocyte contractile performance J Mol Cell Cardiol 2012 53 2 176 186 22587992
- Philipp M Hein L Adrenergic receptor knockout mice: distinct functions of 9 receptor subtypes Pharmacol Ther 2004 101 1 65 74 14729393
- Philipp M Brede M Hein L Physiological significance of alpha(2)-adrenergic receptor subtype diversity: one receptor is not enough Am J Physiol Regul Integr Comp Physiol 2002 283 2 R287 R295 12121839
- Hein L Altman JD Kobilka BK Two functionally distinct alpha2-adrenergic receptors regulate sympathetic neurotransmission Nature 1999 402 6758 181 184 10647009
- Brede M Wiesmann F Jahns R Feedback inhibition of catecholamine release by two different alpha2-adrenoceptor subtypes prevents progression of heart failure Circulation 2002 106 19 2491 2496 12417548
- Brede M Nagy G Philipp M Sorensen JB Lohse MJ Hein L Differential control of adrenal and sympathetic catecholamine release by alpha2-adrenoceptor subtypes Mol Endocrinol 2003 17 8 1640 1646 12764077
- Lymperopoulos A Rengo G Koch WJ Adrenal adrenoceptors in heart failure: fine-tuning cardiac stimulation Trends Mol Med 2007 13 12 503 511 17981507
- Small KM Wagoner LE Levin AM Kardia SL Liggett SB Synergistic polymorphisms of beta1- and alpha2C-adrenergic receptors and the risk of congestive heart failure N Engl J Med 2002 347 15 1135 1142 12374873
- Neumeister A Charney DS Belfer I Sympathoneural and adrenomedullary functional effects of alpha2C-adrenoceptor gene polymorphism in healthy humans Pharmacogenet Genomics 2005 15 3 143 149 15861038
- Cohn JN Levine TB Olivari MT Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure N Engl J Med 1984 311 13 819 823 6382011
- Floras JS The “unsympathetic” nervous system of heart failure Circulation 2002 105 15 1753 1755 11956112
- Rengo G Lymperopoulos A Zincarelli C Myocardial adeno-associated virus serotype 6-betaARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure Circulation 2009 119 1 89 98 19103992
- Port JD Bristow MR Altered beta-adrenergic receptor gene regulation and signaling in chronic heart failure J Mol Cell Cardiol 2001 33 5 887 905 11343413
- Bristow MR Beta-adrenergic receptor blockade in chronic heart failure Circulation 2000 101 5 558 569 10662755
- Cohn JN Pfeffer MA Rouleau J MOXCON Investigators Adverse mortality effect of central sympathetic inhibition with sustained-release moxonidine in patients with heart failure (MOXCON) Eur J Heart Fail 2003 5 5 659 667 14607206
- Kao DP Wagner BD Robertson AD Bristow MR Lowes BD A personalized BEST: characterization of latent clinical classes of nonischemic heart failure that predict outcomes and response to bucindolol PLoS One 2012 7 11 e48184 23144856
- Aggarwal A Esler MD Socratous F Kaye DM Evidence for functional presynaptic alpha-2 adrenoceptors and their down-regulation in human heart failure J Am Coll Cardiol 2001 37 5 1246 1251 11300430
- CONSENSUS Trial Study Group Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group N Engl J Med 1987 316 23 1429 1435 2883575
- Cohn JN Johnson G Ziesche S A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure N Engl J Med 1991 325 5 303 310 2057035
- Francis GS Benedict C Johnstone DE Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD) Circulation 1990 82 5 1724 1729 2146040
- Weber K Extracellular matrix remodeling in heart failure: a role for de novo angiotensin II generation Circuation 1997 96 11 4065 4082
- Schieffer B Wirger A Meybrunn M Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat Circulation 1994 89 5 2273 2282 8181153
- Lang CC Struthers AD Targeting the renin-angiotensin-aldosterone system in heart failure Nat Rev Cardiol 2013 10 3 125 134 23319100
- Kaoukis A Deftereos S Raisakis K The role of endothelin system in cardiovascular disease and the potential therapeutic perspectives of its inhibition Curr Top Med Chem 2013 13 2 95 114 23470073
- Kaluski E Kobrin I Zimlichman R RITZ-5: randomized intravenous TeZosentan (an endothelin-A/B antagonist) for the treatment of pulmonary edema: a prospective, multicenter, double-blind, placebo-controlled study J Am Coll Cardiol 2003 41 2 204 210 12535809
- Mustafa SJ Morrison RR Teng B Pelleg A Adenosine receptors and the heart: role in regulation of coronary blood flow and cardiac electrophysiology Handb Exp Pharmacol 2009 193 161 188 19639282
- Nell PG Albrecht-Küpper B The adenosine A1 receptor and its ligands Prog Med Chem 2009 47 163 201 19328291
- Sabbah HN Gupta RC Kohli S Chronic therapy with a partial adenosine A1-receptor agonist improves left ventricular function and remodeling in dogs with advanced heart failure Circ Heart Fail 2013 6 3 563 571 23564604
- Nishikimi T Kuwahara K Nakagawa Y Kangawa K Nakao K Adrenomedullin in cardiovascular disease: a useful biomarker, its pathological roles and therapeutic application Curr Protein Pept Sci 2013 14 4 256 267 23745694
- Hong Y Hay DL Quirion R Poyner DR The pharmacology of adrenomedullin 2/intermedin Br J Pharmacol 2012 166 1 110 120 21658025
- Yoshizawa T Sakurai T Kamiyoshi A Novel regulation of cardiac metabolism and homeostasis by the adrenomedullin-receptor activity-modifying protein 2 system Hypertension 2013 61 2 341 351 23297372
- Bathgate RA Halls ML van der Westhuizen ET Callander GE Kocan M Summers RJ Relaxin family peptides and their receptors Physiol Rev 2013 93 1 405 480 23303914
- Metra M Cotter G Davison BA Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the Relaxin in Acute Heart Failure (RELAX-AHF) development program: correlation with outcomes J Am Coll Cardiol 2013 61 2 196 206 23273292
- Wang J Li S Corticotropin-releasing factor family and its receptors: tumor therapeutic targets? Biochem Biophys Res Commun 2007 362 4 785 788 17822675
- Emeto TI Moxon JV Rush C Woodward L Golledge J Relevance of urocortins to cardiovascular disease J Mol Cell Cardiol 2011 51 3 299 307 21689660
- Davis ME Pemberton CJ Yandle TG Urocortin 2 infusion in human heart failure Eur Heart J 2007 28 21 2589 2597 17720993
- Gheorghiade M Greene SJ Ponikowski P Haemodynamic effects, safety, and pharmacokinetics of human stresscopin in heart failure with reduced ejection fraction Eur J Heart Fail 2013 15 6 679 689 23471413
- Holmes CL Landry DW Granton JT Science review: Vasopressin and the cardiovascular system part 1. Receptor physiology Crit Care 2003 7 6 427 434 14624682
- Gassanov N Semmo N Semmo M Nia AM Fuhr U Er F Arginine vasopressin (AVP) and treatment with arginine vasopressin receptor antagonists (vaptans) in congestive heart failure, liver cirrhosis and syndrome of inappropriate antidiuretic hormone secretion (SIADH) Eur J Clin Pharmacol 2011 67 4 333 346 21327910
- Ambrosy AP Vaduganathan M Mentz RJ Clinical profile and prognostic value of low systolic blood pressure in patients hospitalized for heart failure with reduced ejection fraction: insights from the Efficacy of Vasopressin Antagonism in Heart Failure: Outcome Study with Tolvaptan (EVEREST) trial Am Heart J 2013 165 2 216 225 23351825
- Kinugawa K Imamura T Komuro I Experience of a vasopressin receptor antagonist, tolvaptan under the unique indication in Japanese heart failure patients Clin Pharmacol Ther Epub July 19 2013
- Ussher JR Drucker DJ Cardiovascular biology of the incretin system Endocr Rev 2012 33 2 187 215 22323472
- Lorber D GLP-1 receptor agonists: effects on cardiovascular risk reduction Cardiovasc Ther 2013 31 4 238 249 23865382
- Brubaker PL Minireview: update on incretin biology: focus on glucagon-like peptide-1 Endocrinology 2010 151 5 1984 1989 20305008
- Surma M Wei L Shi J Rho kinase as a therapeutic target in cardiovascular disease Future Cardiol 2011 7 5 657 671 21929346
- Hattori T Shimokawa H Higashi M Long-term inhibition of Rho-kinase suppresses left ventricular remodeling after myocardial infarction in mice Circulation 2004 109 18 2234 2239 15096457
- McKinsey TA Kass DA Small-molecule therapies for cardiac hypertrophy: moving beneath the cell surface Nat Rev Drug Discov 2007 6 8 617 635 17643091
- Schmidt M Dekker FJ Maarsingh H Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions Pharmacol Rev 2013 65 2 670 709 23447132
- Pereira L Cheng H Lao DH Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia Circulation 2013 127 8 913 922 23363625
- Reiter E Lefkowitz RJ GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling Trends Endocrinol Metab 2006 17 4 159 165 16595179
- Ferguson SS Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling Pharmacol Rev 2001 53 1 1 24 11171937
- Arriza JL Dawson TM Simerly RB The G-protein-coupled receptor kinases beta ARK1 and beta ARK2 are widely distributed at synapses in rat brain J Neurosci 1992 12 10 4045 4055 1403099
- Rockman HA Koch WJ Lefkowitz RJ Seven-transmembrane-spanning receptors and heart function Nature 2002 415 6868 206 212 11805844
- Belmonte SL Blaxall BC G protein coupled receptor kinases as therapeutic targets in cardiovascular disease Circ Res 2011 109 3 309 319 21778435
- Rengo G Lymperopoulos A Leosco D Koch WJ GRK2 as a novel gene therapy target in heart failure J Mol Cell Cardiol 2011 50 5 785 792 20800067
- Ungerer M Böhm M Elce JS Erdmann E Lohse MJ Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart Circulation 1993 87 2 454 463 8381058
- Koch WJ Rockman HA Samama P Cardiac function in mice overexpressing the beta-adrenergic receptor kinase or a beta ARK inhibitor Science 1995 268 5215 1350 1353 7761854
- Thal DM Homan KT Chen J Paroxetine is a direct inhibitor of g protein-coupled receptor kinase 2 and increases myocardial contractility ACS Chem Biol 2012 7 11 1830 1839 22882301
- Eschenhagen T Beta-adrenergic signaling in heart failure: adapt or die Nat Med 2008 14 5 485 487 18463653
- Lymperopoulos A Rengo G Funakoshi H Eckhart AD Koch WJ Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure Nat Med 2007 13 3 315 323 17322894
- Lymperopoulos A Bathgate A Pharmacogenomics of the heptahelical receptor regulators G-protein-coupled receptor kinases and arrestins: the known and the unknown Pharmacogenomics 2012 13 3 323 341 22304582
- Lymperopoulos A Rengo G Gao E Ebert SN Dorn GW2nd Koch WJ Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction J Biol Chem 2010 285 21 16378 16386 20351116
- Lymperopoulos A Beta-arrestin biased agonism/antagonism at cardiovascular seven transmembrane-spanning receptors Curr Pharm Des 2012 18 2 192 198 22229558
- Noor N Patel CB Rockman HA B-arrestin: a signaling molecule and potential therapeutic target for heart failure J Mol Cell Cardiol 2011 51 4 534 541 21074538
- Tilley DG G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function Circ Res 2011 109 2 217 230 21737817
- Zhai P Yamamoto M Galeotti J Cardiac-specific overexpression of AT1 receptor mutant lacking G alpha q/G alpha i coupling causes hypertrophy and bradycardia in transgenic mice J Clin Invest 2005 115 11 3045 3056 16276415
- Rajagopal K Whalen EJ Violin JD Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes Proc Natl Acad Sci U S A 2006 103 44 16284 16289 17060617
- Aplin M Christensen GL Schneider M Differential extracellular signal-regulated kinases 1 and 2 activation by the angiotensin type 1 receptor supports distinct phenotypes of cardiac myocytes Basic Clin Pharmacol Toxicol 2007 100 5 296 301 17448114
- Lymperopoulos A Rengo G Zincarelli C Kim J Soltys S Koch WJ An adrenal beta-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo Proc Natl Acad Sci U S A 2009 106 14 5825 5830 19289825
- Lymperopoulos A Rengo G Zincarelli C Kim J Koch WJ Adrenal Beta-arrestin 1 inhibition in vivo attenuates post-myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels J Am Coll Cardiol 2011 57 3 356 365 21232674