 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
There is a dearth of treatment options for community-acquired and nosocomial Pseudomonas infections due to several rapidly emerging multidrug resistant phenotypes, which show resistance even to combination therapy. As an alternative, developing selective promiscuous hybrid compounds for simultaneous modulation of multiple targets is highly appreciated because it is difficult for the pathogen to develop resistance when an inhibitor has activity against multiple targets.
Methods
In line with our previous work on phytochemical–antibiotic combination assays and knowledge-based methods, using a fragment combination approach we here report a novel drug design strategy of conjugating synergistic phytochemical–antibiotic combinations into a single hybrid entity for multi-inhibition of P. aeruginosa DNA gyrase subunit B (GyrB)/topoisomerase IV subunit B (ParE) and dihydrofolate reductase (DHFR) enzymes. The designed conjugates were evaluated for their multitarget specificity using various computational methods including docking and dynamic simulations, drug-likeness using molecular properties calculations, and pharmacophoric features by stereoelectronic property predictions.
Results
Evaluation of the designed hybrid compounds based on their physicochemical properties has indicated that they are promising drug candidates with drug-like pharmacotherapeutic profiles. In addition, the stereoelectronic properties such as HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbital), and MEP (molecular electrostatic potential) maps calculated by quantum chemical methods gave a good correlation with the common pharmacophoric features required for multitarget inhibition. Furthermore, docking and dynamics simulations revealed that the designed compounds have favorable binding affinity and stability in both the ATP-binding sites of GyrB/ParE and the folate-binding site of DHFR, by forming strong hydrogen bonds and hydrophobic interactions with key active site residues.
Conclusion
This new design concept of hybrid “phyto-drug” scaffolds, and their simultaneous perturbation of well-established antibacterial targets from two unrelated pathways, appears to be very promising and could serve as a prospective lead in multitarget drug discovery.
Introduction
Pseudomonas aeruginosa is one of the most opportunistic and challenging pathogenic bacteria, with constant evolution of resistance and the complexity of multidrug resistant phenotypes due to the extensive use of antibacterial agents by monotherapeutic strategy.Citation1 Multidrug resistant phenotypes in P. aeruginosa occur through the acquisition of multiple imported resistance mechanisms, coupled with chromosomally encoded resistance mechanisms, accumulation of multiple chromosomal changes over time, and/or a single mutational event leading to overexpression of one or more efflux pumps.Citation2 This continuing trend of developing drug resistance can severely limit the therapeutic options for treatment of serious infections caused by pathogens, specifically, P. aeruginosa. Alarmingly, the prospects for novel antipseudomonal drugs in the next few years are quite poor, with most of the drugs in the pipeline being more or less new derivatives of existing families of antimicrobial agents.Citation3 As an alternative strategy, antibacterial combination therapy is being used as a standard practice in many hospitals for a broader empirical coverage by simultaneously administering two different antibacterial agents, leading to a synergistic effect. However there are no clinical data supporting its effectiveness against gram negative infections as well as the issues of drug toxicity, side effects, and selection of multidrug resistant organisms.Citation4,Citation5 Taken together, it is imperative to identify novel chemical entities through effective therapeutic strategy to combat the threat of multidrug resistance (MDR).
Antibacterial polypharmacology is an emerging strategy that overcomes the limitations of both monotherapy and combination therapy by targeting multiple proteins simultaneously in the disease-associated network.Citation6–Citation8 Recently, there has been greater recognition that multi-action hybrid compounds, through fragment combination, can be a potent strategy against robust pathogens because of their ability to delay the onset, decrease the likelihood, and overcome the existing resistance mechanisms with reduced toxicity concerns.Citation6 In a fragment combination approach, two dissimilar entities are combined together with a noncleavable linking group designed to bind independently to two different biological targets and synchronously accumulate at both the target sites.Citation9,Citation10 Furthermore, in comparison to a combination of drugs, multitarget inhibitors have greater predictable pharmacokinetic and pharmacodynamic relationships, as they involve administration of a single drug.Citation11 This fragment combination strategy holds significant promise and has been successfully applied in the design and development of novel, dual-action, hybrid antibacterial agents, such as hybrids, that delay the emergence of bacterial resistance by inhibiting bacterial protein synthesis and fluoroquinolone targets,Citation12 berberine (plant alkaloid, sensitive to NorA efflux) – (2-phenyl-5-nitro-1H-indole, NorA efflux pump inhibitor) hybrid antibacterial, insensitive to MDR efflux,Citation13 trimethoprim–fluoroquinolone hybrids for dual action in dihydrofolate reductase enzyme and fluoroquinolone targets (DNA gyrase subunit A, [GyrA] and topoisomerase IV subunit A, [ParC]).Citation14 Thus, the evidence increasingly shows that discovery of druglike entities with predefined promiscuity is emerging as a productive area of research.
In line with this, we employed a combined knowledge-based and fragment combination approach, focused on the design and development of novel multitarget hybrid compounds that can act by targeting GyrB/ParE and DHFR enzymes through optimization of phytochemicals by conjugating with antibiotics. To the best of our knowledge, this study is the first to successfully combine knowledge from the scientific literature with previous drug combination data and a fragment combination approach towards the identification of novel multi-targeting “phyto-drug” conjugates. In silico-based approaches have always shown promising potential in facilitating drug discovery, and these methods have also been successfully employed for the search and design of selective multitarget compounds.Citation15 Furthermore, we have integrated various effective computational tools to investigate the molecular, electronic, and pharmacokinetic properties, binding affinity and stability of the inhibitor–enzyme complexes of our designed compounds.
Methods and computational details
Ligand data set preparation and properties predictions
Marvin sketch (ChemAxon, Budapest, Hungary) was used to create the SMILES (.smi) format for the designed inhibitors. Open Babel online interface (http://openbabel.sourceforge.net/), through shell script and using .smi as input, was used to generate primary 3D structures, which were then saved in SDF file format.Citation16 The structures were subjected to conformational search calculations in gas phase using the Merck molecular force field method in the SPARTAN’10 program (WaveFunction, Inc, Irvine, CA, USA). The minimum energy conformation was chosen for geometry-optimization using the Austin model 1 (AM1) semiempirical algorithm. The optimized structures were reoptimized using the density functional theory hybrid, B3LYP/6-31G** basis set functional.Citation17 The stereoelectronic properties such as HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) energies, band gap (HOMO–LUMO), MEP, dipole moment and polarizability, physicochemical properties (molecular volume, polar surface area [PSA]), and absolute energies were calculated from single-point energy calculations on the complete geometry-optimized structures of the designed compounds using B3LYP/6-31G** basis set. In addition, percentage of absorption (%ABS) was calculated by using the following equation,
based on the computed PSA values.Citation18
To obtain additional insight on the toxicity risks (mutagenicity, tumorigenicity, irritation, and reproductive effects) and physicochemical properties (molecular weight, cLogP, solubility, PSA, number of hydrogen bond donors and acceptors, number of rotatable bonds, drug-likeness, and overall drug score) of the designed hybrid compounds, GyrB/ParE inhibitors (cyclothialidine and novobiocin) and DHFR inhibitors (methotrexate and trimethoprim), phytochemicals (protocatechuic acid and gallic acid), and sulfonamides (sulfamethoxazole and sulfadiazine), were calculated using OSIRIS property explorerCitation19 and Molinspiration property prediction toolkit (Molinspiration Cheminformatics, Slovensky Grob, Slovak Republic) (http://www.molinspiration.com/cgi-bin/properties). The toxicity risk assessment indicated potential risks in the drawn structure concerning the risk category specified, and the process relied on the precomputed set of structural fragments that give toxicity alerts if they were encountered in the drawn 2D structures. Properties with high risks of undesired effects, like mutagenicity or a poor intestinal absorption, were shown as a negative sign, whereas a positive sign indicated drug-conform behavior.
Molecular docking
The MolDock docking engine in Molegro virtual docker (CLC bio, Aarhus N, Denmark) was used for studying the binding modes of the designed hybrid compounds in the active site cavities of the modeled P. aeruginosa GyrB/ParE and DHFR enzymes (see Supplementary materials), which was based on a new heuristic search algorithm (MolDock score) that combines differential evolution with a cavity prediction algorithm.Citation20 In our docking experiments, a MolDock grid scoring function using template docking with default values: −500 overall strength and 0.4 Å energy grid resolution was used to evaluate the energy between the ligand and the target enzyme. Grid resolution, number of runs, population size, maximum iterations, pose generation energy threshold, simplex evolution max steps, and neighbor distance factor were set as 0.30 Å, 20, 50, 1500, 100, 1.00 for each run, respectively, using the MolDock SE algorithm. The ligands from the crystal structures of Escherichia coli GyrB/ParE and DHFR were transferred into the workspace, keeping the orientation as a control and were kept as the reference ligand. The complete geometry-optimized structures of the hybrid compounds and the generated protein homologs were also transferred, and hydrogen molecules were added to both ligands and protein molecules using the preparation wizard in the Molegro workspace. During import of the 3D structures of the ligands, charges and bond orders were assigned, the torsional angle of the 3D structures was also determined, and all acyclic single bonds were set as flexible. Binding sites in the electrostatic surface of the protein were identified using the grid-based cavity prediction algorithm. A total of five cavities were detected, the prepositioned reference ligand in the active site cavity was identified, and the docking was constrained to the predicted active site cavity. Multiple poses were returned for each run with the root mean square deviation (RMSD) threshold set to 1.00 Å. The pose with the highest rerank and MolDock score was retained in the workspace for detailed evaluation of the ligand binding at the active site cavity. The rerank score uses a weighted combination of the terms used by the MolDock score mixed with a few additional terms (the rerank score includes the steric terms which are Lennard–Jones approximations to the steric energy; the MolDock score uses a piece-wise linear potential to approximate the steric energy).Citation20 The rerank scoring function improved the docking accuracy by identifying the most promising docking solution from the solutions obtained by the MolDock docking algorithm.Citation20 The rerank score provided an estimate of the strength of the interaction. It was not calibrated in chemical units, and it did not take complex contributions such as entropy into account. Even though the rerank score might be successful in ranking different poses of the same ligand, it might be less successful in ranking poses of different ligands.Citation20 Along with both MolDock and reranking scores, we also predicted binding affinities using a calibrated model that is included in the Molegro virtual docker. The binding affinity model was trained using a data set of more than 200 structurally diverse complexes from Protein Data Bank (PDB) with known binding affinities.Citation21 Hence, in our docking experiments we used this recommended strategy of ranking the docking results by their rerank scores and subsequently the binding affinity measure to get high ranked poses. The validation of the docking protocol was carried out by redocking the imported reference ligands from their respective experimental PDB structures in the predicted active site cavity of the model using the RMSD measure. The RMSD (Å) values for the redocked inhibitor methotrexate (MTX) and substrate NADP in the modeled P. aeruginosa DHFR active site were 0.3968 Å and 0.5794 Å, respectively, wherein the substrate, ADPNP (5′–adenylyl-β-γ-imidodiphosphate, the nonhydrolyzable analog of ATP) in the P. aeruginosa GyrB/ParE were 0.2529 Å/0.2052 Å, indicating high similarity between predicted and experimental binding mode. Furthermore, the binding conformations of ligands in the enzyme active site cavity were analyzed by visualizing the H-bond and electrostatic interactions formed between the ligands and the active site residues.
Molecular dynamics simulations
The molecular dynamics simulations for the corresponding substrate bound enzymes and top ranked docked inhibitor in both the enzymes were performed with the YASARA dynamics package version 10 (YASARA Biosciences, Vienna, Austria).Citation22 A periodic simulation cell of at least 20 Å larger than the protein was employed with explicit solvent around the P. aeruginosa enzyme complexes (I–GyrB, IV–DHFR VII–ParE, II–ADPNP–GyrB, V–MTX-NADP-DHFR, VIII–ADPNP-ParE), and top ranked hybrid compound-bound enzyme complexes (III-phyto-drug [PD]_2a-GyrB, VI–PD_2a-NADP-DHFR, and IX–PD_2a-ParE) were energy minimized to correct the covalent geometry and to remove bumps. The AMBER03 force field was used with Van der Waals pairs cut-off distance 7.86 Å and long range statistics algorithm calculated using the Particle Mesh Ewald method.Citation23 The automatic force field parameter assignment was carried out by AutoSMILES,Citation24 which assigned pH-dependent fractional bond orders and protonation patterns. This was followed by geometry-optimization of the structures with the COSMO solvation modelCitation24 using semiempirical AM1 Mulliken point charge calculations and assignment of AM1BCC (AM1 bond charge correction),Citation25 atom, and bond types, with further refinement using known restrained electrostatic potential charges. And finally, GAFF (General AMBER force field)Citation26 atom types and remaining force field parameters were assigned. The hydrogen bonding network of the complexes were optimized by a method that positions polar hydrogen atoms in protein structures by optimizing the total hydrogen bond energy.Citation27 The simulation cell was filled with TIP3 water, and Na+ and Cl− counter ions were added at the most favorable positions to neutralize the ion strength in the cell. A few water molecules were deleted to readjust the solvent density to 0.997 g/mL, and the pressure was controlled by rescaling the simulation box dimensions to maintain the water density. The pKa values were predicted using the empirical pKa prediction equation that is approximated as a function of electrostatic potential, hydrogen bonds, and accessible surface area by Ewald summation implemented in YASARA.Citation28 A short molecular dynamics simulation was run on the solvent only, and the entire system was energy minimized by steepest descent minimization to remove conformational stress. This was followed by simulated annealing minimization using a time step of 2 femtoseconds and atom velocities scaled down by 0.9 every 10th step until convergence (energy improved by less than 0.1% during 200 steps). The molecular dynamics simulation was then initiated, with temperature fixed at 298 K and multiple time steps set for intramolecular and intermolecular forces at 1.33 femtoseconds and 4 femtoseconds, respectively. Each complex was subjected to 3000 picoseconds (ps) of molecular dynamics simulations, and snapshots were saved every 1 ps for data analysis. The trajectories of the molecular dynamics simulations were analyzed for the equilibrium stability by measuring the RMSD of the complexes and the root mean square fluctuation (RMSF) of residues around the ligand-binding active sites.
Results
Multi-action hybrid compounds design hypothesis
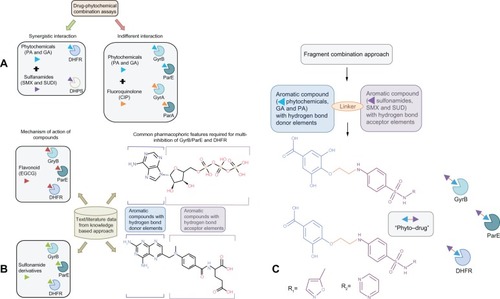
As a first step, we surveyed the biomedical literature to establish a link between the targets (GyrB/ParE and DHFR) based on the fact that if the same compound has activity against different targets they might be related ().Citation29 It has been reported that the plant flavonoid, epigallocatechin gallate and its derivatives access the active site cavities of GyrB, ParE and DHFR) enzymes,Citation30–Citation32 which is illustrated in . In support of this, we proposed earlier that phytochemicals (structural analogs of epigallocatechin gallate, protocatechuic acid and gallic acid) are synergistic in combination with the antifolate drugs, sulfamethoxazole and sulfadiazine (which target dihydropteroate synthase enzyme in the folic acid biosynthesis pathway) by binding to DHFR, and are additive in combination with ciprofloxacin (which targets GyrA and ParC) by binding to GyrB and ParE against P. aeruginosa isolatesCitation33,Citation34 (). It has also been reported that derivatives of benzene sulfonamide groups also bind to the target enzymes,Citation35,Citation36 which is illustrated in . Additionally, important interactions for the inhibition of the enzymes GyrB/ParE and DHFR of E. coli have been identified based on their individual pharmacophore models from molecular dynamics simulations.Citation37,Citation38 Using these pharmacophore models we derived a common pharmacophoric feature model for multiinhibition of these enzymes (), which are (1) an aromatic element with hydrogen-bond donor elements mapping the adenine moiety of ATP in GyrB/ParE and the pterin/pteridine moiety of dihydrofolate/methotrexate in DHFR,Citation37,Citation38 and (2) ribose and phosphate groups of ATP in GyrB as well as p-aminobenzoate linker and glutamate tail region of folate, mapping for aromatic/hydrophobic moiety with hydrogen-bond acceptor elements.Citation37,Citation38 Thus, we embarked on using the simple phenols (gallic acid and protocatechuic acid), which are hydroxyl-substituted aromatic ring compounds identified from our previous work, to satisfy the common pharmacophoric feature: (1) conjugated with a noncleavable linking group (hydrophobic element) to another aromatic element with hydrogen bond acceptor moieties and (2), the sulfonamide groups (sulfamethoxazole and sulfadiazine) for the design of multitargeting “phyto-drug” hybrid compounds ().
Figure 1 How did we connect the dots? Hypothesis and design of multitargeting compounds. (A) Drug–phytochemical interactions identified using combination assays. Green arrows signify synergistic interaction of phytochemicals (protocatechuic acid and gallic acid, represented as blue arrowhead) in combination with sulfonamide antibiotics (sulfamethoxazole and sulfadiazine, represented as violet arrowhead). Sulfonamides block the dihydropteroate synthase enzyme, while the phytochemicals target the subsequent enzyme dihydrofolate reductase (DHFR) in the folate biosynthesis pathway leading to a synergistic effect. Red arrows signify indifferent interaction of phytochemicals (protocatechuic acid and gallic acid, represented as blue arrowhead) in combination with fluoroquinolone antibiotics (ciprofloxacin, represented as orange arrowhead). Ciprofloxacin binds to DNA gyrase subunit A (GyrA) and topoisomerase IV subunit A (ParC), while phytochemicals target the subunit B (GyrB and ParE) of both these enzymes leading to an indifferent effect via equivalent action of DNA replication inhibition; (B) Knowledge-based approach was used to establish relationship between therapeutic targets (GyrB/ParE and DHFR) identified based on our previous drug–phytochemical interaction network. Mechanism of action of epigallocatechin gallate (plant flavonoid, red arrowhead) and previously reported sulfonamide derivatives targeting GyrB/ParE and DHFR enzymes, gives an illustration of the ability of these compounds to bind to the identified target sets. Derived common pharmacophoric features for multi-inhibition of GyrB/ParE and DHFR enzymes based on the previously reported individual pharmacophore models; (C) Schematic illustration of the fragment combination method used to derive the hybrid compounds, phytochemicals (blue arrowhead) linked with sulfonamide groups (violet arrowhead) using noncleavable linker (orange line) that can simultaneously bind to GyrB/ParE and DHFR enzymes.
In silico calculations of physicochemical, stereoelectronic properties, drug likeness, and toxicity risks
In order to identify the ultimate drug candidate it is first necessary to start with right lead compounds of drug likeness and ligand efficiency.Citation39 Thus in recent years, various in silico-based predictions of physicochemical properties with the consideration of drug likeness have gained importance due to their potential to rapidly eliminate drug candidates prior to synthesis. It is also evident that several physicochemical properties are useful indicators for drug likeness of a compound with an impact on specific pharmacokinetic processes like permeation, intestinal absorption, reversible binding, distribution, and excretion of a compound.Citation40
As a preliminary step in our study, we investigated relevant toxicity risks, physicochemical properties, bioavailability, drug likeness, and overall drug scores of the designed hybrid compounds using various in silico-based prediction toolkits. The predicted toxicity risks shown in indicate that all the hybrid compounds are nonmutagenic, nontumorigenic, nonirritating, nonreproductive, and are comparable to standard traded drugs. For comparison, we evaluated the toxicity risks for commercially available GyrB/ParE inhibitors (cyclothialidine and novobiocin) and DHFR inhibitors (methotrexate and trimethoprim) of which cyclothialidine and trimethoprim showed nontoxic effects, whereas novobiocin and methotrexate showed risks concerning reproductive and tumorigenic effects, respectively. Also, the predicted toxicity risks for phytochemicals (protocatechuic acid and gallic acid) showed mutagenic effects for protocatechuic acid, while gallic acid showed both mutagenic and reproductive effects. Among sulfonamides (sulfamethoxazole and sulfadiazine), sulfamethoxazole showed tumorigenic risk effects, and sulfadiazine was found to be associated with a reproductive toxicity alert. Overall, the proposed hybrid structures (PD_1a, PD_1b, PD_2a, and PD_2b) in showed nontoxic behavior with highly desirable physicochemical parameters disclosing their potential as promising agents for antimicrobial therapy.
Table 1 Calculated toxicity risks, molecular properties, drug likeness, and overall drug score of the compounds investigated
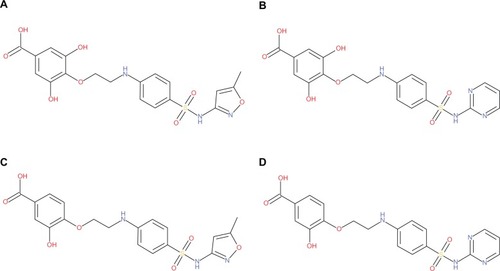
Figure 2 Structures of the designed hybrid compounds, “phyto-drug” (phytochemicals–antibiotics conjugates, (A) PD_1a; (B) PD_1b; (C) PD_2a; (D) PD_2b.
The Lipinski’s RO5, used as a filter for drug likeness, defines four simple physicochemical parameter ranges (molecular weight ≤ 500, cLogP ≤ 5, H-bond donors ≤ 5, H-bond acceptors ≤ 10) associated with acceptable aqueous solubility and intestinal permeability. Molecules violating more than one of these rules may have problems with oral bioavailability.Citation41 The physicochemical properties calculated, such as molecular weight of the proposed compounds are below 450 (), and thus are more likely to have higher absorption. The cLogP value is the logarithm of its partition coefficient between n-octanol and water, measuring the compound’s hydrophilicityCitation19 The estimated cLogP values for the proposed compounds are less than 5.0, indicating high lipophilicity and higher absorption and permeation. The aqueous solubility (log S) measure significantly affects the drug absorption/distribution characteristics, and is a unit stripped logarithm of the solubility (moles/L). It is suggested that more than 80% of the drugs in the market have aqueous solubility values greater than −4.Citation19 The solubility measures for the proposed compounds in are in the expected range of −2.95 to −3.97 and are comparable to that of the standard commercially available drugs. The number of Lipinski’s RO5 violations is one each for designed hybrid compounds PD_1a, PD_1b and none for PD_2a and PD_2b, which are within the range sets defined for oral bioavailability. The number of rotatable bonds and PSA are now widely used filters for drug likeness, and those compounds which meet only the two criteria of: (1) ≤ 10 rotatable bond counts, and (2) PSA ≤ 140 ÅCitation2 (or ≤ 12 H-bond donors and acceptors) will have increased oral bioavailability in the rat.Citation42 The calculated rotatable bond counts () of the proposed compounds are equal to 9, and PSA are within the range for compounds PD_2a and PD_2b, and just above for compounds PD_1a and PD_1b (). On the other hand using PSA, we calculated the %ABS according to Equationequation (1)(1) : %ABS = 109 − (0.354 × PSA) as reported by Zhao et al,Citation18 the calculated absorption percentages are in the range of 55%–65% for all the proposed compounds (). Drug likeness scores are calculated by summing up score values, partially based on topological descriptors, and fingerprints of Molfile MDL (Accelrys, San Diego, CA, USA) structure keys including properties, such as cLogP and molecular weight, are estimated to be in the positive range for 80% of the traded drugs.Citation19 The drug likeness scores for the proposed compounds are positive (), indicating that the molecules predominantly contain fragments which are frequently present in commercial drugs, and hence have the possibility of being ‘drug-like’. Finally, the overall drug score (), combining drug likeness, cLogP, aqueous solubility, molecular weight, and toxicity risks describe the potential to qualify for a drug on 0 to 1 scale.Citation19 The overall drug scores were in the range of 0.71–0.77 (), indicating the proposed compounds potential to qualify as suitable drug candidates.
Table 2 Calculated absolute energies, stereoelectronic, and molecular properties at B3LYP/6-3IG** basis set levels, and percentage of absorption of the designed hybrid compounds
In order to gain more insight into the molecular recognition through ligand-receptor interactions and structural factors that are involved in the multispecific activity of the designed hybrid compounds, stereoelectronic properties such as molecular orbital energies (HOMO and LUMO), band gap (difference between HOMO and LUMO), and MEP profiles were calculated using the B3LYP/631G** density functional theory on the geometry-optimized structures. Calculated ab initio quantum chemical B3LYP/6-31G** calculations and electronic properties of the compounds are listed in . HOMO and LUMO orbital energies are related to ionization potential and electron affinity and their respective frontier orbitals are associated to the molecule’s reactivity ie, HOMO energy is susceptible to electrophilic attack (donate e−) and LUMO energy susceptible to nucleophilic attack (accept e−).Citation44 The reactivity index (band gap) of the compounds with small difference implies high reactivity, and large difference implies low reactivity in reactions.Citation45 The energy gap (HOMO – LUMO, shown in ) of the proposed compounds was found to be within a narrow range of −4.35 to −5.03, indicating that these energies permit electron transfer and exchange making those compounds very highly reactive and similar in nature.
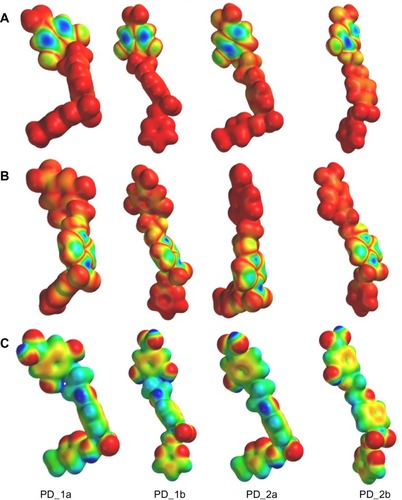
Three dimensional HOMO and LUMO isosurfaces over the Van der Waals surface and the MEP mapped onto total electron density surface of all the compounds are shown in –. For the three dimensional HOMO orbitals of all the compounds (), the predominant HOMO (electron-rich) orbitals were consistently observed on the hydroxyl substituted aromatic ring of the phytochemicals (gallic acid and protocatechuic acid), indicating strong electrophilic affinity, and the most significant LUMO (electron-poor) orbitals () of all the compounds were consistently observed on the sulfonamide rings, indicating the localization of strong electron acceptor elements, and is thereby susceptible to strong nucleophilic attack. Additionally, the MEP is a useful descriptor for understanding electrophilic/nucleophilic sites as well as hydrogen bonding interactions. In general, the negative potential sites (red color in ) of MEP represent regions of electrophilic reactivity and interaction through π-π bonding with the aromatic systems of interacting enzyme residues, while positive potential sites (blue color in ) represent regions of nucleophilic reactivity.Citation46 As expected, the MEP maps of all the compounds (), clearly show the presence of both a positive potential site and a negative potential site around the hydroxyl groups in the aromatic ring of the phytochemicals, suggesting that these hydroxyl groups can act as both hydrogen bond donor and acceptor elements, indicating their participation in hydrogen bond interactions with hydroxyl and carbonyl groups of key active site residues in GyrB (Asp75, Thr167, Tyr111, and water molecule), ParE (Asp68, Glu45, and water molecule) and DHFR (Asp30, Ile8, Tyr110, and Ile104) enzymes. The predominant red regions (negative potential sites) in the MEP profiles of all the compounds are localized around the sulfonyl functional groups (oxygen atoms) of the sulfonamide rings, nitrogen (N7) and oxygen (O8) atom in the oxazole ring of sulfamethoxazole, nitrogen (N7) atoms in the pyrimidine ring of sulfadiazine, oxygen (O8) atom and the carbonyl oxygen (O8) atom in phytochemicals, suggesting that these oxygen and nitrogen atoms can act as hydrogen bond acceptors and possibly participate in hydrogen bonding interaction with donor groups of key active site residues in GyrB (Tyr111, Asn48, Val122, Lys105, Leu117, His118, and Gly119), ParE (Tyr104, Asn41, Ile115, Gly114, Gly112, His111, and Leu110), and DHFR (Arg55 and Arg60) enzymes. Also, regions at the aromatic rings in all the compounds show red regions with decreasing intensity, indicating weak positive electrostatic potential and thereby implying a hydrophobic nature of the aromatic rings. The above mentioned molecular orbitals and MEP profile features appear to be consistent with the key pharmacophoric derivatives required for potent multi-inhibition of GyrB/ParE and DHFR enzymes.
Figure 3 Representations of (A) HOMO (B) LUMO and (C) Molecular electrostatic potential (MEP) isodensity surfaces calculated at B3LYP/6-31G** basis set levels on the optimized geometry of the hybrid compounds.
Molecular docking
To better understand the binding modes of the designed hybrid compounds in both GyrB/ParE and DHFR enzymes, molecular docking studies were carried out using the Molegro virtual docker program. The docking results of all the proposed compounds along with ADPNP (GyrB/ParE substrate)/methotrexate (DHFR inhibitor) in P. aeruginosa GyrB/ParE and DHFR are ranked based on their binding affinity scores ( and , and ).
Table 3 Docking results of ADPNP and designed hybrid compounds (ranked on the basis of binding affinity scores) at the ATP binding site in P. aeruginosa GyrB
Table 4 Docking results of methotrexate and designed hybrid compounds (ranked on the basis of binding affinity scores) at the folate binding site in P. aeruginosa DHFR
Docking of ADPNP and proposed hybrid compounds at the ATP binding site of GyrB
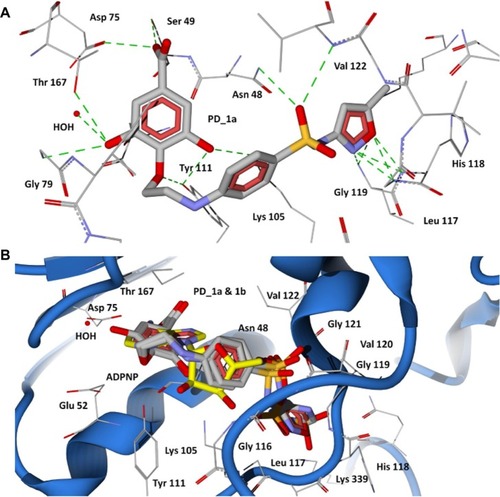
The 43-kDa domain of P. aeruginosa GyrB has two distinct subdomains, the N-terminal subdomain comprising an eight-stranded β sheet and four α helices containing the ATP-binding site, and the C-terminal subdomain consisting of a four-stranded β sheet and four α helices.Citation47 The GyrB in complex with the ATP analog, ADPNP, shows that the conformation is stabilized by H-bonds with residues Asn48, Asp75, Gly104, Lys105, Tyr111, Leu117, His118, Gly119, Val120, Gly121, Val122, Thr167, Lys 339, and Ile80, Ile96, and Val122 forming favorable hydrophobic interactions with the adenine and ribose moieties (), which are similar to the crystal structure of the E. coli GyrB43-ADPNP complex. In addition to these interactions, another important H-bond network was formed by the residues Asp75, Gly79, and Thr167 and the adenine ring of ADPNP with a conserved water molecule.Citation47 The residues that interact with the adenine ring of the ATP include Asp75, Tyr111, and Thr167. The ribose and phosphate groups are stabilized by Asn48, Lys105, Leu117, His118, Gly119, and Asn48 is one of the key residues required for binding of inhibitors.Citation38 Hence, a common feature can be rationalized in that the majority of the inhibitors of GyrB maintain a hydrogen bond network with Asp75, a highly conserved water molecule, and Asn48 residues, which is very important for binding of any new inhibitor class.Citation38,Citation47 The docking results show that the proposed hybrid compounds have good binding affinities at the ATP binding site of GyrB, maintaining contacts with all the key residues required for inhibition. The major residues participating in hydrogen bond interactions with the designed inhibitors in the GyrB active site are tabulated in . Considering the top ranked hybrid compounds PD_2a and 1a, both having similar binding conformations and interactions (), the hydroxyl moiety in the carboxylic group of the protocatechuic acid forms hydrogen bonds with Asp75 and Ser49 and the carbonyl moiety forms a hydrogen bond with Asn48, similar to the adenine ring interaction of ATP. The 3′-OH and 4′-O group of the protocatechuic acid in PD_2a forms additional hydrogen bond contacts with Thr167, Gly79, a conserved water molecule, and Tyr 111. The additional hydroxyl group in the gallic acid moiety of PD_1a forms hydrogen bond interactions with Tyr111 and Lys105 (). The phenyl ring of sulfamethoxazole in both PD_2a and 1a sits in the ribose binding region forming hydrophobic contacts with residues Ile80, Ile96, and Val122, while the sulfonyl group positioning similar to the α-phosphate group of ADPNP forms hydrogen bond acceptor contacts with Asn48 and Val122 residues, and the oxazole ring orienting in between the β and γ-phosphate groups of ADPNP forms similar hydrogen bond acceptor interactions with Leu117, His118, and Gly119 (). Compounds PD_2b and 1b were found to bind, preferably maintaining the important interactions in a similar way to PD_2a and 1a compounds (). However, in comparison to PD_2a and 1a, the interactions established by compounds PD_2b and 1b are weaker, and the main reason for this effect may be attributed to the pyrimidine ring positioned directly over the γ-phosphate moiety, thereby limiting the hydrogen bond interactions with Gly119 and His118 residues. Supporting our claim, it has previously been demonstrated that phenolic groups have favorable binding energy at the adenine binding pocket if ATP and in the phosphate group binding site, oxazole has favorable clustering compared to benzene groups.Citation38
Figure 4 Binding modes of ADPNP and methotrexate in P. aeruginosa GyrB and DHFR active site cavities obtained from docking studies. (A) The nonhydrolyzable analog of ATP (ADPNP) (yellow stick model) docked at ATP binding site of GyrB; (B) DHFR inhibitor methotrexate (yellow stick model) docked at folate binding site of DHFR and the cofactor NADP is shown as aqua stick model.
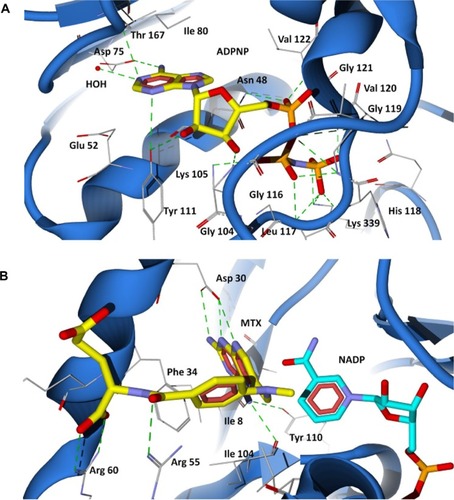
Figure 5 Docking of designed hybrid compounds at ATP binding site of P. aeruginosa GyrB. (A) Binding pose of the top scored compound PD_2a (grey stick model); (B) PD_2a and PD_2b (stick model colored by their element) superimposed with ADPNP (yellow stick model).
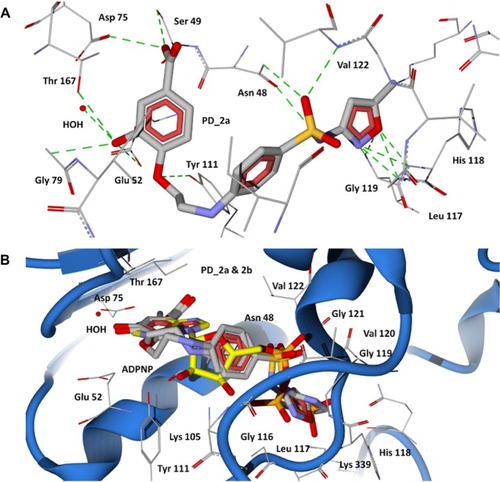
Figure 6 Docking of designed hybrid compounds at ATP binding site of P. aeruginosa GyrB. (A) Binding pose of the top scored compound PD_1a (grey stick model); (B) PD_1a and PD_1b (stick model colored by their element) superimposed with ADPNP (yellow stick model).
The 43 kDa E. coli GyrB subunit and the 43 kDa topoisomerase IV ParE subunit structures are strikingly similar in their conserved active sites, suggesting that inhibitors might simultaneously target both type II topoisomerase enzymes with a possibility for broad spectrum action.Citation48 Hence, we modeled the topoisomerase IV enzyme of P. aeruginosa using the E. coli template structure (1S16) ( and ), and we performed similar docking calculations of the designed hybrid compounds in the ADPNP bound active site cavity of ParE. The docked conformations obtained were similar to GyrB, indicating a highly conserved binding mode ( and ), and suggesting that the designed compounds also bind to the ParE subunit of Topoisomerase IV.
Docking of methotrexate and proposed hybrid compounds at the folate binding site of DHFR
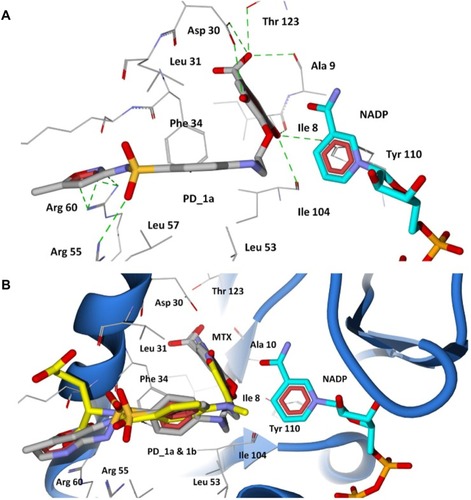
The 2,4-diaminopteridine ring of methotrexate is situated into a deep hydrophobic pocket and interacts strongly with the P. aeruginosa DHFR through five major hydrogen bonds (). The N1 and the 2-amino group of methotrexate form a pair of hydrogen bonds with Asp30. The N1 is protonated and the carboxylate of the acidic residue Asp30 is ionized, facilitating a favorable ionic interaction.Citation49 The 4-amino group forms strong hydrogen bond interactions with the main chain carbonyl oxygen atom of Ile8, Ile104, and the hydroxyl group of Tyr110. The interaction pattern of diaminopteridine ring structures of all antifolates are highly preserved among the known DHFR complexes in various species.Citation49 In addition to the hydrogen bonding, the diaminopteridine ring forms Van der Waals interactions with Phe34, Ile8, and Ile104. The residues Phe34, Leu53, and Leu57 provide Van der Waals contacts to the phenyl group in the p-amino benzoate ring of methotrexate. The carbonyl group of p-aminobenzoate forms a hydrogen bond with Arg55, and the glutamate tail of methotrexate lies on the protein surface with its α-carboxylic group forming strong salt bridge interactions with the positively charged Arg60 residue (). The docking results show that all the proposed hybrid compounds have a good fit into the cavity of the DHFR active site and are positioned in a way similar to the inhibitor (methotrexate). The major residues participating in hydrogen bond interactions with the designed inhibitors in the DHFR active site are tabulated in . The docked conformation of the top ranked hybrid compounds, PD_2a and PD_1a, are shown in and . The hybrid compounds PD_2a and 1a docks at the active site cavity of P. aeruginosa DHFR with its hydroxyl substituted aromatic rings being held in the interior of a deep cleft, while the sulfonamide moiety of sulfamethoxazole extends out towards the entrance of the binding pocket forming hydrogen bonding and hydrophobic interactions. The hydroxyl groups in the phenyl ring of phytochemicals are involved in hydrogen bonding interactions with six residues located interior of the active site Asp30, Thr123, Ile8, Ala9, Tyr110, and Ile104, while the ring contacts residues Phe34, Ile8, Ile104, and Leu31 by Van der Waals interactions. The phenyl ring of sulfamethoxazole adopts a similar orientation to that of the phenyl ring of methotrexate and forms Van der Waals contacts with Phe34 Leu31, Leu53, and Leu57 residues, which results in good ligand-enzyme affinity. The sulfonyl group of sulfamethoxazole forms hydrogen bond acceptor contacts with Arg55 by overlaying the carbonyl group of the p-aminobenzoate moiety in methotrexate ( and ). The oxazole ring of the sulfamethoxazole demonstrates hydrogen bond acceptor interactions with the guanidium group of Arg60 at the entrance of the pocket in an orientation similar to that of the α-carboxylic group in methotrexate. In the case of PD_1b and 2b ( and ), they are positioned in a similar way to that of PD_1a and 2a and also maintain similar interactions with the residues located in the interior of the active site. The phenyl ring of phytochemicals in both PD_1b and 2b are well adjusted in the hydrophobic pocket, situated deep inside the active site cavity, while the benzene sulfonamide ring adopts an orientation extended towards the Phe34 residue forming π-π contact and thereby disallowing the hydrogen bond interactions between the sulfonyl group and the Arg55 residue. On the other hand, the pyrimidine ring of sulfadiazine moieties in PD_1b and 2b establishes hydrogen bond interactions with both Arg55 and Arg60 residues.
Figure 7 Docking of designed hybrid compounds at folate binding site of P. aeruginosa DHFR. (A) Binding pose of the top scored compound PD_2a (grey stick model); (B) PD_2a and PD_2b (stick model colored by their element) superimposed with methotrexate (yellow stick model), and cofactor NADP is represented as aqua stick model.
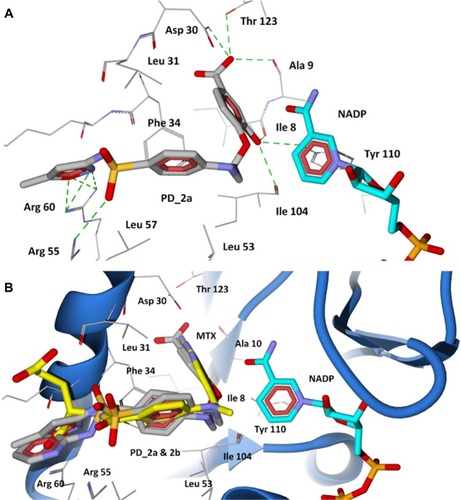
Figure 8 Docking of designed hybrid compounds at folate binding site of P. aeruginosa DHFR. (A) Binding pose of the top scored compound PD_1a (grey stick model); (B) PD_1a and PD_1b (stick model colored by their element) superimposed with methotrexate (yellow stick model), and cofactor NADP is represented as aqua stick model.
Molecular dynamics simulations
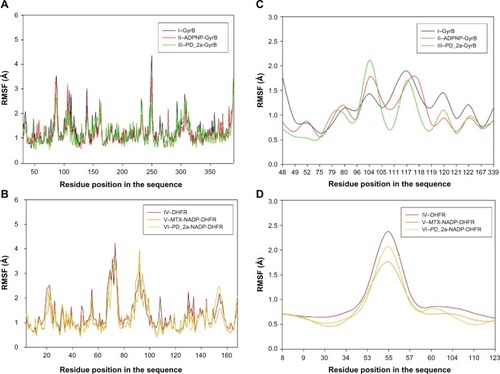
To evaluate the stability of the unbound enzyme complexes (I–GyrB, IV–DHFR and VII–ParE), ligand-bound enzyme complexes (II–ADPNP-GyrB, V–MTX-NADP-DHFR and VIII–ADPNP-ParE) and top ranked hybrid compound-bound enzyme complexes (III–PD_2a-GyrB, VI–PD_2a-NADP-DHFR and IX–PD_2a-GyrB) were analyzed for the conformational changes with respect to the initial structure by RMSD during 3000 ps simulation time ( and ). The average RMSD values obtained for complex I, IV and VII were approximately 2.060 Å, 1.97 Å, and 2.001 Å, respectively; for complex II, V and VIII they were 1.924 Å, 2.095 Å, and 2.181 Å, respectively; and for complex III, VI and XI they were 2.087 Å, 1.854 Å and 2.113 Å, respectively. As can be seen in the plots, after 500 ps, all the complexes tend to converge with low values of deviation of the molecules with respect to the original atomic positions, indicating the systems are stable and equilibrated. Furthermore, analyses of the RMSF of key active site residues versus the residue number for all the complexes are shown in –. The RMSF value illustrates the average displacement of each residue in relation to their average backbone structure over the whole simulation and indicates the relative flexibility that describes each of these residue elements. As expected, the RMSF plots of all the complexes indicate that the main backbone residues with high fluctuations are those in the flexible loops. The residue positions between 78 and 88, and the turn which includes residues in positions between 248 and 251 of complexes I–GyrB, II– ADPNP-GyrB, and III–PD_2a-GyrB shows high fluctuations (). Similarly, the complexes IV–DHFR, V–MTX-NADP-DHFR, and VI–PD_2a-NADP-DHFR show loops that include residues between 67 and 78, and the α-helix structure which includes residues in positions between 90 and 96, which is located away from the binding site cavity, show high fluctuations (). On the other hand, the complexes VII–ParE, VIII–ADPNP-ParE, and IX–PD_2a-ParE indicate similar RMSF distributions and dynamic features, except in coils located between residue positions 71 and 81, 93 and 103, and turns 233–236, 299 and 303, which are further away from the active binding site areas (). Both the systems with complexes I–III and VII–IX correspond to similar high fluctuations, and closer visual inspection reveals that conformational changes, due to the binding of respective similar substrates and inhibitors, produce displacement of the flanking loops/coils and turns, away from the binding site. The analysis of the mobile flexibilities of key active site residues RMSF values of active site residues in each complex fluctuates widely from 0.5 Å to 2.4 Å, indicating that the active site residue elements maintain their positions in relation to their corresponding enzyme complexes (, and S9). The comparison between the initial unbound and ligand-bound and inhibitor-bound complexes clearly shows the conformational changes favoring the binding of the substrates designed hybrid compounds in the active site cavities of the enzyme complexes. Thus, the low RMSD and RMSF deviations indicate that the inhibitor-enzyme complexes are stable and preserve the inhibitor interactions shown in the molecular docking experiments.
Figure 9 Root mean square deviation (RMSD) of backbone Cα atoms of the complexes versus time in picoseconds. (A) Unbound GyrB complex – I, ADPNP bound GyrB complex – II and top scored hybrid compound PD_2a bound GyrB complex-III; (B) Unbound DHFR complex – IV, methotrexate and cofactor NADP bound DHFR complex – V and top scored hybrid compound PD_2a and cofactor NADP bound DHFR complex – VI.
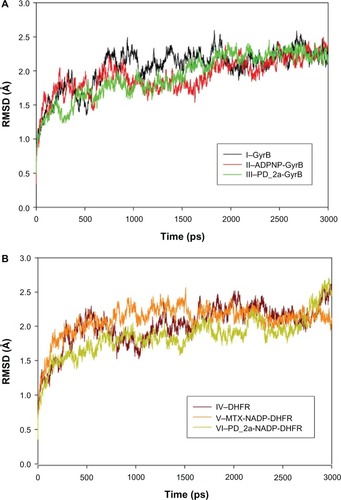
Figure 10 Root mean square fluctuation (RMSF) of backbone Cα atoms of the complexes versus residue number in the sequence. (A) Unbound GyrB complex – I, ADPNP bound GyrB complex – II and top-scored hybrid compound PD_2a bound GyrB complex – III; (B) Unbound DHFR complex – IV, methotrexate and cofactor NADP bound DHFR complex – V and top-scored hybrid compound PD_2a and cofactor NADP bound DHFR complex – VI; (C) Closer look of GyrB active site residues RMSF during the simulation of three complexes; (D) Closer look of DHFR active site residues RMSF during the simulation of three complexes.
Discussion
As a first step in the multitarget therapeutics design, the most crucial part is to identify or prioritize possible combination of targets that can be targeted by a single agent to achieve optimal therapeutic benefit.Citation8 The network-based knowledge from scientific literature is increasingly gaining interest for the insights it offers into identification of relevant target sets and development of multitarget therapeutics.Citation8 The knowledge-based methods employ text mining of biological data from the scientific literature ie, exploit information from literature to generate novel hypotheses, and connecting concepts by new and indirect associative techniques to contribute to drug discovery.Citation50,Citation51 In light of the meager antibiotic pipeline for gram-negative bacteria and the threat of MDR, the motivation for rational design of multitarget inhibitors came from one of the best studied and investigated plant polyphenol, epigallocatechin gallate (from green tea). Epigallocatechin gallate and its derivatives have numerous biological activities including interactions with multiple enzymes, such as GyrB/ParE DHFR.Citation30–Citation32 It is reported that ligands with exact and similar structures are more likely to have similar properties and therefore often bind proteins with similar active sites, and based on this, proteins can be connected for multitarget drug design.Citation52,Citation53 Previously, using drug–phytochemical combination assays, we proposed that phytochemicals (protocatechuic acid, gallic acid, quercetin and myricetin), which are structural analogs and fragments of epigallocatechin gallate, bind to the folate binding site of DHFR and ATP-binding site of GyrB/ParE enzymes of P. aeruginosa and are synergistic in combination with sulfonamides, such as sulfamethoxazole and sulfadiazine.Citation33,Citation34,Citation54 More importantly, the major concern in our study was achieving higher minimum inhibitory concentration (MIC) values of the chosen phytochemicals compared to that of the antibiotics used. In support of this, several studies have reported that phytochemicals have less stability and selectivity, adverse phytochemical–antibiotic interactions, less bioavailability, and are less potent due to their MIC ranges (<1,000 μg/mL), in comparison to the compounds from microbial origin (antibiotics).Citation55,Citation56 Interestingly, phytochemicals can be converted to pharmacologically acceptable antimicrobial agents by optimization through structural modifications.Citation57
Natural products have long been a source of highly diverse lead structures for the drug development process, representing almost half of the drugs currently in clinical use.Citation58 The existing natural antibiotics are either derived directly or have been successfully modified into broad spectrum compounds such as ampicillin, derived from penicillin and azithromycin derived from erythromycin.Citation59 Similarly, structural modification of plant-derived antibacterials may help to develop more potent, target-specific, pharmacologically acceptable, broad spectrum compounds.Citation57,Citation59 In continuation of our previous herb–drug combination studies, the present study used 3D structure-based pharmacophore information for two different enzymes (GyrB/ParE and DHFR) from published literature, coupled with our previous phytochemical–drug combination assay data. We designed and validated a novel class of hybrid compounds (“phyto-drug”) for simultaneous inhibition of these enzymes using various in silico methods.
The target enzymes GyrB/ParE and DHFR are independently essential from two pharmacologically important pathways in the biological network whose perturbation could result in the desired therapeutic outcome. DNA gyrase and topoisomerase IV are type II topoisomerase enzymes involved in bacterial DNA replication and cell division, and their structural similarity typically allows ligands to inhibit both the enzymes.Citation32 Despite being highly homologous enzymes, DNA gyrase composed of two heterodimeric subunits GyrA/GyrB primarily acts in initiation of DNA replication and elongation of nascent DNA, while topoisomerase IV (also consisting of two heterodimeric subunits ParC/ParE) functions to relax DNA supercoils and decatenates daughter chromosomal DNA.Citation47 Aminocoumarins, novobiocin and cyclothialidines, are the known inhibitors of ATPase catalytic region in the N-terminal domains of both GyrB/ParE. However, novobiocin has been withdrawn from clinical use due to toxicity reasons, and cyclothialidines are reported to have weak antibacterial activity due to poor penetration effects.Citation47 Similarly, DHFR is another important enzyme from the folic acid biosynthesis pathway that is involved in the reduction of dihydrofolate to tetrahydrofolate and is inhibited by diaminopyrimidine antibiotics such as trimethoprim and methotrexate.Citation60 Currently, the standard course of treatment for uncomplicated forms of urinary tract infections as well as prophylaxis and Pneumocystis carinii pneumonia is a combination of trimethoprim and sulfonamide drugs resulting in a synergistic action.Citation61 Sulfonamides act as structural analogs of p-aminobenzoic acid, competitively inhibiting the additional enzyme dihydropteroate synthase in the folate pathway, which catalyzes the addition of p-aminobenzoic acid to dihydropterin pyrophosphate to form pteroic acid.Citation61 Due to the low outer membrane permeability, poor affinity, and characteristic intrinsic resistance by efflux pumps, the antibacterial potency of trimethoprim and sulfonamides are limited (with MICs usually in the resistance range).Citation62 Thus, the rational design and development of novel hybrid entities that can inhibit both GyrB/ParE and DHFR enzymes would be beneficial in treating various multidrug resistant phenotypes.
The designed hybrid compounds or “phyto-drug” evaluated for its physicochemical, drug likeness, and toxicity risk profiles using in silico-based calculations resulted in nontoxic behavior, highly drug-like with desirable physicochemical properties in comparison to that of individual phytochemicals and antibiotics (see ). In addition, the structural factors involved in designed hybrids–receptor interactions evaluated using molecular orbitals and MEP profiles, based on stereoelectronic propertie appear to be in good agreement with the key pharmacophoric features required for multi-inhibition. An important aspect in our hybrid design framework is the approach for “revival of older antibiotics” and “molecular tinkering of phytochemicals” through structural combination design by linking common inhibitors (phytochemicals, protocatechuic acid and gallic acid) of DHFR and GyrB/ParE with synergistic sulfonamide antibiotics, which may help improve the properties and also bypass the mechanism of resistance of these compounds. Plant-derived phenolic acids (phytochemicals) have the potential to modify bacterial resistance by evading the efflux mechanisms in multidrug resistant gramnegative bacteria.Citation63 Also, the semisynthetic tailoring and repositioning of known classes of antibiotics will not only increase the efficacy by expanding the spectrum of use but will also offer incremental improvements over existing therapies in reducing resistance and toxicity to antibiotics.Citation64 Another, novel strategy used here is the integration of multispecificity in enzymes of unrelated pathways, as they are highly advantageous in overcoming resistance of targets in different pathways by significantly delaying resistance development. The binding modes of the designed hybrids at ATP-binding sites of GyrB/ParE and the folate-binding site of DHFR were calculated using docking and dynamics simulation and were shown to have conserved and stable binding affinity maintaining key molecular interactions necessary for multienzyme inhibition. These compounds are potential drug candidates, and hence identification and modification of the most biologically active promiscuous plant-derived natural products/antibiotics with the view to improve and balance their drug-like properties will offer novel agents with superior therapeutic properties against infectious diseases.
Conclusion
In summary, our findings, for the first time, have demonstrated the integration of knowledge-based methods, phytochemical–antibiotic interaction network data, and a fragment combination approach in the design and development of novel hybrid compounds, based on the conjugation of phytochemicals and antibiotics for blockage of enzymes (GyrB/ParE and DHFR), in order to restrain the MDR capability of P. aeruginosa. The biological rationale of our hybrid design framework is that it is much more difficult for a pathogen to develop resistance when an inhibitor has activity against multiple targets in two pharmacologically important pathways coupled with restriction of unwanted, off-target effects of the phytochemicals, due to its conjugation with antibiotics through structural modifications. It is also significant that our design strategy has opened an opportunity for revival of older antibiotics to bypass the resistance mechanisms through fragment combination. Finally, the identification of targets with common phytochemicals inhibitory profile through systemic interaction patterns, and network analysis through phytochemical–antibiotic combination assays may help to cluster common drug targets and form a base for accelerating the process of antibacterial multitarget drug discovery.
Supplementary materials
Sequence analysis and homology modeling
The amino acid sequences of the target enzymes P. aeruginosa DNA Gyrase subunit B (GyrB, Locus ID – PA0004), topoisomerase subunit B (ParE, Locus ID – PA4967) and dihydrofolate reductase (FolA, Locus ID – PA0350) were retrieved from Pseudomonas genome database (http://www.pseudomonas.com/). The amino acid sequences were subjected to BLASTP search against Protein Data Bank (PDB) and SWISS-MODEL template selection tool to identify the suitable templates for comparative modeling. Based on the template selection analysis, E. coli GyrB (PDB code: 1EI1, 2.3 Å), E. coli ParE (PDB code: 1S16, 2.1 Å) and E. coli DHFR (PDB code: 1RX3, 2.2 Å) were selected as suitable templates with sequence identity of 74%, 75%, and 45%, respectively. The alignment of P. aeruginosa GyrB/ParE and DHFR protein sequences and template structures – E. coli GyrB (1EI1), ParE (1S16), and DHFR (1RX3) protein sequences, were performed using CLUSTALW2 (http://www.ebi.ac.uk/Tools/clustalw2/index.html). The pair-wise alignment () was carefully examined for any potential alignment errors and the 3D model building of GyrB/ParE and DHFR was performed by two automated modeling programs: SWISS-MODELCitation1 and MODELLER 9v7.Citation2 The generated homology models were submitted to WHATIF server (http://swift.cmbi.ru.nl/servers/html/index.html) to model missing side chains and remove atomic clashes (bumps).Citation3 The RMSD values were obtained by structural superimposition of all the GyrB/ParE and DHFR homology models from both SWISS-MODEL and MODELLER with their respective E. coli templates (1EI1, 1S16, and 1RX3) using SUPERPOSECitation4 web server (http://wishart.biology.ualberta.ca/SuperPose/). Out of all the models estimated, the SWISS-MODEL constructed models resulted in low RMSD values (), 0.46 Å (GyrB), 0.36 (ParE), and 0.39 Å (DHFR), respectively. RMSD values of ≤2 Å from the experimentally determined structures are considered as “highly accurate” models, while models with Cα RMSD above this threshold and ≤4 Å were termed “reliable” models, and such models are not fit for drug design and binding affinity analysis.Citation5 Thus, low RMSD values obtained shows the selected homology models are acceptable and quite similar to their respective templates.
The quality of obtained P. aeruginosa GyrB/ParE and DHFR models were further subjected to evaluations based on stereochemical analysis, absolute measures on the geometry, and energy profiles independently using the structure assessment tools, PROCHECK, ANOLEA, and QMEAN available in the SWISS-MODEL Workspace,Citation1 Verify3DCitation6 and ProSA.Citation7 The Ramachandran plot produced from PROCHECK analysis (, , and ) showed 84.3% (GyrB), 90.5% (ParE), and 86.5% (DHFR) amino acid residues within the most favored regions, 15.7% (GyrB), 9.5% (ParE), and 12.7% (DHFR) within additionally allowed regions indicating that the models have good overall stereochemical quality. The QMEAN analysis used for the estimation of the global quality of the entire model as well as for the local per residue analysis of different regions within the models resulted in the QMEAN6 global score values 0.707 (GyrB), 0.598 (ParE), and 0.752 (DHFR), which are well within the quality range (0–1) with higher values for better models.Citation8 In addition, estimated QMEAN Z-scores represent a measure of the absolute quality of the models by providing an estimate of scores obtained for high-resolution reference structures solved experimentally by X-ray crystallography models. Low quality are expected to have strong negative Z-scores, and “good structures” have (mean Z-scores = −0.65).Citation9 The observed Z-score values for the predicted models were −0.718 (GyrB), −0.53 (ParE), and −0.077 (DHFR), suggesting these models are of comparable quality to high-resolution experimental structures. The energy profiles of the models were estimated using both ANOLEA and ProSA analysis showing regions with predominant low energy (negative values). The ProSA tool is widely used to check 3D models of protein structures for potential errors by calculating z-score and a plot of its residue energies, z-score value, is displayed in a plot that contains the z-scores of all experimentally determined protein chains in current PDB and models with z-scores outside the range characteristic for native proteins indicate erroneous structures, while the energy plot shows the local model quality by plotting residue energies as a function of amino acid sequence position in which positive values correspond to problematic or erroneous parts of a model.Citation7 The calculated z-scores −8.45 (GyrB), −8.7 (ParE), and −5.19 (DHFR) of the modeled structures are within the range of scores typically found for native proteins of similar size and the energy plot showed all the residues in GyrB/ParE have negative energies, while residues in DHFR showed maximum residues with negative energy with very few residues displaying positive energy values (, , and ). Finally, Verify3D was used to test the accuracy of a 3D model by comparing the model to its own amino acid sequence using a 3D profile.Citation6 The average 3D-1D profile scores for the model (GyrB, 0.67) and that of its template (1EI1, 0.71), model (ParE, 0.73) and its template (1S16, 0.71) similarly, (DHFR, 0.69) and its template (1RX3, 0.71) are very close. Hence, the validation based on the geometry, energy profiles and overall stereo-chemical quality suggests that the homology models obtained are appropriate for further protein-ligand interaction studies.
Figure S1 Alignment of target (P. aeruginosa) sequence and E. coli template protein sequence. (A) DNA Gyrase subunit B (GyrB) with template (PDB: 1EI1). (B) Topoisomerase IV subunit B (ParE) with template (PDB: 1S16). (C) Dihydrofolate reductase (DHFR) with template (PDB: 1RX3).
Figure S2 Superimposition between P. aeruginosa homology models and the template crystal structures. (A) GyrB model (blue) and template structure E. coli (1EI1, red) bound with ADPNP (yellow stick model). (B) ParE model (blue) and template structure E. coli (1S16, green) bound with ADPNP (magenta stick model). (C) DHFR model (blue) and template structure E. coli (1RX3, green) bound with the inhibitor methotrexate (yellow stick model), and NADP cofactor (aqua stick model).
Note: Active site residues are shown as wireframe colored by their element.
Figure S3 Validation of P. aeruginosa GyrB homology model (A) Ramachandran plot and (B) ProSA (protein structure analysis) energy plot.
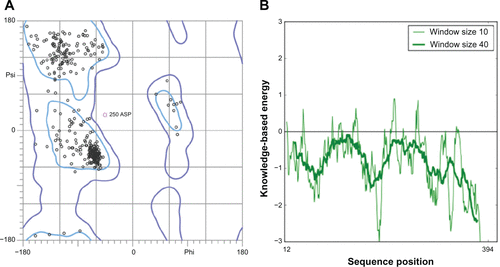
Figure S4 Validation of P. aeruginosa ParE homology model (A) Ramachandran plot and (B) ProSA (protein structure analysis) energy plot.
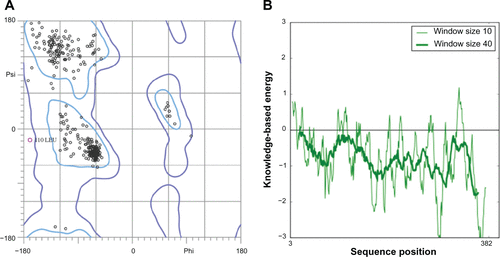
Figure S5 Validation of P. aeruginosa DHFR homology model (A) Ramachandran plot and (B) ProSA (protein structure analysis) energy plot.
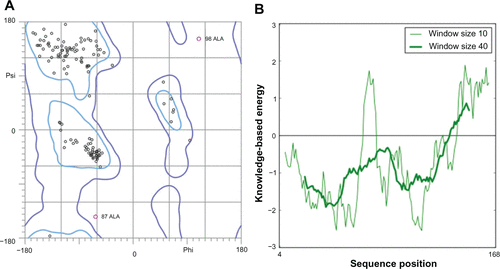
Figure S6 Docking of designed hybrid compounds at ATP binding site of P. aeruginosa ParE. (A) Binding pose of the top-scored compound PD_2a (grey stick model) (B) PD_2a and PD_2b (stick model colored by their element) superimposed with ADPNP (magenta stick model).
Notes: The residues are shown as wireframe colored by their element and labeled in white. The hydrogen bonds are illustrated as dotted green lines. The water molecule in both (A) and (B) is shown as a red colored ball model.
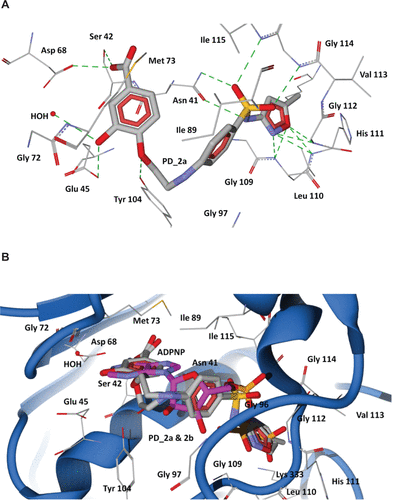
Figure S7 Docking of designed hybrid compounds at ATP binding site of P. aeruginosa ParE. (A) Binding pose of the top-scored compound PD_1a (grey stick model). (B) PD_1a and PD_1b (stick model colored by their element) superimposed with ADPNP (magenta stick model).
Notes: The residues are shown as wireframe colored by their element and labeled in white. The hydrogen bonds are illustrated as dotted green lines. The water molecule in both (A) and (B) is shown as a red colored ball model.
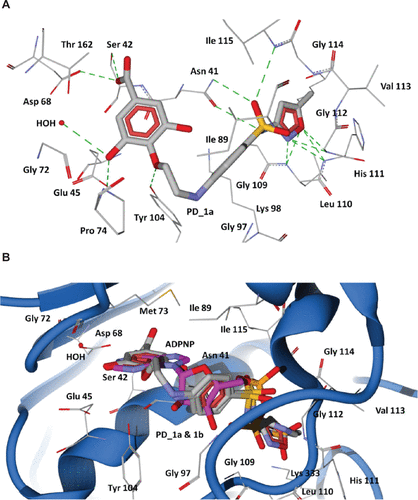
Figure S8 Root mean square deviation (RMSD) of backbone Cα atoms of the complexes versus time in picoseconds. Unbound ParE complex – VII, ADPNP bound ParE complex – VIII and top-scored hybrid compound PD_2a bound ParE complex – IX.
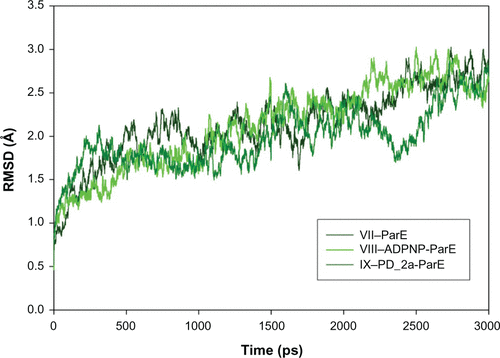
Figure S9 Root mean square fluctuation (RMSF) of backbone Cα atoms of the complexes versus residue number in the sequence. (A) Unbound ParE complex – VII, ADPNP bound ParE complex – VIII and top-scored hybrid compound PD_2a bound ParE complex – IX; (B) Closer look of ParE active site residues RMSF during the simulation of three complexes.
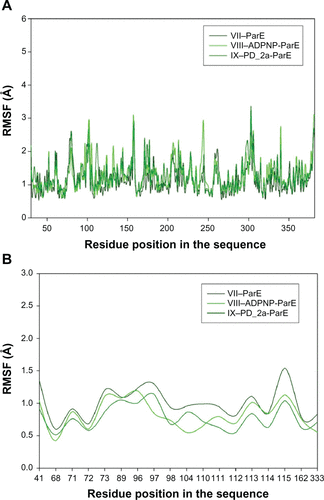
Table S1 Docking results of ADPNP and designed hybrid compounds (ranked on the basis of binding affinity scores) at the ATP binding site in P. aeruginosa ParE
References
- Arnold K Bordoli L Kopp J Schwede T The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling Bioinformatics 2006 22 2 195 201 16301204
- Sali A Blundell TL Comparative protein modelling by satisfaction of spatial restraints J Mol Biol 1993 234 3 779 815 8254673
- Rodriguez R Chinea G Lopez N Pons T Vriend G Homology modeling, model and software evaluation: three related resources Bioinformatics 1998 14 6 523 528 9694991
- Maiti R Van Domselaar GH Zhang H Wishart DS SuperPose: a simple server for sophisticated structural superposition Nucleic Acids Res 2004 32 Web Server issue W590 W594 15215457
- Rayan A New tips for structure prediction by comparative modeling Bioinformation 2009 3 6 263 267 19255646
- Eisenberg D Lüthy R Bowie JU VERIFY3D: assessment of protein models with three-dimensional profiles Methods Enzymol 1997 277 396 406 9379925
- Wiederstein M Sippl MJ ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins Nucleic Acids Res 2007 35 Web Server issue W407 W410 17517781
- Benkert P Tosatto SC Schomburg D QMEAN: a comprehensive scoring function for model quality assessment Proteins 2008 71 1 261 277 17932912
- Benkert P Biasini M Schwede T Toward the estimation of the absolute quality of individual protein structure models Bioinformatics 2011 27 3 343 350 21134891
Acknowledgments
Premkumar Jayaraman, Kishore R Sakharkar, and Meena K Sakharkar conceived the study, designed the methods, carried out the data analysis, and drafted the manuscript. Meena K Sakharkar supervised the research and contributed to drafting the manuscript, Lim ChuSing Daniel, Kishore R Sakharkar and Sarinder K Dhillon, and Mohammad Imran Siddiqi gave useful suggestions and improved on the manuscript. All authors have read and approved the final manuscript.
This work was funded by a seed grant from Biopharmaceutical Engineering Cluster, Nanyang Technological University, Singapore and an AcRF Tier1 grant from Nanyang Technological University, Singapore.
Disclosure
The authors report no conflicts of interest.
References
- Breidenstein EB de la Fuente-Núñez C Hancock RE Pseudomonas aeruginosa: all roads lead to resistance Trends Microbiol 2011 19 8 419 426 21664819
- Lister PD Wolter DJ Hanson ND Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms Clin Microbiol Rev 2009 22 4 582 610 19822890
- Mesaros N Nordmann P Plésiat P Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium Clin Microbiol Infect 2007 13 6 560 578 17266725
- Traugott KA Echevarria K Maxwell P Green K Lewis JS2nd Monotherapy or combination therapy? The Pseudomonas aeruginosa conundrum Pharmacotherapy 2011 31 6 598 608 21923444
- Johnson SJ Ernst EJ Moores KG Is double coverage of gramnegative organisms necessary? Am J Health Syst Pharm 2011 68 2 119 124 21200057
- Pokrovskaya V Baasov T Dual-acting hybrid antibiotics: a promising strategy to combat bacterial resistance Expert Opin Drug Discov 2010 5 9 883 902 22823262
- Zimmermann GR Lehár J Keith CT Multi-target therapeutics: when the whole is greater than the sum of the parts Drug Discov Today 2007 12 1–2 34 42 17198971
- Hopkins AL Network pharmacology: the next paradigm in drug discovery Nat Chem Biol 2008 4 11 682 690 18936753
- Bremner JB Ambrus JI Samosorn S Dual action-based approaches to antibacterial agents Curr Med Chem 2007 14 13 1459 1477 17584056
- Morphy R Rankovic Z Designing multiple ligands – medicinal chemistry strategies and challenges Curr Pharm Des 2009 15 6 587 600 19199984
- Morphy R Kay C Rankovic Z From magic bullets to designed multiple ligands Drug Discov Today 2004 9 15 641 651 15279847
- Pokrovskaya V Belakhov V Hainrichson M Yaron S Baasov T Design, synthesis, and evaluation of novel fluoroquinolone-aminoglycoside hybrid antibiotics J Med Chem 2009 52 8 2243 2254 19301822
- Ball AR Casadei G Samosorn S Conjugating berberine to a multidrug efflux pump inhibitor creates an effective antimicrobial ACS Chemi Biol 2006 1 9 594 600
- Labischinski H Cherian J Calanasan C Boyce RS Inventors Hybrid antimicrobial compounds and their use United States patent US WO/2010/025906 3 11 2010
- Ma XH Shi Z Tan C In-silico approaches to multi-target drug discovery: computer aided multi-target drug design, multi-target virtual screening Pharm Res 2010 27 5 739 749 20221898
- O’Boyle NM Banck M James CA Morley C Vandermeersch T Hutchison GR Open Babel: an open chemical toolbox J Cheminform 2011 3 33 21982300
- Koch W Holthausen MC A Chemist’s Guide to Density Functional Theory Weinheim, Germany Wiley-VCH 2000
- Zhao YH Abraham MH Le J Rate-limited steps of human oral absorption and QSAR studies Pharm Res 2002 19 10 1446 1457 12425461
- Sander T Freyss J von Korff M Reich JR Rufener C OSIRIS, an entirely in-house developed drug discovery informatics system J Chem Inf Model 2009 49 2 232 246 19434825
- Thomsen R Christensen MH MolDock: a new technique for high-accuracy molecular docking J Med Chem 2006 49 11 3315 3321 16722650
- Wang R Fang X Lu Y Wang S The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures J Med Chem 2004 47 12 2977 2980 15163179
- Krieger E Darden T Nabuurs SB Finkelstein A Vriend G Making optimal use of empirical energy functions: force-field parameterization in crystal space Proteins 2004 57 4 678 683 15390263
- Essmann U Perera L Berkowitz ML Darden T Lee H Pedersen LG A smooth particle mesh Ewald method J Chem Phys 1995 103 19 8577 8593
- Klamt A Conductor-like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena J Phys Chem 1995 99 7 2224 2235
- Jakalian A Jack DB Bayly CI Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. parameterization and validation J Comput Chem 2002 23 16 1623 1641 12395429
- Wang J Wolf RM Caldwell JW Kollman PA Case DA Development and testing of a general amber force field J Comput Chem 2004 25 9 1157 1174 15116359
- Hooft RW Sander C Vriend G Positioning hydrogen atoms by optimizing hydrogen-bond networks in protein structures Proteins 1996 26 4 363 376 8990493
- Krieger E Nielsen JE Spronk CA Vriend G Fast empirical pKa prediction by Ewald summation J Mol Graph Model 2006 25 4 481 486 16644253
- Xie L Xie L Kinnings SL Bourne PE Novel Computational approaches to polypharmacology as a means to define responses to individual drugs Annu Rev Pharmacol Toxicol 2012 52 1 361 379 22017683
- Spina M Cuccioloni M Mozzicafreddo M Mechanism of inhibition of wt-dihydrofolate reductase from E. coli by tea epigallocatechin-gallate Proteins 2008 72 1 240 251 18214969
- Gradisar H Pristovsek P Plaper A Jerala R Green tea catechins inhibit bacterial DNA gyrase by interaction with its ATP binding site J Med Chem 2007 50 2 264 271 17228868
- Bradbury BJ Pucci MJ Recent advances in bacterial topoisomerase inhibitors Curr Opin Pharmacol 2008 8 5 574 581 18555745
- Jayaraman P Sakharkar KR Sing LC Chow VT Sakharkar MK Insights into antifolate activity of phytochemicals against Pseudomonas aeruginosa J Drug Target 2011 19 3 179 188 20429775
- Sakharkar MK Jayaraman P Soe WM Chow VT Sing LC Sakharkar KR In vitro combinations of antibiotics and phytochemicals against Pseudomonas aeruginosa J MicrobioI Immunol Infect 2009 42 5 364 370
- Marques SM Enyedy EA Supuran CT Krupenko NI Krupenko SA Santos MA Pteridine-sulfonamide conjugates as dual inhibitors of carbonic anhydrases and dihydrofolate reductase with potential antitumor activity Bioorg Medicinal Chem 2010 18 14 5081 5089
- Brvar M Perdih A Oblak M Masic LP Solmajer T In silico discovery of 2-amino-4-(2,4-dihydroxyphenyl)thiazoles as novel inhibitors of DNA gyrase B Bioorg Med Chem Lett 2010 20 3 958 962 20045642
- Lerner MG Bowman AL Carlson HA Incorporating dynamics in E. coli dihydrofolate reductase enhances structure-based drug discovery J Chem Inf Model 2007 47 6 2358 2365 17877338
- Schechner M Sirockin F Stote RH Dejaegere AP Functionality maps of the ATP binding site of DNA gyrase B: generation of a consensus model of ligand binding J Med Chem 2004 47 18 4373 4390 15317451
- Manly CJ Chandrasekhar J Ochterski JW Hammer JD Warfield BB Strategies and tactics for optimizing the hit-to-lead process and beyond – a computational chemistry perspective Drug Discov Today 2008 13 3–4 99 109 18275907
- Matter H Baringhaus KH Naumann T Klabunde T Pirard B Computational approaches towards the rational design of drug-like compound libraries Comb Chem High Throughput Screen 2001 4 6 453 475 11562252
- Lipinski CA Lead- and drug-like compounds: the rule-of-five revolution Drug Discov Today Technol 2004 1 4 337 341
- Veber DF Johnson SR Cheng HY Smith BR Ward KW Kopple KD Molecular properties that influence the oral bioavailability of drug candidates J Med Chem 2002 45 12 2615 2623 12036371
- Maiti R Van Domselaar GH Zhang H Wishart DS SuperPose: a simple server for sophisticated structural superposition Nucleic Acids Res 2004 32 Web Server issue W590 W594 15215457
- Lessa JA Mendes IC da Silva PR 2-Acetylpyridine thiosemicarbazones: cytotoxic activity in nanomolar doses against malignant gliomas Eur J Med Chem 2010 45 12 5671 5677 20884094
- Liu XH Chen PQ Wang BL Li YH Wang SH Li ZM Synthesis, bioactivity, theoretical and molecular docking study of 1-cyano-N-substituted-cyclopropanecarboxamide as ketol-acid reductoisomerase inhibitor Bioorg Med Chem Lett 2007 17 13 3784 3788 17512731
- Nascimento EC Martins JB Electronic structure and PCA analysis of covalent and non-covalent acetylcholinesterase inhibitors J Mol Model 2011 17 6 1371 1379 20839017
- Oblak M Kotnik M Solmajer T Discovery and development of ATPase inhibitors of DNA gyrase as antibacterial agents Curr Med Chem 2007 14 19 2033 2047 17691945
- Bellon S Parsons JD Wei Y Crystal structures of Escherichia coli topoisomerase IV ParE subunit (24 and 43 kilodaltons): a single residue dictates differences in novobiocin potency against topoisomerase IV and DNA gyrase Antimicrob Agents Chemother 2004 48 5 1856 1864 15105144
- Cody V Schwalbe CH Structural characteristics of antifolate dihydrofolate reductase enzyme interactions Crystallogr Rev 2006 12 4 301 333
- Agarwal P Searls DB Literature mining in support of drug discovery Brief Bioinform 2008 9 6 479 492 18820304
- Yang Y Adelstein SJ Kassis AI Target discovery from data mining approaches Drug Discov Today 2009 14 3–4 147 154 19135549
- Park K Kim D Binding similarity network of ligand Proteins 2008 71 2 960 971 18004762
- Durrant JD Amaro RE Xie L A multidimensional strategy to detect polypharmacological targets in the absence of structural and sequence homology PLoS Comput Biol 2010 6 1
- Jayaraman P Sakharkar MK Lim CS Tang TH Sakharkar KR Activity and interactions of antibiotic and phytochemical combinations against Pseudomonas aeruginosa in vitro Int J Biol Sci 2010 6 6 556 558 20941374
- Gibbons S Moser E Kaatz GW Catechin gallates inhibit multidrug resistance (MDR) in Staphylococcus aureus Planta Med 2004 70 12 1240 1242 15643566
- Hemaiswarya S Kruthiventi AK Doble M Synergism between natural products and antibiotics against infectious diseases Phytomedicine 2008 15 8 639 652 18599280
- Cushnie TP Lamb AJ Antimicrobial activity of flavonoids Int J Antimicrob Agents 2005 26 5 343 356 16323269
- Harvey AL Natural products in drug discovery Drug Discov Today 2008 13 19–20 894 901 18691670
- Lewis K Ausubel FM Prospects for plant-derived antibacterials Nat Biotechnol 2006 24 12 1504 1507 17160050
- Bermingham A Derrick JP The folic acid biosynthesis pathway in bacteria: evaluation of potential for antibacterial drug discovery Bioessays 2002 24 7 637 648 12111724
- Hawser S Lociuro S Islam K Dihydrofolate reductase inhibitors as antibacterial agents Biochem Pharmacol 2006 71 7 941 948 16359642
- Köhler T Kok M Michea-Hamzehpour M Multidrug efflux in intrinsic resistance to trimethoprim and sulfamethoxazole in Pseudomonas aeruginosa Antimicrob Agents Chemother 1996 40 10 2288 2290 9036831
- Chusri S Villanueva I Voravuthikunchai SP Davies J Enhancing antibiotic activity: a strategy to control Acinetobacter infections J Antimicrob Chemother 2009 64 6 1203 1211 19861335
- Rogers GB Carroll MP Bruce KD Enhancing the utility of existing antibiotics by targeting bacterial behaviour? Brit J Pharmacology 2012 165 4 845 857