
Abstract
Purpose
Over the last few years, covalent fragment-based drug discovery has gained significant importance. Thus, striving for more warhead diversity, we conceived a library consisting of 20 covalently reacting compounds. Our covalent fragment library (CovLib) contains four different warhead classes, including five α-cyanoacacrylamides/acrylates (CA), three epoxides (EO), four vinyl sulfones (VS), and eight electron-deficient heteroarenes with a leaving group (SNAr/SN).
Methods
After predicting the theoretical solubility of the fragments by LogP and LogS during the selection process, we determined their experimental solubility using a turbidimetric solubility assay. The reactivities of the different compounds were measured in a high-throughput 5,5'-dithiobis-(2-nitrobenzoic acid) DTNB assay, followed by a (glutathione) GSH stability assay. We employed the CovLib in a (differential scanning fluorimetry) DSF-based screening against different targets: c-Jun N-terminal kinase 3 (JNK3), ubiquitin-specific protease 7 (USP7), and the tumor suppressor p53. Finally, the covalent binding was confirmed by intact protein mass spectrometry (MS).
Results
In general, the purchased fragments turned out to be sufficiently soluble. Additionally, they covered a broad spectrum of reactivity. All investigated α-cyanoacrylamides/acrylates and all structurally confirmed epoxides turned out to be less reactive compounds, possibly due to steric hindrance and reversibility (for α-cyanoacrylamides/acrylates). The SNAr and vinyl sulfone fragments are either highly reactive or stable. DSF measurements with the different targets JNK3, USP7, and p53 identified reactive fragment hits causing a shift in the melting temperatures of the proteins. MS confirmed the covalent binding mode of all these fragments to USP7 and p53, while additionally identifying the SNAr-type electrophile SN002 as a mildly reactive covalent hit for p53.
Conclusion
The screening and target evaluation of the CovLib revealed first interesting hits. The highly cysteine-reactive fragments VS004, SN001, SN006, and SN007 covalently modify several target proteins and showed distinct shifts in the melting temperatures up to +5.1 °C and −9.1 °C.
Graphical Abstract
Introduction
Targeted covalent inhibitors (TCIs) have gained increasing attention in the drug discovery community in recent years.Citation1–7 A number of TCIs have received FDA approval, such as covalent protein kinase inhibitors (e.g., Afatinib, Osimertinib)Citation8–10 or the KRASG12C inhibitor Sotorasib.Citation11,Citation12 A detailed account of all FDA-approved TCIs and those undergoing clinical trials is beyond the scope of this paper. However, such information is outlined in comprehensive reviews.Citation5,Citation13
The development of intrinsically reactive covalent drugs has historically been considered challenging due to their potential for off-target reactions and idiosyncratic toxicity.Citation3,Citation14,Citation15 However, a covalent binding mechanism offers numerous advantages over the classical non-covalent mechanism, such as improved (yet time-dependent) potency, a long duration of action with a possible decoupling of pharmacodynamics from pharmacokinetics but also improved selectivity if properly implemented.Citation3,Citation5,Citation16,Citation17 A TCI design that provides a balanced reactivity profile can reduce toxicity and maximize the benefits from the covalent mode of action.Citation5,Citation18
TCIs with their electrophilic covalent reactive groups (CRGs, often referred to as “warheads”) target nucleophilic amino acid residues, mainly the thiol group of cysteines, which is highly polarizable and a soft nucleophile being the strongest among the proteinogenic amino acids.Citation19–21 Most TCIs have an attenuated Michael acceptor, typically an acrylamide derivative, as the warhead to target non-catalytic cysteine residues.Citation22,Citation23
The binding of the TCI to the target can generally be described by a two-step process involving an initial non-covalent, reversible binding, followed by the formation of a (irreversible) covalent bond between the TCI and the targeted amino acid residue, and is best described by the second order rate constant kinact/KI governing both steps.Citation24–27 Therefore, the binding of the TCI to the target is a time-dependent process and increased incubation time results in increased occupancy and thus (apparent) potency.Citation5,Citation11,Citation26
The discovery process of TCIs involves several strategies. The most popular approach is the structure-based design. In this approach, the warhead is attached to an optimized non-covalently binding ligand at an optimal position with respect to the amino acid of interest in the binding pocket.Citation5,Citation7,Citation28,Citation29 Alternative approaches to TCI discovery include re-design of off-target hits,Citation30,Citation31 screening of covalent fragment librariesCitation7,Citation28,Citation32–34 or DNA-encoded libraries with covalent inhibitors,Citation35–37 and virtual screening with covalent docking.Citation21,Citation38–40
Covalent fragment libraries have recently gained prominence with several covalent fragment-based drug discovery (FBDD) success stories for challenging and previously undruggable targets such as the oncogenic G12C mutant of KRAS.Citation7,Citation11,Citation12,Citation41 Compared to the structure-based approach, covalent FBDD does not require the availability of a non-covalent ligand binding near a targetable reactive amino acid residue.Citation7,Citation28 Therefore, this strategy has also been denominated as “electrophile first approach”.Citation1 In addition, the advantages of fragment-based approaches can be exploited to broadly cover chemical space, offering a unique potential for identifying new binding modes and “cryptic” pockets as well as addressing new targets.Citation7,Citation42–44
In our group, we have developed a halogen-enriched fragment library (HEFLib) to explore the potential of halogen bonds as protein–ligand interactions using the principle of FBDD.Citation45–47 Halogen bonds are non-classical interactions between the electron deficient part of chlorine, bromine, or iodine, the so-called σ-hole, with electron-rich Lewis-bases. The strength of a halogen bond depends on the size of the halogen’s σ-hole and is characterized by the Vmax value.Citation48 It is possible to tune this strength by changing the (hetero-)aromatic scaffold or by adding electron-withdrawing groups to the system bound to the interacting halogen.Citation49 However, tuning of the Vmax of halogens bound to (hetero-)aromatic systems could lead to unintended nucleophilic aromatic substitution (SNAr) reactions at the arene core.
The characterization and screening of our previously established HEFLib with different targets confirmed that one of the fragments (compound 4482) undergoes a chemical reaction with the target protein to establish a covalent bond instead of forming σ-hole interactions as intended.Citation45,Citation50–52
HEFLib compound 4482 was identified as a reactive fragment, achieving general stabilization of the tumor suppressor protein p53 by covalent modification of surface-exposed cysteines via SNAr reaction.Citation52 These surprising but promising observations, in addition to the previously mentioned advantages of TCIs and FBDD, encouraged us to design a covalent fragment library (CovLib). When designing covalent fragment libraries, warhead properties such as stability, reactivity, and positioning of the warhead should be considered in combination with classical FBDD criteria (“rule of three”Citation53).Citation28,Citation29,Citation54 For maximum diversity in library development, the selection should include fragments with a wide range of reactivities and different warheads.Citation7,Citation28 Hence, we selected an initial small set of covalently reactive fragments (CovLib) with different warhead classes to represent different parts of the wide chemical space. The CovLib consists of 20 fragments with four different warhead classes including five α-cyanoacyrlamides/acrylates (CA), three epoxides (EO), four vinyl sulfones (VS), and eight electron-deficient heteroarenes with leaving group (SNAr/SN) warheads. Herein, we characterized various properties of the CovLib that are relevant to the drug design process. First, we evaluated the solubility of the fragments using computational LogP and LogS calculations and the experimental solubility with a turbidimetric solubility assay. In the next step, we assessed the reactivity towards cysteine surrogates in a high-throughput DTNB assay, followed by a more detailed investigation with a physiologically more relevant GSH stability assay. In addition, the CovLib was experimentally screened by DSF against the targets c-Jun N-terminal kinase 3 (JNK3, residues 39–402), ubiquitin-specific protease 7 (USP7, residues 208–560, catalytic domain), and the tumor suppressor p53 (residues 94–312, core domain). Finally, intact protein mass spectrometry was performed to validate the covalent binding of the identified fragment hits, yielding interesting starting points for further development.
Materials and Methods
Materials
The compounds studied were purchased from Aldrich Market Select (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) at purity levels of 90% or higher. Purity was confirmed by high performance liquid chromatography (HPLC) on an Ultimate 3000 HPLC-System (Thermo Fisher Scientific, Dreieich, Germany). Results of the purity confirmation are summarized in Table S1.1. The corresponding chromatograms are presented in the Figures S1.1–S1.7, S1.9, S1.10, and S1.12–S1.22. For two compounds, additional 1H nuclear magnetic resonance (NMR) spectroscopy (Bruker Avance III HD 400 MHz) was performed. NMR spectra are reported as chemical shifts (δ) in parts per million (ppm) in relation to tetramethylsilane (TMS) and were calibrated using the residual peak of the used solvent. Coupling constants (J) are reported in units of hertz (Hz). The following abbreviations are used to describe multiplicities: s (singlet), d (doublet), q (quartet), and m (multiplet). The corresponding spectra and chemical shifts are provided in Figures S1.8 and S1.11. For the compound EO002 the identity and purity could not be confirmed. The HPLC chromatogram and the NMR spectrum indicated a mixture of several components that were present in small amounts in the stock of the compound. These could be caused by degradation during transport or storage at the vendor. For the compound VS001, the identity but not the purity could be verified by NMR spectroscopy.
Structural Filtering Process
The Aldrich Market Select Data Bank (~18 million compounds as of April 2023) was filtered employing a customized python script using the RDKit package. Compounds larger than 22 non-hydrogen atoms and smaller than six non-hydrogen atoms were removed. Further, compounds that show a logP > 2 were excluded. LogP values were calculated by RDKit according to Wildman et al.Citation55
QPLogP and QPLogS Calculations
LogS and logP values (denoted QPLogS and QPLogPo/w) were calculated using the QikProp module of Schrödinger suite version 2021−1.Citation56 Molecules were protonated and preprocessed using Schrödingers’ LigPrep module with default parameters.Citation57 All calculations were then carried out using the default parameters and the normal processing mode of QikProp.
Turbidimetric Solubility Assay
The turbidimetric solubility assay was performed using phosphate buffered saline (PBS) pH 7.4 at 25 °C in a 200 µL scale. The 100 mM fragment stocks in dimethyl sulfoxide (DMSO) were diluted by a factor of 4/5 with DMSO in a dilution series. Diluted compounds were added to the buffer, resulting in fragment concentrations of 5 mM, 4 mM, 3.2 mM, 2.56 mM, 2.048 mM, 1.638 mM, 1.311 mM, 1.049 mM, 0.839 mM, 0.671 mM, 0.537 mM, and 0.429 mM and 5% DMSO. Extinction at 600–800 nm was measured in a Lumox® multiwell, 96-well-plate (SARSTEDT, Nürnbrecht, Germany) using the CLARIOstar Plate Reader (BMG Labtech, Ortenberg, Germany) for 2 h with 300 rpm double orbital shaking for 60s every 2 min. The concentration-dependent extinction spectra for all compounds, measured at the beginning (0 min), an intermediate cycle (57 min), and at the end (117 min), are depicted in the Figures S2.1–S2.19.
DTNB Assay
The thiol-reactivity assay was performed according to a previously published protocol by Resnick et alCitation34 with some modifications. The cysteine surrogate 2-nitro-5-thiobenzoate anion (TNB2-) was prepared in situ by reduction of 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) in the presence of tris(2-carboxyethyl) phosphine hydrochloride (TCEP). Reaction conditions were 200 µL buffer (20 mM NaPi pH 7.4, 150 mM NaCl), 10% acetonitrile, 100 µM fragment, 25 µM DTNB, and 100 µM TCEP (yielding to 50 µM TNB2-) at 37° C. The reaction was performed in a Lumox® multiwell, 96-well-plate (SARSTEDT, Nürnbrecht, Germany) covered with a lid. TNB2- absorbance at 412 nm was monitored every 5 min for 4 h using the CLARIOstar Plate Reader (BMG Labtech, Ortenberg, Germany). Measurements were performed in triplicate, and a parallel experiment without DTNB was conducted to determine the background absorption of the compounds. A measurement without fragment was performed to calculate the extinction coefficient of TNB2- from the absorption of the first time point. The compound background absorbance was subtracted from each measurement, and the remaining TNB2- and compound concentrations were calculated for each time point. The data were then fitted in OriginPro2020 (OriginLab, Northampton, MA, USA) and the second order rate constant k2 was calculated using the integrated rate equation of second-order kinetics with multiple reactants:
[A0] and [B0] are the initial concentration of the fragment and TNB2- respectively, and [A]t is the remaining compound concentration as a function of time. The [A0] value for the initial compound concentration was set on the calculated compound concentration at 0 min. [B0] was set by default on the experimentally used concentration of 50 µM. For reactive compounds (iodoacetamide, SN001, SN002) it was necessary to set [B0] as a variable for the calculation. For the very reactive compounds (SN006, SN007), a considerable difference was found between the calculated compound concentration at 0 min and the employed compound concentration of 100 µM. The reason for this could be the short time delay in the measurement procedure of approximately 5 min between the start of the reaction by addition of TNB2- and the first measurement cycle of the plate reader. Due to the very high reaction rates of these compounds, significant degradation can take place during this period. To overcome this problem in the evaluation, we have introduced the time delay variable td for very reactive compounds. All parameters used for the fitting calculations and the computed results can be found in Table S3.1–S3.3. The k2 values are given as the mean value of the triplicate determination with the respective standard deviation, which was calculated according to the rules of error propagation. Figures S3.1–S3.20 show the measured fragment concentrations [A]t as a function of time and the calculated fitting curves.
Glutathione Assay
Glutathione (GSH) reactivity studies were performed according to a protocol established for heterocyclic electrophilic fragments by Keeley et al.Citation32 Reaction conditions were PBS pH 7.4, 10% acetonitrile, 100 µM ketoprofen as internal standard, 250 µM fragment, and 5 mM GSH excess at 37 °C. Measurement times were after 0, 1, 2, 4, 8, 12, and 24 h. For very reactive fragments (t1/2 < 1 h) analysis was performed every 20 min. The mixture was analyzed with an Ultimate 3000 HPLC-System (Thermo Fisher Scientific, Dreieich, Germany) with UV-detection. The reaction of the compounds was detected by measuring the decreasing area under the curve (AUC) of the fragment relative to the internal standard. The relative declining area was fitted in OriginPro2020 (OriginLab, Northampton, MA, USA) to the integrated rate equation of pseudo-first order kinetics:
Half-life t1/2 was calculated from the pseudo-first order rate constant k following the equation:
Measurements were performed as duplicates with 5 mM GSH and once without GSH in PBS to check for hydrolytic degradation. The GSH t1/2 values are given as the mean value of the duplicate determination with the respective standard deviation, which was calculated according to the rules of error propagation. In contrast to the calculations by Keeley et al,Citation32 the rate constants kGSH and the corresponding half-lives were not corrected for the degradation reaction in pure buffer. For comparison, the half-lives for degradation in PBS (t1/2 PBS) are also given. Figures S4.1–S4.19 show the measured relative AUC of the fragments as a function of time and the calculated fitting curves.
Molecular Biology
The expression and purification of JNK3 (39–402), using a pET24a(+)_HLT_JNK3 construct, were carried out as previously described.Citation58 The purity of the protein was analyzed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and ultra-high-performance liquid chromatography electrospray ionization mass spectrometry (UHPLC-ESI-MS).
His-tagged USP7CD (208–560) was transformed into E. coli BL 21 (DE3) pLysS cells (Novagen, Merck, Darmstadt, Germany). The sequence was also inserted into the pET24a(+)_HLT vector. Cultures were grown in 2xYT media containing kanamycin and chloramphenicol at 37 °C until an OD600 of 0.6–0.8 was reached. The bacteria were induced with 0.7 mM isopropyl-β-D-thiogalactoside and incubated overnight at 18 °C. The cells were harvested by centrifugation at 4000 g at 4 °C for 30 min. Harvested cell pellets were resuspended in lysis buffer (20 mM HEPES pH 8.0, 150 mM NaCl, 2 mM TCEP) and cracked using Bandelin Sonopuls HD 3200 with KE76 sonicator tip (Bandelin, Berlin, Germany). The lysate was clarified by centrifugation for 1 h at 18,500 rpm, filtered through a 0.22 µm membrane and then loaded onto 2×5 mL HisTrapTM FF Ni2+-columns (Cytiva, Marlborough, MA, USA) pre-equilibrated with 20 mM HEPES pH 8.0, 150 mM NaCl, 2 mM TCEP, 20 mM imidazole. The column was washed to baseline with lysis buffer plus 20 mM imidazole before the protein was eluted with lysis buffer plus 250 mM imidazole. The six His tags were removed using TEV protease while dialyzing against lysis buffer overnight at 4 °C. The cleaved sample was then loaded again onto 2×5 mL HisTrapTM FF Ni2+-columns (Cytiva, Marlborough, MA, USA) pre-equilibrated with lysis buffer plus 20 mM imidazole and washed with lysis buffer plus 20 mM imidazole. Fractions containing protein were concentrated down to 10 mL and loaded onto a size-exclusion HiLoadTM 26/60 SuperdexTM 75 prep grade column (Cytiva, Marlborough, MA, USA) pre-equilibrated with 25 mM Tris pH 8.0, 150 mM NaCl and 5 mM TCEP. Peak fractions visualized by SDS-PAGE were pooled and concentrated before they were flash-frozen in liquid nitrogen and stored at −80 °C. In addition, the correct protein mass was verified by UHPLC-ESI-MS.
The pET24a(+)_HLT_T-p53C plasmid, containing the stabilized core domain of p53, T-p53C (94–312, M133L/V203A/N239Y/N268D), was transformed into E. coli BL 21 (DE3) pLysS cells (Novagen, Merck, Darmstadt, Germany). Expression and purification were performed as previously described.Citation52 The purity of T-p53C was monitored by SDS-PAGE, and the correct protein mass was confirmed by UHPLC-ESI-MS. All used protein sequences are depicted in .
Table 1 All Used Protein Sequences
Differential Scanning Fluorimetry (DSF)
The melting temperatures of JNK3, USP7, and T-p53C in presence or absence of fragments were determined by DSF. Experiments were performed on a Qiagen Rotor-Q Model-5-Plex HRM real-time PCR instrument (Qiagen, Hilden, Germany). SYPRO Orange (Life Technologies Corporation, Eugene, OR, USA) served as a fluorescent dye, and a final concentration of 5x was used. The experiments were performed with 8 µM protein, and a final compound concentration of 1 mM was applied, corresponding to a protein-to-compound ratio of 1:125.Citation59 For USP7 Tris buffer (25 mM Tris pH 8.0, 150 mM NaCl, 5 mM TCEP, 5% (v/v) DMSO), and for JNK3 and T-p53C phosphate buffer (25 mM KPi pH 7.2, 150 mM NaCl, 1 mM TCEP, 5% (v/v) DMSO) were used.
All measurements were performed after 30 min of incubation. DSF experiments were performed with a constant heating rate of 270 °C/h.Citation60 The temperature was ramped from 28 °C to 60–70 °C and the excitation and emission filters were set to 470 nm and 610 nm, respectively. The melting temperatures (Tm) of JNK3, USP7, and T-p53C were determined from the maxima of the first derivatives of the melting curves in OriginPro2020 (OriginLab, Northampton, MA, USA). ∆Tm was calculated by subtracting the resulting Tm of the protein samples from the Tm of the samples containing protein and fragment. All samples were measured at least in triplicate.
Intact Protein Mass Spectrometry
T-p53C in phosphate buffer (25 mM KPi pH 7.2, 150 mM NaCl, 5 mM DTT) was mixed with fragments dissolved in DMSO, yielding a protein-to-compound ratio of 1:100 and 5% (v/v) DMSO.Citation61 USP7 was in Tris buffer (25 mM Tris pH 8.0, 150mM NaCl, 5 mM TCEP) with a protein-to-compound ratio of 1:100 and 5% (v/v) DMSO. In certain cases (SN002, VS004) reactions with TCEP and DTT occurred in the UHPLC-ESI-MS experiments and were therefore excluded from measurements. The mixtures of USP7 and T-p53C were both incubated at 20 °C for 4 h on a rotating shaker. Intact protein mass analysis was performed using UHPLC-ESI-MS measurements using a C4-bonded 400 Å 2.7 µm superficially porous particle reversed-phase column and a time-of-flight mass analyzer. Data acquisition and data analysis were performed as previously described.Citation52 The deconvoluted MS spectra of the native proteins are depicted in Figure S6.1 and Figure S6.2.
Results
Selection Process
Our aim was to create a diverse fragment library with covalently binding fragments to engage structurally diverse protein targets. We decided to use different types of warheads to obtain a broad range of reactivity, reaction geometry, and interaction patterns to generate more diverse covalent binding modes than with a library of fragments containing only a single warhead chemotype.
We chose α-cyanoacrylamides/acrylates and vinyl sulfones as Michael-type reacting warheads. They are both higher reactivity derivatives of the commonly used and well-studied acrylamide warheads,Citation5,Citation13 which may give rise to higher hit rates. For α,β-unsaturated carbonyls and sulfones, the reactivity depends among others on substitution at the α- and β- position enabling to a broad spectrum of intrinsic reactivities. Moreover, α-cyanoacrylamide compounds typically undergo a reversible cysteine modification through their high αC-H acidity, which facilitates proton abstraction and the reverse ß-elimination.Citation13 This could avoid potential off-target modification but also create difficulties in the experimental adduct determination. Additionally, we purchased epoxides as very small-sized reactive groups. Moreover, they could be attacked by nucleophiles at two vicinal positions, making them very versatile warheads.Citation5 Epoxides are used as a covalent reactive group in approved and widely used drugs like the antibiotic Fosfomycin or the proteasome inhibitor Carfilzomib, demonstrating the clinical applicability of the epoxide-moiety.Citation62–64 As last type of warhead, we used electron-deficient heteroarenes with leaving groups, which can undergo SNAr. They are less studied as covalent reactive groups in drug design, although heterocycles in general are very present in approved drugs.Citation65–68 They have several advantages over commonly used structures, such as structural rigidity and broad tunability by varying the leaving groups, electron-withdrawing groups, and the (hetero)aromatic scaffold. Moreover, their synthesis and reactions are well studied and established in medicinal chemistry.Citation5,Citation13
A dataset of commercially available compounds was provided by Aldrich Market Select (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany). Compounds larger than 22 non-hydrogen atoms, smaller than six non-hydrogen atoms, and a logP >2 were removed. SMARTs were created for the selected warhead types, representing the core scaffolds of the corresponding reactive groups, and used as a structural filter. Filtered compounds were clustered, and fragments were manually selected considering structural and steric properties, assumed reactivities, as well as prices and available quantities.
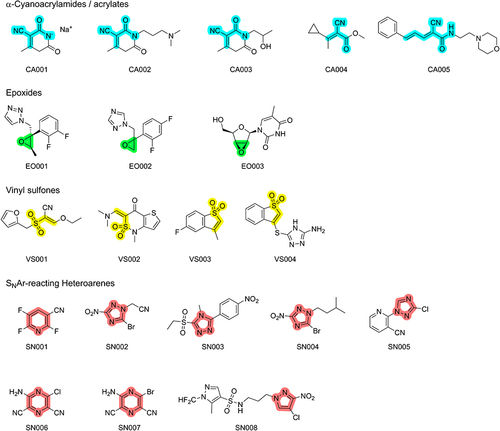
In total, we purchased 20 fragments including five α-cyanoacrylamides/acrylates, four vinyl sulfones, three epoxides, and eight SNAr-type electrophiles. The structures are depicted in . We are aware that 20 fragments and four different warhead classes are still far from the optimal quantity and diversity of fragments, we initially aimed for. Restricted by the pandemic, delivery difficulties, and financial resources, we initially used this selection of representative structures as a first starting point to engage in the topic.
Figure 1 Chemical structures of the 20 fragments purchased for the CovLib. The scaffolds for the respective warhead types are highlighted in cyan for α-cyanoacrylamides/acrylates, in green for epoxides, in yellow for vinyl sulfones, and in light red for SNAr-type electrophiles.
Reactivity and Solubility Characterization
When selecting the library, we used the logP value ≤2 calculated by RDKit as the threshold for our compound set. The partition coefficient is an indicator for the hydrophilicity/lipophilicity of a compound and therefore a key characteristic for drug molecules and compounds during the drug design and development process. It is an important indicator of the solubility of a substance and influences both the pharmacodynamic and the pharmacokinetic parameters of a molecule.
For our studies, the threshold was introduced to select compounds that are soluble enough for our screening procedures and to ensure a good starting point for eventual further drug development.
To further investigate the influence of logP and compare it with the experimentally determined values, we used Schrödinger suite version 2021-1 to predict QPLogPo/w and QPlogS values with the well-established QikProp module. The investigated compounds show QPlogP values from - 0.68 to 2.3. In addition, this broad logP range is also covered within the warhead classes.
For a detailed evaluation of the sufficient fragment solubility, we measured the kinetic solubility in a turbidimetric assay in a PBS buffer pH 7.4 containing 5% (v/v) DMSO to mimic typical assay conditions. The highest concentration up to which no scattering is observed is reported as “minimal instant solubility” (MIS) based on the first measurement cycle and as “minimal final solubility” (MFS) based on the last measurement cycle. MIS and MIC values can be found in . Fragment CA001 was not soluble in DMSO at a concentration of 100 mM, which was required for the first dilution series. A possible reason for this is that the fragment was purchased as sodium salt, which is likely to polar for sufficient solubility in organic solvents such as DMSO. The DMSO suspension at 100 mM was soluble in the PBS buffer and did not result in light scattering, indicating a sufficient solubility in aqueous media. Spectra could not be recorded for fragment VS002 because it was not soluble in DMSO at a concentration of 100 mM. Compound VS002 is a jet-black solid that becomes a black suspension when DMSO is added. When the suspension is mixed with buffer and evaluated in the assay, this resulted in a high absorbance, although it is unclear whether this is due to precipitation of fragments or the high background absorbance of the black solution. All other compounds were soluble at a concentration of at least 4 mM. This is sufficient solubility for typical fragment-screening assays. We observed no differences between minimal instant and final solubility. In contrast, the QPLogS solubility predicted by QikProp was notably lower for some of the fragments. In particular, CA005 and SN008 deviate significantly with predicted solubilities of 0.77 and 0.054 mM, respectively. This could be due to their relatively large size and some degree of flexibility interconnecting two submoieties. The prediction model relies on a database of experimentally derived solubility data on which the model is trained using a combination of diverse approaches. Furthermore, QikProp is independent of the 3D structure of the fragment. The difference in solubility may therefore be explained by structural differences between the actual fragment and the fragments used to train the QikProp model.Citation69
Table 2 Results of the Experimental Turbidimetric Solubility Characterization, Computational QPlogPo/w and QPLogS Calculations, and the Reactivity Assessment via GSH and DTNB Assay
In the next step, we characterized the thiol reactivity of our CovLib to determine its intrinsic reactivity. It is important to investigate whether the biological activity depends on a specific covalent or non-covalent binding event or on the high intrinsic reactivity of the fragment. Further, the thiol reactivity indicates general chemical stability of the compounds, which is a key property for in vitro studies and potential clinical use. We performed a high-throughput DTNB assay to estimate the fragments’ reactivity, followed by a detailed investigation using a GSH assay. Since this DTNB assay is a high-throughput measurement with absorbance at 412 nm as sole readout, it has some sources of error. For example, side reactions of the compounds with TCEP or the buffer substances cannot be detected. In addition, it is difficult to characterize molecules with high intrinsic absorption at 412 nm. Therefore, we performed the HPLC-UV-based GSH assay, which is well established in TCI development, as a second step to validate and further characterize the thiol reactivity.Citation70 Monitoring degradation by chromatography allowed us to characterize almost any compound present in the solution under study, with limitations for very low concentration and weakly absorbing compounds. Thus, it is possible to detect side-reactions or multiplereaction products in the assay. On the other hand, HPLC detection is much more time-consuming compared to the DTNB assay.
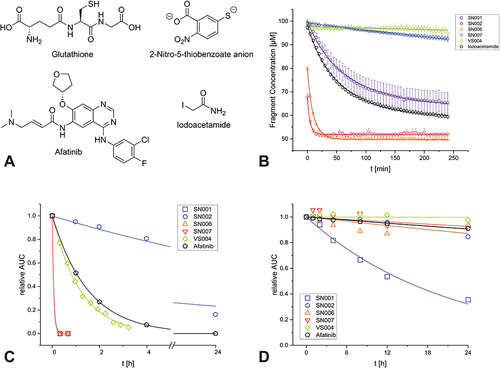
We examined iodoacetamide as a reference compound in the DTNB assay, which is known as a nonselective thiol labeling reagent and often used for comparison in this type of assay.Citation71,Citation72 We determined a second order rate constant of 2.6 M−1s−1. Comparable publications measured values of 2.5 M−1s−1Citation71 and 2.8 M−1s−1.Citation72
As a reference for the GSH assay we characterized the reactivity of Afatinib, an approved covalently acting tyrosine kinase inhibitor used for the treatment of non-small cell lung carcinoma.Citation73 We determined a pseudo-first order half-life of 1.1 h. In studies by Gerstenecker et al,Citation74 a half-life of 5 h was measured using a similar experimental set-up, but the acetonitrile concentration differed with 50% (v/v) instead of the 10% (v/v) we used.
In general, we observed several reactivity trends among warhead classes. All epoxides were evaluated as unreactive in both assays. Although known for their reactivity, there are examples in the literature of biologically active epoxy compounds with low reactivity.Citation75 All the oxiranes studied are substituted on both ring-carbons, which could lead to steric hindrance of the attacking nucleophile and thus relatively low reactivity in the assays. EO002 could not be studied because HPLC and NMR data indicated degradation prior to use (Figure S1.7, Figure S1.8). Since the compound has an unsubstituted oxirane carbon, it seems plausible that the compound is more reactive than the other two epoxy fragments, which could have led to the decomposition.
The α-cyanoacrylamides/acrylates showed no GSH reactivity with t1/2 >100 h (CA001, CA002, CA003) or low reactivity (CA004, CA005) with half-lives of 27 h and 59 h, respectively. None of them showed significant reactivity in the DTNB assay. CA001 was not soluble in ACN at 3.33 mM and was therefore added as a suspension. The compound was completely dissolved in the ACN/PBS mixture, but the concentration remains imprecise. It is counterintuitive that the stability of CA004 in PBS buffer (25 h) appears to be slightly lower than in GSH-containing PBS buffer (27 h). One possible explanation is that the substance precipitates slowly during the 24 h measurement, caused by the slow evaporation of ACN through the punctured septum of the HPLC vial. This would result in a decrease in AUC for both measurements. Kinetic solubility determinations, QPLogPo/w and QPLogS calculations showed intermediate but sufficient solubility for the compound. It is noteworthy that ACN was used as a co-solvent for the reactivity evaluation compared to DMSO in the turbidimetric solubility measurements, which could explain this discrepancy. In addition, no additional peaks occurred in the HPLC chromatogram with GSH, supporting the hypothesis that the apparent fragment degradation is not caused by a reaction with glutathione. In principle, the α-cyano substitution should increase the reactivity of the fragments compared to an unsubstituted Michael acceptor. Moreover, the nitrile increases the C-H acidity of the thiol adduct, favoring the reversal reaction, which could lead to covalent-reversible bonding. All the α-cyanoacrylamides/acrylates studied exhibit a double substitution or a single substitution with a bulky residue at the β-position, which could cause the relatively low reactivity toward thiols.
We encountered various problems when evaluating the four vinyl sulfones. VS002 was not soluble in ACN at 3.33 mM, so we could only evaluate the supernatant of the suspension. Therefore, the concentration of the compound was significantly lower compared to the standard method. In the DTNB assay, we observed a distinct reactivity of 2.6 M−1s−1. Although we could clearly detect the compound in the fragment mixture of the GSH assay, we measured only negligible degradation of VS002 with a half-life of >100 h. Due to this discrepancy and the fundamental problems with solubility, the exact reactivity remains unclear. VS001 showed sufficient solubility, but purity confirmation by HPLC was not possible. The chromatogram showed several peaks that blurred together. All chromatograms of the purity confirmation can be found in the SI. One possible explanation is the intermediate on-column degradation during the HPLC run, caused by the aqueous acidic eluent system. Therefore, assessment by GSH assay was not possible. The identity of the compound was confirmed by 1H-NMR in DMSO-d6. Additionally, the spectrum (Figure S11.11) shows two small peaks, which could not be assigned to the structure of VS001. These peaks could be caused by potential unknown reversible or irreversible adduct formation. Reactivity determination with DTNB showed a slight increase in absorbance at 412 nm, indicating some form of interference with the assay. Regarding the activating α-cyano substitution of the fragment, such high reactivity seems possible. The ethoxy group in the β-position also opens the possibility of a substitution reaction. It should be noted, however, that alcoholates are significantly worse leaving groups than thiolates. The reactivity of VS003 and VS004 could be well characterized in the GSH assay with half-lives of >100 h and 0.82 h, respectively. Both have the same hetero-bicyclic scaffold but differ by a substituent in β-position. The heteroaromatic sulfide of VS004 seems to strongly increase the thiol reactivity compared to the methyl substituent of VS003. Moreover, the heteroaromatic scaffold enables thiol exchange as a substitution reaction in addition to hetero-Michael addition. VS004 does not appear to be reactive in the DTNB assay. One possible reason for this discrepancy is the possibility of a reversible reaction with TNB2-. However, this does not appear to be the case in the reaction with GSH, which is probably due to the excess of GSH.
For the SNAr reacting compounds, a significant difference between 5- and 6-membered heterocycles stands out. All 5-membered arenes are evaluated as unreactive with half-lives >100 h, except SN002, which is medium reactive with a half-life of 11 h with GSH. A comparison of SN002 with its structural analogue SN004 shows that the cyanomethyl group of SN002 increases the SNAr reactivity in contrast to the alkyl substitution of SN004. In contrast to that, all three 6-membered heterocycles were found to be highly reactive. One reason for this could be the higher electron density at 5-membered arenes compared to 6-membered heterocycles resulting in lower SNAr reactivity. For SN001, SN006, and SN007, no remaining fragment could be detected from the second measurement point after 20 min in the GSH assay. Thus, their GSH half-life was estimated to be ≪0.33 h. Due to the HPLC runtime of 20 min, we could not test for the remaining fragment before the 20 min time point. Considering pseudo-first order kinetics, the half-life could be ~ 2.5 min or lower. Accordingly, the GSH assay could not discriminate between these highly reactive compounds. In the DTNB assay, the reactivity of SN001 was measured similar to that of iodoacetamide at 2.6 M−1s−1. The reaction rates of SN006 and SN007 are even higher with more than ten-fold reactivity of 26 M−1s−1 and 65 M−1s−1, respectively. This highlights the capability of the DTNB assay to discriminate the reactivities of highly reactive compounds, which the GSH assay could not achieve. A possible reason is the lower ratio of compound to nucleophile, which could generally result in slower reaction rates. Comparing the structures of SN001 to SN006 and SN007, the additional ring nitrogen and the additional nitrile group appear to lead to a higher reactivity. It is notable that for SN001 an intermediate aqueous stability was detected with a half-life of 15 h in PBS, whereas the higher thiol-reactive compounds SN006 and SN007 were found to be stable in the aqueous medium. A possible explanation for this could be found in the Pearson’s theory of hard and soft acids and bases (HSAB).Citation76 The strongly electron-withdrawing fluorine as a leaving group at SN001 makes the bound carbon less polarizable or harder than the carbon at the chlorine or bromine leaving groups in SN006 or SN007. This should favor the reaction with harder nucleophiles such as water compared to soft sulfur nucleophiles in GSH or TNB2-. Vice versa, the less polarizable carbons on chlorine/bromine in SN006/SN007 should favor the reaction with softer sulfur nucleophiles compared to hard oxygen bases.
Compared to the reference compounds, SN001, SN006, and SN007 should be assessed as rather nonselective arylating agents, as their DTNB reactivity is similar or even higher than the reactivity of iodoacetamide, which is known for its non-selective thiol labeling ability. VS004 was evaluated as reactive with a GSH half-life slightly below that of the clinically approved Afatinib, but the potentially interfered DTNB assay showed no reactivity. SN002 showed very interesting intermediate reactivity towards the thiols used, highlighting its potential as a selective, covalently modifying fragment. Chemical structures of the cysteine surrogates and reference compounds and time-dependent degradation plots of selected compounds measured in the reactivity assay are depicted in .
Figure 2 Chemical structures of the cysteine surrogates and reference compounds used for the reactivity assessment (A) Time-dependent depletion of selected compounds and the reference compound Iodoacetamide in the DTNB assay. The measurement points are shown as the mean value of the triplicate determination with the respective standard deviation as error bars. The second order fit function is shown as colored line (B) Time-dependent decrease of the relative AUC of selected compounds and the reference compound Afatinib in the GSH assay (C), and in PBS as a reference measurement (D) For GSH, the measurement points are shown as the mean of the duplicate determination, and the pseudo-first order fit function is shown as a colored line.
Evaluation of the CovLib Against Different Targets
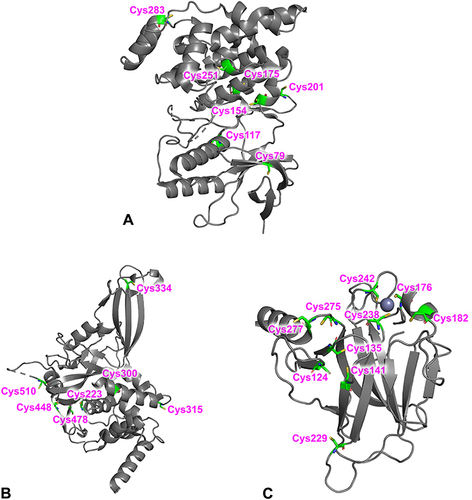
The CovLib was screened against several targets, including the protein kinase JNK3, the cysteine protease USP7, and the tumor suppressor p53, with different reactive non-catalytic (JNK3, p53, USP7) or catalytic (USP7) cysteine residues ().
Figure 3 Overall structures of JNK3 (Protein Data Bank (PDB) code 8BZP)Citation77 (A), USP7 (PDB code 4M5W)Citation78 (B), and T-p53C (PDB code 1UOL)Citation79 (C) All cysteines in the protein structures are highlighted.
The serine-threonine kinase JNK3 belongs to the mitogen-activated kinases (MAPK) family and is involved in various signal transduction, cell differentiation, and inflammatory processes.Citation80–83 Whereas the other members of the highly conserved JNK family (JNK1 and JNK2) are ubiquitously expressed, the expression of JNK3 is primarily restricted to central neuronal system, testis, and cardiac myocytes.Citation84,Citation85 Thus, the tissue contribution of JNK3 in combination with knockout mice experiments demonstrated that JNK3 is a promising target for the treatment of neurodegenerative disorders, such as Parkinson’s, Huntington’s, and Alzheimer’s diseases.Citation86–90
All members of the JNK family have a highly conserved and unique non-catalytic cysteine (Cys154, JNK3 numbering) that was covalently addressed in previous studies by a variety of acrylamide containing kinase inhibitors.Citation31,Citation91,Citation92 In addition to Cys154 in the ATP-binding pocket, Cys283 in the C-terminal domain of JNK3 was identified as another reactive residue.Citation93–95
The deubiquitinase ubiquitin-specific protease 7 (USP7), also known as herpes-associated ubiquitin-specific protease (HAUSP), is one of the approximately 100 deubiquitinating enzymes (DUB) that removes ubiquitin and protects substrate protein from degradation. Over the last few years, USP7 has emerged as a potential therapeutic target in cancer therapy due to its stabilizing effect on the oncogenic ligase mouse double minute 2 homolog (MDM2), which acts as a negative regulator of p53.Citation96–98 USP7 is a multi-domain cysteine protease consisting of 1102 amino acids (aa), an N-terminal TRAF-like domain (NTD), a catalytic domain (CD) with the catalytic triad (C223, H464, and D481) and five C-terminal ubiquitin-like domains (CTD).Citation99 In addition to the catalytic cysteine 223, there are six more cysteines (C300, C315, C334, C478, C488, C510) in the catalytic domain (aa 208–560) of USP7. The catalytic domain has an architecture that is similar to an open hand with fingers, palm, and thumb subdomains.Citation100 Thereby, the catalytic triad is located between the palm and thumb subdomains characterized by a highly conserved apoenzyme form.Citation101 When ubiquitin binds, the conformation changes to an active state so that the catalytic triad comes closer together. This proximity is essential for an efficient ubiquitin cleavage.Citation78,Citation102 There exist examples of targeting the catalytic cleft in USP7: A thiophene scaffold (based on, eg, P5091)Citation103 showed a covalent bond to Cys223 (as a 5-acetyl-3-nitro-thiophen-2-yl substituent). MS data indicate that the sesquiterpene lactone parthenolide binds up to thirteen cysteines covalently in USP7. However, the catalytic Cys223 does not seem to be modified by parthenolide, despite its inhibition of USP7.Citation104 Another covalent inhibitor is the 4-hydroxypiperidine FT827 with a vinyl sulfonamide as warhead which reacts with Cys223.Citation105 This scaffold is also found in non-covalent inhibitors like FT671. There is also the opportunity to target USP7 through allosteric inhibitors like GNE-6776Citation106 or thienopyridine derivatives.Citation107 These examples demonstrate various possibilities to target USP7, but nevertheless, there are currently no relevant inhibitors in clinical trials.Citation101
The tumor suppressor p53 plays an essential role in the cell cycle. Activation of p53 leads to the transcription of many genes involved in cell cycle arrest, apoptosis, and DNA repair.Citation108–111 Mutations of p53 are found in many tumors.Citation112 Oncogenic mutations, mainly located in the core-domain, can be divided into DNA contact mutants that result in the loss of an important DNA contact and structural mutants that destabilize the protein.Citation113,Citation114 Hence, there has been a long-standing interest in finding stabilizing small molecules to restore the p53 activity.Citation46,Citation60,Citation115–117 Reactive fragments like the 2-sulfonylpyrimidine PK11000Citation59 and the methylene quinuclidinone PRIMA-1Citation118–121 achieve general stabilization of p53 through modification of surface-exposed cysteines and thus represent a rescue strategy for destabilized p53 cancer mutants.Citation52,Citation59,Citation61 The cysteines C124, C182, C229, C277, and C275 in the core domain of p53 are fully or partially surface-exposed and thus amenable to covalent modification.Citation61,Citation122 The thiol groups of cysteines C277 and C182 have the highest solvent accessibility.Citation123 Arylations of the surface-exposed Cys277 and Cys182 were found for mild arylating 2-sulfonylpyrimidine-containing compounds.Citation59,Citation123,Citation124 Methylene quinuclidinone, the bioactive compound of PRIMA-1 and its methylated analog APR-246, is a Michael acceptor and modifies multiple cysteines on the surface of the p53 core domain.Citation119,Citation121,Citation125
DSF Measurements
The influence of the CovLib compounds on the melting temperatures of JNK3, USP7, and T-p53C and a possible fragment binding was investigated by DSF.Citation126–128 DSF is an efficient and fast primary screening method for ligand identification, even for kinases such as JNK3.Citation58,Citation129–131 Typically, ligand binding in the ATP-binding pocket significantly increases the Tm of kinases with a correlation between ligand binding and stabilization within a structural series.Citation58,Citation129 Due to the large number of thermodynamically destabilized structural mutants, the thermal stabilization of p53 in the DSF experiment is a main indicator for a fragment hit.Citation46,Citation60,Citation112,Citation116,Citation117,Citation132
An incubation time of 30 min was chosen for the CovLib screening. According to the reactivity screening, the CovLib fragments are either highly reactive or chemically stable. As fragment SN002 is the only fragment with intermediate GSH reactivity, extended incubation times were also tested. shows the DSF screening results of the CovLib with JNK3, USP7, and T-p53C. The corresponding melting curves are depicted in Figure S5.1, Figure S5.2, Figure S.5.3, and the first derivatives of the melting curves are shown in . The DSF results show similar trends as the determined reactivity of the compounds.
Figure 4 First derivatives of the melting curves of α-cyanoacylamides/acrylates (A), epoxides (B), vinyl sulfones (C), and SNAr (D) fragments (1 mM) with JNK3 (8 µM protein) after 30 min of incubation.
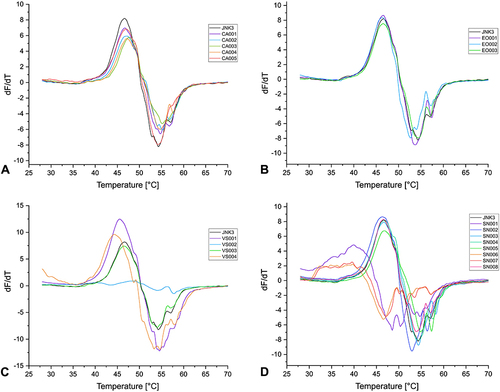
Figure 5 First derivatives of the melting curves of α-cyanoacrylamides/acrylates (A), epoxides (B), vinyl sulfones (C), and SNAr (D) fragments (1 mM) with USP7 (8 µM protein) after 30 min of incubation.
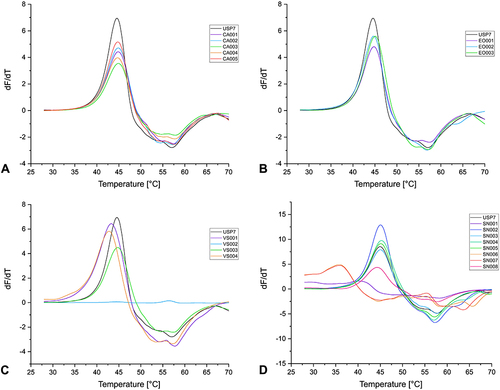
Figure 6 First derivatives of the melting curves of α-cyanoacrylamides/acrylates (A), epoxides (B), vinyl sulfones (C), and SNAr (D) fragments (1 mM) with T-p53C (8 µM protein) after 30 min of incubation.
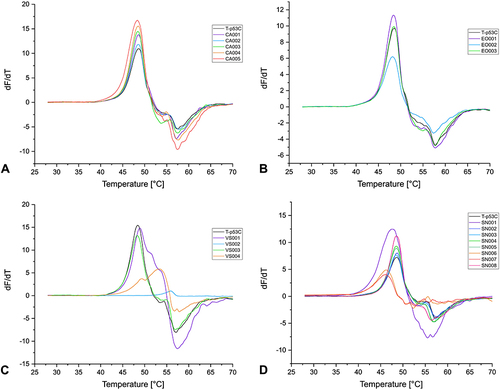
Table 3 ∆Tm ± SD of JNK3, USP7, and T-p53C (8 µM Protein) Incubated with 1 mM Compound (Protein-to-Compound Ratio 1:125) for 30 Min at 20 °C
All the tested compounds with a covalent reversible α-cyanoacrylamide/acrylate warhead had only a small effect on the Tm of the proteins. Only CA003 stabilized JNK3 by approximately 1.10 °C. However, this compound was not subjected to further characterization as kinase inhibitors usually exhibit a larger shift in the Tm.Citation58,Citation129,Citation131
The GSH reactivity study showed that the two fragments with an epoxide warhead are chemically stable, and DSF measurements also revealed that these CovLib compounds had no significant effect on the thermal stability of the proteins.
The reactivity studies suggest that the vinyl sulfones VS002 and VS003 represent stable compounds, while VS004 shows significant reactivity. VS001 was also classified as reactive, yet the characterization of purity and reactivity was associated with several problems, including Pan-Assay Interference Compounds (PAINS)Citation133 characteristics, as already mentioned. Fragment VS002 could not be screened by DSF due to the extremely low fluorescence intensity, and no protein melting curves were obtained. Reduced fluorescence signal intensity can result from interactions of the compound with the optical properties of the fluorescent dye SYPRO Orange, leading to partial fluorescence quenching.Citation116,Citation126 As expected, due to its relatively low thiol reactivity and currently no non-covalent binding mechanism, VS003 did not significantly affect the Tm of all three proteins.
The compound VS001 slightly decreased the Tm of JNK3 and USP7, while the Tm of T-p53C was slightly increased. The reactive compound VS004 destabilized JNK3 by about −2.40 °C and USP7 by −1.75 °C. However, the vinyl sulfone VS004 exhibited a significant increase in the Tm of T-p53C (∆Tm 5.06 °C), indicating fragment binding. In the melting curve of T-p53C with compound VS004, two transitions appeared (). The first derivative displayed a minor shoulder and a major peak, possibly indicating differently modified protein species or contributions from non-covalently bound fragments with distinct melting points.Citation127,Citation134,Citation135
Fragments SN003, SN004, SN005, and SN008, which would react via SNAr, are stable in the presence of thiol(ate) nucleophile, and no effect on Tm of the proteins was detected. Fragment SN002 showed an intermediate reactivity; however, the DSF data after 30 min incubation did not indicate fragment binding. This compound was also examined with T-p53C and USP7 after 4 h and 24 h of incubation, but no effect on Tm was observed with increasing incubation time (Figure S5.4, Figure S5.5, Table S5.1).
Fragments SN001, SN006, and SN007 showed a very short t1/2 in the GSH assay and strongly influenced the Tm of the proteins. Fragment SN001 largely destabilized JNK3 as well as USP7, but T-p53C was only slightly destabilized by about −0.57 °C. No melting curves for JNK3 were obtained with fragments SN006 and the bromine analog SN007. High fluorescence intensity was measured even at low temperatures, indicating denaturation, aggregation, or misfolding.Citation127 The melting curves of USP7 for these compounds indicated a high destabilization of about −9.10 °C. The Tm of T-p53C was strongly decreased with SN006 and SN007. This suggests that the high reactivity of the fragments leads to multiple arylations, which result in a strong loss of protein stability, in the case of JNK3 even to denaturation or binding in the unfolded state of the proteins, causing a reduction of Tm.Citation136,Citation137 However, the negative shifts do not additionally exclude binding in the native state.Citation127
In summary, the DSF measurements revealed the first interesting hits with the reactive fragments VS004, SN001, SN006, and SN007. The vinyl sulfone VS004 led to a strong stabilization of T-p53C. The fragment SN001 destabilized JNK3 and USP7 enormously, whereas the Tm of T-p53C was only slightly reduced. These first interesting DSF hits now require further investigation.
Intact Protein Mass Spectrometry
Fragment hits identified by DSF were validated by UHPLC-ESI-MS. Intact protein mass spectrometry is a principal screening method in covalent FBDD to directly identify the covalent binding of the fragments to the proteins.Citation7,Citation12,Citation33,Citation34,Citation138–140 However, covalent adduct formation with α-cyanoacrylamides cannot be observed by protein mass spectrometry due to rapid reversibility of binding, especially if the structural integrity of the target protein and thus stabilization of the covalent complex is lost.Citation13,Citation62,Citation141
The covalent modifications of USP7 and T-p53C were confirmed by the deconvoluted MS spectra for the six CovLib fragments VS001, VS004, SN001, SN002, SN006, and SN007 ( and ). The deconvoluted MS spectra of the reactive fragments indicated multiple modifications of the targets USP7 and T-p53C.
Figure 7 Deconvoluted MS spectra of VS001 (A), VS004 (measured without TCEP) (C), SN001 (E) with USP7 (protein-to-compound ratio 1:100, theoretical mass of unmodified USP7: 41,145.61 Da) and VS001 (B), VS004 (measured without DTT) (D), and SN001 (F) with T-p53C (protein-to-compound ratio 1:100, theoretical mass of unmodified T-p53C: 24,756.03 Da) after 4 h of incubation at 20 °C.
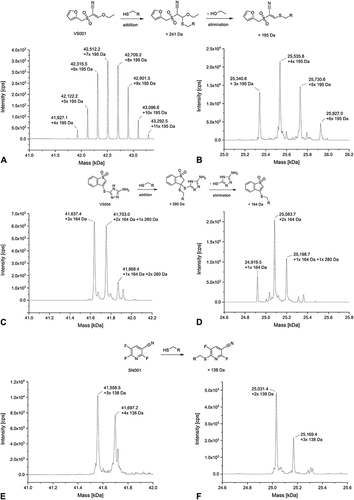
Figure 8 Deconvoluted MS spectra of SN002 (measured without TCEP) (A), SN006 (C), SN007 (E) with USP7 (protein-to-compound ratio 1:100, theoretical mass of unmodified USP7: 41,145.61 Da) and SN002 (measured without DTT) (B), SN006 (D), and SN007 (F) with T-p53C (protein-to-compound ratio 1:100, theoretical mass of unmodified T-p53C: 24,756.03 Da) after 4 h of incubation at 20 °C.
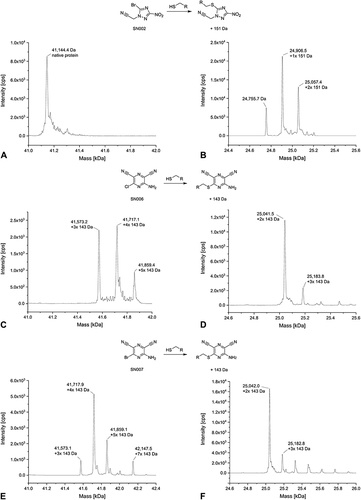
In the deconvoluted MS spectra of the vinyl sulfone VS001 with USP7 and T-p53C, multiple peaks with an increase in the native protein mass were detected ( and ). The number of VS001 molecules bound to USP7 exceeded the number of cysteines in the protein (7 cysteines), indicating binding to other aa than cysteines. Consistent with the HSAB concept,Citation76 vinyl sulfones are also known to target the ε-amino group of lysine.Citation5,Citation13,Citation21,Citation142 The mass difference between each peak was approximately 195 Da, which can be explained by the addition of cysteine and the removal of an ethanolate from VS001.
Not unexpectedly, the MS spectrum of USP7 with VS004 showed several peaks (). The main peak showed attachment of three VS004 molecules with elimination of heteroaryl thiolate (i.e., thiol exchange). The second highest peak detected showed a mixture of addition and thiol exchange products with two equivalents of the thiol exchange product plus one equivalent of the addition product detected. In the last smaller signal, one equivalent of addition product plus two modifications by thiol exchange were found. The spectrum of VS004 with T-p53C () showed also both nucleophilic addition to the α,ß-unsaturated vinyl sulfone and the product of subsequent elimination of the heteroaryl thiolate leaving group. The protein species with two VS004 molecules attached and loss of heteroaryl thiolate represented the highest peak.
For the SNAr reactive compounds, multiple arylations of USP7 and T-p53C were detectable after 4 h of incubation and a protein-to-compound ratio of 1:100. In the deconvoluted MS spectrum of compound SN001 with USP7 the main peak showed triple arylated protein and a smaller peak with four times arylated protein (). In comparison, in the deconvoluted MS spectrum of T-p53C (), the double arylated protein species represented the highest signal. A small peak corresponding to the three times modified protein was also visible.
A difference in the covalent modifications between the two targets USP7 and T-p53C exists for SN002. For USP7, no modified masses were detectable with SN002 (). The only peak represented the mass of the native protein. However, compound SN002 was identified as a hit for T-p53C through cross-validation by MS, in contrast to the primary screening by DSF. In the deconvoluted MS spectrum, covalent modifications of T-p53C were observed with the single arylated protein species displaying the highest signal (). However, native T-p53C was also detected. This can be explained by the lower reactivity and longer t1/2 of SN002 in the GSH assay, compared to the other CovLib hits. The incubation time of 4 h before the MS experiment might be too short for the complete reaction of the fragment. Therefore, increasing the incubation time might result in an increased modification of T-p53C.
In the deconvoluted MS spectra of compound SN006 () and its bromo analog SN007 (), USP7 arylated with three and four molecules represented the highest signal. Another small signal corresponding to the five times arylated protein was detected in both spectra, whereas SN007 arylated USP7 seven times. MS analysis of T-p53C revealed two peaks, the main peak represented the protein species with two attached molecules SN006 () and SN007 (), respectively. Additionally, triple modified T-p53C was also detected in both spectra. Furthermore, in the spectrum of SN007 peaks with lower intensity indicating the four- and five-times modified protein were detected. It is possible that the increased overall intensity of the SN007 spectrum compared to SN006 contributed to the detection of these additional peaks.
In summary, the MS experiments confirmed that the hits VS001, VS004, SN001, SN006, and SN007 identified by DSF covalently modify USP7 and T-p53C. The CovLib hits found in this context represent highly reactive compounds that multiply and non-specifically modify both targets. Still, they can be utilized as starting points for identification of covalently ligandable amino acids and pockets and subsequent compound optimization by increasing non-covalent binding affinity and specificity, while decreasing reactivity. Furthermore, MS analysis revealed SN002 as another CovLib hit. Although this fragment was covalently bound to T-p53C, it did not interact with USP7 indicating a certain level of specificity.
Conclusion
For a covalent FBDD approach, we have assembled a library of covalent fragments (CovLib) with the goal of achieving a wide range of diversity and reactivity. Initially, 20 fragments with four different warhead classes were selected as a first test set, which we describe here. Therefore, it should be noted that the study has limitations due to the small size of the library. We calculated QPLogP and QPLogS as theoretical parameters for the solubility. In addition, we experimentally evaluated the turbidimetric solubility of the fragments. Two fragments (CA001, VS002) showed insufficient solubility in DMSO, with sodium salt CA001 probably being too polar for adequate solubility in aprotic organic solvents without the addition of water. This shows that it could be useful to consider a minimum logP value in addition to the maximum value when selecting fragments for a screening library.
The reactivity of the fragments was analyzed using a high-throughput DTNB assay and a GSH stability assay. Strong reactivity differences were found among the warhead classes. All selected α-cyanoacrylamides/acrylates and epoxides were evaluated as less reactive compounds, with steric hindrance most likely being a major contributor to this unexpected behavior. The selected SNAr and vinyl sulfone fragments were either highly reactive (SN001, SN006, SN007, VS001, VS004) or stable (SN003, SN004, SN005, SN008, VS002, VS003) towards sulfur nucleophiles. SN002 was the only fragment of moderate reactivity. Notably, the covalent reactivity of a fragment is also influenced by its non-covalent scaffold, in addition to the respective warhead type.
Screening of the CovLib against three different protein targets, i.e., JNK3, USP7, and p53, provided first promising results. DSF measurements identified the fragments VS004, SN001, SN006, and SN007 as hits. Notably, compound VS001 impacted the Tm of the proteins. Nevertheless, this compound emerged as potential PAINS. Vinyl sulfone VS004 only resulted in a significant stabilization of T-p53C (∆Tm 5.06 °C). The fragment SN001 strongly destabilized JNK3 and USP7, whereas the melting temperature of T-p53C was only slightly lowered. Compounds SN006 and SN007 destabilized all tested targets, and no melting curve could be detected for JNK3. The covalent attachment of the fragments to their target proteins was confirmed by MS, which revealed multiple modifications of T-p53C and USP7. Fragment SN002, which is the only fragment with intermediate reactivity, exclusively modified T-p53C in the MS experiment. However, this compound did not affect the Tm of T-p53C even after increased incubation time. In principle, the hit rates in both DSF and MS correlated with the results from reactivity assessment, i.e. reactive compounds strongly influenced the targets, while non-reactive compounds showed no effect.
The characterization and screening of the CovLib yielded the first interesting hits, including the fragments VS004, SN001, SN006, and SN007. The hits and their different influences on the melting temperature and specific binding to the tested proteins need further investigation. The knowledge gained from the characterization and target evaluation of the first CovLib fragments will serve as a basis for the selection of additional compounds to integrate more fragments of moderate reactivity into the library and to find more selective hits for the respective proteins and labeling sites or other interesting targets. These may then provide a starting point for optimization using common FBDD strategies and rational fine-tuning of reactivity to enable the discovery of drug-like covalent modulators with high potency and selectivity.
Abbreviations
aa, amino acids; AUC, area under the curve; CD, catalytic domain; CRG, covalent reactive group; DSF, differential scanning fluorimetry; DTNB, 5,5'-dithiobis-(2-nitrobenzoic acid), Ellman’s Reagent; FBDD, fragment-based drug discovery; GSH, glutathione; HEFLib, Halogen-Enriched Fragment Library; HPLC, high performance liquid chromatography; HSAB, hard and soft acid and base; JNK3, c-Jun N-terminal kinase 3; KPi, potassium phosphate; MDM2, mouse double minute 2 homolog; n.a., not available; NMR, nuclear magnetic resonance; NaPi, sodium phosphate; PBS, phosphate-buffered saline; PDB, Protein Data Bank; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; SNAr, nucleophilic aromatic substitution; TCI, targeted covalent inhibitor; t1/2, half-life; Tm, melting temperature; TNB2-, 2-nitro-5-thiobenzoate anion; T-p53C, quadrupole thermostable mutant of the p53 core-domain, 94–312, M133L/V203A/N239Y/N268D; UHPLC-ESI-MS, ultra-high-performance liquid chromatography electrospray ionization mass spectrometry; USP7, ubiquitin-specific protease 7; 2xYT, 2x yeast extract tryptone medium.
Author Contributions
F.M.B. envisioned the research. T.K., L.E., M.S., and F.M.B. conceptualized the experiments and designed the study. M.S. performed the Turbidimetric Solubility Assay, DTNB Assay, and Glutathione Assay. T.K. and L.E. prepared the proteins by heterologous expression and performed the DSF studies. S.J., B.M., C.K., and M.L. performed and analyzed the UHPLC-ESI-MS experiments. M.E. performed all computational investigation including the customized fragment filtering process. T.K., M.S., F.M.B., and M.G. selected the final compounds. T.K., L.E., and M.S. conducted data analysis and reprocessing of the experimental data. T.K., L.E., M.S., and F.M.B. wrote the manuscript. M.E., S.J., B.M., C.K., M.L., and M.G. critically reviewed the manuscript. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
The authors acknowledge support from the state of Baden–Württemberg through bwHPC and the German Research Foundation (DFG) through Grant No. INST 40/575-1 FUGG (JUSTUS 2 cluster). In addition, support is acknowledged from the High Performance and Cloud Computing Group at the Zentrum für Datenverarbeitung of the University of Tübingen and the German Research Foundation (DFG) through Grant No. INST 37/935-1 FUGG (BinAC cluster).
References
- Boike L, Henning NJ, Nomura DK. Advances in covalent drug discovery. Nat Rev Drug Discov. 2022;21(12):881–898. doi:10.1038/s41573-022-00542-z
- Baillie TA. Targeted Covalent Inhibitors for Drug Design. Angew Chem Int Ed. 2016;55(43):13408–13421. doi:10.1002/anie.201601091
- Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10(4):307–317. doi:10.1038/nrd3410
- De Cesco S, Kurian J, Dufresne C, Mittermaier AK, Moitessier N. Covalent inhibitors design and discovery. Eur J Med Chem. 2017;138:96–114. doi:10.1016/j.ejmech.2017.06.019
- Gehringer M, Laufer SA. Emerging and re-emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J Med Chem. 2019;62(12):5673–5724. doi:10.1021/acs.jmedchem.8b01153
- Chaikuad A, Koch P, Laufer SA, Knapp S. The cysteinome of protein kinases as a target in drug development. Angew Chem Int Ed Engl. 2018;57(16):4372–4385. doi:10.1002/anie.201707875
- Keeley A, Petri L, Ábrányi-Balogh P, Keserű GM. Covalent fragment libraries in drug discovery. Drug Discov Today. 2020;25(6):983–996. doi:10.1016/j.drudis.2020.03.016
- Butterworth S, Cross DAE, Finlay MRV, Ward RA, Waring MJ. The structure-guided discovery of osimertinib: the first U.S. FDA approved mutant selective inhibitor of EGFR T790M. . MedChemComm. 2017;8(5):820–822. doi:10.1039/C7MD90012K
- Finlay MRV, Anderton M, Ashton S, et al. Discovery of a Potent and Selective EGFR Inhibitor (AZD9291) of Both Sensitizing and T790M Resistance Mutations That Spares the Wild Type Form of the Receptor. J Med Chem. 2014;57(20):8249–8267. doi:10.1021/jm500973a
- Zhao Z, Bourne PE. Progress with covalent small-molecule kinase inhibitors. Drug Discov Today. 2018;23(3):727–735. doi:10.1016/j.drudis.2018.01.035
- Lanman BA, Allen JR, Allen JG, et al. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. J Med Chem. 2020;63(1):52–65. doi:10.1021/acs.jmedchem.9b01180
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:7477):548–551. doi:10.1038/nature12796
- Hillebrand L, Liang XJ, Serafim RAM, Gehringer M. Emerging and re-emerging warheads for targeted covalent inhibitors: an update. J Med Chem. 2024;67(10): 7668–7758. doi:10.1021/acs.jmedchem.3c01825
- Uetrecht J. Idiosyncratic Drug Reactions: current Understanding. Annu Rev Pharmacol Toxicol. 2007;47(1):513–539. doi:10.1146/annurev.pharmtox.47.120505.105150
- Baillie TA. Approaches to mitigate the risk of serious adverse reactions in covalent drug design. Expert Opin Drug Discov. 2021;16(3):275–287. doi:10.1080/17460441.2021.1832079
- Bauer RA. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov Today. 2015;20(9):1061–1073. doi:10.1016/j.drudis.2015.05.005
- Lonsdale R, Ward RA. Structure-based design of targeted covalent inhibitors. Chem Soc Rev. 2018;47(11):3816–3830. doi:10.1039/c7cs00220c
- Barf T, Kaptein A. Irreversible protein kinase inhibitors: balancing the benefits and risks. J Med Chem. 2012;55(14):6243–6262. doi:10.1021/jm3003203
- McKenna SM, Fay EM, McGouran JF. Flipping the switch: innovations in inducible probes for protein profiling. ACS Chem Biol. 2021;16(12):2719–2730. doi:10.1021/acschembio.1c00572
- Liu Q, Sabnis Y, Zhao Z, et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20(2):146–159. doi:10.1016/j.chembiol.2012.12.006
- Serafim RAM, Haarer L, Pedreira JGB, Gehringer M. Covalent chemical probes for protein kinases. Curr Res Chem Biol. 2023;100040. doi:10.1016/j.crchbi.2022.100040
- Lu X, Smaill JB, Patterson AV, Ding K. Discovery of cysteine-targeting covalent protein kinase inhibitors. J Med Chem. 2022;65(1):58–83. doi:10.1021/acs.jmedchem.1c01719
- Hillebrand L, Gehringer M. Never gonna give you up – current developments in covalent protein kinase inhibitors. scientific articles. Chimia. 2022;76(5):435. doi:10.2533/chimia.2022.435
- Strelow JM. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discov. 2017;22(1):3–20. doi:10.1177/1087057116671509
- Li KS, Quinn JG, Saabye MJ, et al. High-Throughput Kinetic Characterization of Irreversible Covalent Inhibitors of KRASG12C by Intact Protein MS and Targeted MRM. Anal Chem. 2022;94(2):1230–1239. doi:10.1021/acs.analchem.1c04463
- Mons E, Roet S, Kim RQ, Mulder MPC. A comprehensive guide for assessing covalent inhibition in enzymatic assays illustrated with kinetic simulations. Curr Protoc. 2022;2(6):e419. doi:10.1002/cpz1.419
- Craven GB, Affron DP, Kösel T, et al. Multiparameter kinetic analysis for covalent fragment optimization by using quantitative irreversible tethering (qIT). ChemBioChem. 2020;21(23):3417–3422. doi:10.1002/cbic.202000457
- McAulay K, Bilsland A, Bon M. Reactivity of covalent fragments and their role in fragment based drug discovery. Pharmaceuticals. 2022;15(11):1366. doi:10.3390/ph15111366
- Flanagan ME, Abramite JA, Anderson DP, et al. Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J Med Chem. 2014;57(23):10072–10079. doi:10.1021/jm501412a
- Forster M, Liang XJ, Schröder M, et al. Discovery of a novel class of covalent dual inhibitors targeting the protein kinases BMX and BTK. Int J Mol Sci. 2020;21(23):9269.
- Zhang T, Inesta-Vaquera F, Niepel M, et al. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol. 2012;19(1):140–154. doi:10.1016/j.chembiol.2011.11.010
- Keeley A, Ábrányi-Balogh P, Keserű GM. Design and characterization of a heterocyclic electrophilic fragment library for the discovery of cysteine-targeted covalent inhibitors. MedChemComm. 2019;10(2):263–267. doi:10.1039/C8MD00327K
- Jöst C, Nitsche C, Scholz T, Roux L, Klein CD. Promiscuity and selectivity in covalent enzyme inhibition: a systematic study of electrophilic fragments. J Med Chem. 2014;57(18):7590–7599. doi:10.1021/jm5006918
- Resnick E, Bradley A, Gan J, et al. Rapid covalent-probe discovery by electrophile-fragment screening. J Am Chem Soc. 2019;141(22):8951–8968. doi:10.1021/jacs.9b02822
- Zambaldo C, Daguer J-P, Saarbach J, Barluenga S, Winssinger N. Screening for covalent inhibitors using DNA-display of small molecule libraries functionalized with cysteine reactive moieties. MedChemComm. 2016;7(7):1340–1351.
- Clark MA, Acharya RA, Arico-Muendel CC, et al. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat Chem Biol. 2009;5(9):647–654.
- Zimmermann G, Rieder U, Bajic D, et al. A specific and covalent JNK-1 Ligand selected from an encoded self-assembling chemical library. Chemistry. 2017;23(34):8152–8155. doi:10.1002/chem.201701644
- Scarpino A, Ferenczy GG, Keserű GM. Covalent docking in drug discovery: scope and limitations. Curr Pharm Des. 2020;26(44):5684–5699. doi:10.2174/1381612824999201105164942
- Sotriffer C. Docking of covalent ligands: challenges and approaches. Mol Inform. 2018;37(9–10):1800062. doi:10.1002/minf.201800062
- Scarpino A, Ferenczy GG, Keserű GM. Comparative evaluation of covalent docking tools. J Chem Inf Model. 2018;58(7):1441–1458. doi:10.1021/acs.jcim.8b00228
- Shraga A, Resnick E, Gabizon R, London N. Chapter Eight - Covalent Fragment Screening. Ward RA, Grimster NP, eds.. Academic Press: 2021. 243–265.
- Murray CW, Rees DC. The rise of fragment-based drug discovery. Nat Chem. 2009;1(3):187–192. doi:10.1038/nchem.217
- Kuntz ID, Chen K, Sharp KA, Kollman PA. The maximal affinity of ligands. Proc Natl Acad Sci U S A. 1999;96(18):9997–10002. doi:10.1073/pnas.96.18.9997
- Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9(10):430–431. doi:10.1016/S1359-6446(04)03069-7
- Heidrich J, Sperl LE, Boeckler FM. Embracing the diversity of halogen bonding motifs in fragment-based drug discovery-construction of a diversity-optimized halogen-enriched fragment library. Front Chem. 2019;7:9. doi:10.3389/fchem.2019.00009
- Wilcken R, Liu X, Zimmermann MO, et al. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J Am Chem Soc. 2012;134(15):6810–6818. doi:10.1021/ja301056a
- Zimmermann MO, Lange A, Wilcken R, et al. Halogen-enriched fragment libraries as chemical probes for harnessing halogen bonding in fragment-based lead discovery. Future Med Chem. 2014;6(6):617–639. doi:10.4155/fmc.14.20
- Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem. 2013;56(4):1363–1388. doi:10.1021/jm3012068
- Lange A, Heidrich J, Zimmermann MO, Exner TE, Boeckler FM. Scaffold effects on halogen bonding strength. J Chem Inf Model. 2019;59(2):885–894. doi:10.1021/acs.jcim.8b00621
- Dammann M, Kramer M, Zimmermann MO, Boeckler FM. Quadruple Target Evaluation of Diversity-Optimized Halogen-Enriched Fragments (HEFLibs) reveals substantial ligand efficiency for AP2-Associated Protein Kinase 1 (AAK1). Original Research. Front Chem. 2022;9:815567. doi:10.3389/fchem.2021.815567
- Dammann M, Stahlecker J, Zimmermann MO, et al. Screening of a halogen-enriched fragment library leads to unconventional binding modes. J Med Chem. 2022;65(21):14539–14552. doi:10.1021/acs.jmedchem.2c00951
- Stahlecker J, Klett T, Schwer M, et al. Revisiting a challenging p53 binding site: a diversity-optimized HEFLib reveals diverse binding modes in T-p53C-Y220C.. RSC Med Chem. 2022;13(12):1575–1586. doi:10.1039/D2MD00246A
- Congreve M, Carr R, Murray C, Jhoti H. A ‘Rule of Three’ for fragment-based lead discovery? Drug Discov Today. 2003;8(19):876–877. doi:10.1016/S1359-6446(03)02831-9
- Martin JS, MacKenzie CJ, Fletcher D, Gilbert IH. Characterising covalent warhead reactivity. Biorg Med Chem. 2019;27(10):2066–2074. doi:10.1016/j.bmc.2019.04.002
- Wildman SA, Crippen GM. Prediction of Physicochemical Parameters by Atomic Contributions. J Chem Inf Comput Sci. 1999;39(5):868–873. doi:10.1021/ci990307l
- Schrödinger, LCC. Schrödinger Release 2021-1: QikProp. New York: Schrödinger, LCC; 2021.
- Schrödinger, LCC. Schrödinger Release 2021-1: LigPrep. New York: Schrödinger, LCC; 2021.
- Lange A, Günther M, Büttner FM, et al. Targeting the Gatekeeper MET146 of C-Jun N-terminal kinase 3 induces a bivalent halogen/chalcogen bond. J Am Chem Soc. 2015;137(46):14640–14652. doi:10.1021/jacs.5b07090
- Bauer MR, Joerger AC, Fersht AR. 2-Sulfonylpyrimidines: mild alkylating agents with anticancer activity toward p53-compromised cells. Proc Natl Acad Sci U S A. 2016;113(36):E5271–E5280. doi:10.1073/pnas.1610421113
- Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci U S A. 2008;105(30):10360–10365. doi:10.1073/pnas.0805326105
- Kaar JL, Basse N, Joerger AC, Stephens E, Rutherford TJ, Fersht AR. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010;19(12):2267–2278. doi:10.1002/pro.507
- Jackson PA, Widen JC, Harki DA, Brummond KM. Covalent modifiers: a chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-Michael addition reactions. J Med Chem. 2017;60(3):839–885. doi:10.1021/acs.jmedchem.6b00788
- Kaur B, Singh P. Epoxides: developability as active pharmaceutical ingredients and biochemical probes. Bioorg Chem. 2022;125:105862. doi:10.1016/j.bioorg.2022.105862
- Harshbarger W, Miller C, Diedrich C, Sacchettini J. Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure. 2015;23(2):418–424. doi:10.1016/j.str.2014.11.017
- Vitaku E, Smith DT, Njardarson JT. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem. 2014;57(24):10257–10274. doi:10.1021/jm501100b
- Heravi MM, Zadsirjan V. Prescribed drugs containing nitrogen heterocycles: an overview. RSC Adv. 2020;10(72):44247–44311. doi:10.1039/d0ra09198g
- Wu Y-J. Chapter 1 - heterocycles and medicine: a survey of the heterocyclic drugs approved by the U.S. FDA from 2000 to present. In: Gribble GW, Joule JA, editors. Prog Heterocycl Chem. Elsevier; 2012:1–53.
- Hossain M, Habib I, Singha K, Kumar A. FDA-approved heterocyclic molecules for cancer treatment: synthesis, dosage, mechanism of action and their adverse effect. Heliyon. 2024;10(1):e23172. doi:10.1016/j.heliyon.2023.e23172
- Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Adv Drug Deliv Rev. 2002;54(3):355–366. doi:10.1016/s0169-409x(02)00008-x
- Ábrányi-balogh P, Petri L, Imre T, et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur J Med Chem. 2018;160:94–107. doi:10.1016/j.ejmech.2018.10.010
- Grabrijan K, Hrast M, Proj M, et al. Covalent inhibitors of bacterial peptidoglycan biosynthesis enzyme MurA with chloroacetamide warhead. Eur J Med Chem. 2022;243:114752. doi:10.1016/j.ejmech.2022.114752
- Proj M, Knez D, Sosič I, Gobec S. Redox active or thiol reactive? Optimization of rapid screens to identify less evident nuisance compounds. Drug Discov Today. 2022;27(6):1733–1742. doi:10.1016/j.drudis.2022.03.008
- Cee VJ, Volak LP, Chen Y, et al. Systematic Study of the Glutathione (GSH) reactivity of N-Arylacrylamides: 1. Effects of aryl substitution. J Med Chem. 2015;58(23):9171–9178. doi:10.1021/acs.jmedchem.5b01018
- Gerstenecker S, Haarer L, Schröder M, et al. Discovery of a potent and highly isoform-selective inhibitor of the neglected ribosomal protein S6 kinase beta 2 (S6K2). Cancers. 2021;13(20):1.
- Porter NJ, Christianson DW. Binding of the microbial cyclic tetrapeptide trapoxin A to the class I histone deacetylase HDAC8. ACS Chem Biol. 2017;12(9):2281–2286. doi:10.1021/acschembio.7b00330
- Pearson RG. Hard and soft acids and bases. J Am Chem Soc. 1963;85(22):3533–3539. doi:10.1021/ja00905a001
- Vaas S, Zimmermann MO, Schollmeyer D, et al. Principles and applications of CF(2)X moieties as unconventional halogen bond donors in medicinal chemistry, chemical biology, and drug discovery. J Med Chem. 2023;66(15):10202–10225. doi:10.1021/acs.jmedchem.3c00634
- Molland K, Zhou Q, Mesecar AD. A 2.2 Å resolution structure of the USP7 catalytic domain in a new space group elaborates upon structural rearrangements resulting from ubiquitin binding. Acta Crystallogr F Struct Biol Commun. 2014;70(Pt 3):283–287. doi:10.1107/s2053230x14002519
- Joerger AC, Allen MD, Fersht AR. Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J Biol Chem. 2004;279(2):1291–1296. doi:10.1074/jbc.M309732200
- Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50–83. doi:10.1128/mmbr.00031-10
- Barr RK, Bogoyevitch MA. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int J Biochem Cell Biol. 2001;33(11):1047–1063.
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–252. doi:10.1016/s0092-8674(00)00116-1
- Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23(16):2838–2849. doi:10.1038/sj.onc.1207556
- Bogoyevitch MA. The isoform‐specific functions of the c‐Jun N‐terminal Kinases (JNKs): differences revealed by gene targeting. Bioessays. 2006;28(9):923–934.
- Cui J, Zhang M, Zhang YQ, Xu ZH. JNK pathway: diseases and therapeutic potential. Acta Pharmacol Sin. 2007;28(5):601–608. doi:10.1111/j.1745-7254.2007.00579.x
- Antoniou X, Falconi M, Di Marino D, Borsello T. JNK3 as a therapeutic target for neurodegenerative diseases. J Alzheimers Dis. 2011;24(4):633–642. doi:10.3233/jad-2011-091567
- Hunot S, Vila M, Teismann P, et al. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101(2):665–670. doi:10.1073/pnas.0307453101
- Wityak J, McGee KF, Conlon MP, et al. Lead optimization toward proof-of-concept tools for Huntington’s disease within a 4-(1H-pyrazol-4-yl)pyrimidine class of pan-JNK inhibitors. J Med Chem. 2015;58(7):2967–2987. doi:10.1021/jm5013598
- Braithwaite SP, Schmid RS, He DN, et al. Inhibition of c-Jun kinase provides neuroprotection in a model of Alzheimer’s disease. Neurobiol Dis. 2010;39(3):311–317. doi:10.1016/j.nbd.2010.04.015
- Zhang G-Y, Zhang Q-G. Agents targeting c-Jun N-terminal kinase pathway as potential neuroprotectants. Expert Opin Investig Drug. 2005;14(11):1373–1383. doi:10.1517/13543784.14.11.1373
- Muth F, El-Gokha A, Ansideri F, et al. Tri- and tetrasubstituted pyridinylimidazoles as covalent inhibitors of c-Jun N-Terminal Kinase 3. J Med Chem. 2017;60(2):594–607. doi:10.1021/acs.jmedchem.6b01180
- Hoffelner BS, Andreev S, Plank N, Koch P. Photocaging of pyridinylimidazole-based covalent JNK3 inhibitors affords spatiotemporal control of the binding affinity in live cells. Pharmaceuticals. 2023;16(2):264. doi:10.3390/ph16020264
- Senkane K, Vinogradova EV, Suciu RM, et al. The proteome‐wide potential for reversible covalency at cysteine. Angew Chem. 2019;131(33):11507–11511. doi:10.1002/anie.201905829
- Liu R, Verma N, Henderson JA, Zhan S, Shen J. Profiling MAP kinase cysteines for targeted covalent inhibitor design. RSC Med Chem. 2022;13(1):54–63. doi:10.1039/d1md00277e
- Liu R, Zhan S, Che Y, Shen J. Reactivities of the FRONT POCket N-terminal cap cysteines in human kinases. J Med Chem. 2022;65(2):1525–1535. doi:10.1021/acs.jmedchem.1c01186
- Tavana O, Gu W. Modulation of the p53/MDM2 interplay by HAUSP inhibitors. J Mol Cell Biol. 2017;9(1):45–52. doi:10.1093/jmcb/mjw049
- Leger PR, Hu DX, Biannic B, et al. Discovery of potent, selective, and orally bioavailable inhibitors of USP7 with in vivo antitumor activity. J Med Chem. 2020;63(10):5398–5420. doi:10.1021/acs.jmedchem.0c00245
- Reverdy C, Conrath S, Lopez R, et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem Biol. 2012;19(4):467–477. doi:10.1016/j.chembiol.2012.02.007
- Faesen AC, Dirac AM, Shanmugham A, Ovaa H, Perrakis A, Sixma TK. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol Cell. 2011;44(1):147–159. doi:10.1016/j.molcel.2011.06.034
- Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–397. doi:10.1146/annurev.biochem.78.082307.091526
- Oliveira RI, Guedes RA, Salvador JAR. Highlights in USP7 inhibitors for cancer treatment. Front Chem. 2022;10:1005727. doi:10.3389/fchem.2022.1005727
- Hu M, Li P, Li M, et al. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111(7):1041–1054. doi:10.1016/s0092-8674(02)01199-6
- Chauhan D, Tian Z, Nicholson B, et al. Deubiquitylating enzyme USP-7, a novel therapeutic target in multiple myeloma. Blood. 2009;114(22):610. doi:10.1182/blood.V114.22.610.610
- Li X, Kong L, Yang Q, et al. Parthenolide inhibits ubiquitin-specific peptidase 7 (USP7), Wnt signaling, and colorectal cancer cell growth. J Biol Chem. 2020;295(11):3576–3589. doi:10.1074/jbc.RA119.011396
- Turnbull AP, Ioannidis S, Krajewski WW, et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550(7677):481–486. doi:10.1038/nature24451
- Kategaya L, Di Lello P, Rougé L, et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature. 2017;550(7677):534–538. doi:10.1038/nature24006
- Ohol YM, Sun MT, Cutler G, et al. Novel, selective inhibitors of USP7 uncover multiple mechanisms of antitumor activity in vitro and in vivo. Mol Cancer Ther. 2020;19(10):1970–1980. doi:10.1158/1535-7163.Mct-20-0184
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi:10.1038/35042675
- Lane DP. p53, guardian of the genome. Nature. 1992;358(6381):15–16. doi:10.1038/358015a0
- Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13(6):1027–1036. doi:10.1038/sj.cdd.4401910
- Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–582. doi:10.1146/annurev.biochem.77.060806.091238
- Joerger AC, Fersht AR. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu Rev Biochem. 2016;85:375–404. doi:10.1146/annurev-biochem-060815-014710
- Joerger AC, Fersht AR. Structure–function–rescue: the diverse nature of common p53 cancer mutants. Oncogene. 2007;26(15):2226–2242. doi:10.1038/sj.onc.1210291
- Joerger AC, Ang HC, Fersht AR. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc Natl Acad Sci U S A. 2006;103(41):15056. doi:10.1073/pnas.0607286103
- Liu X, Wilcken R, Joerger AC, et al. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013;41(12):6034–6044. doi:10.1093/nar/gkt305
- Baud MGJ, Bauer MR, Verduci L, et al. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur J Med Chem. 2018;152:101–114. doi:10.1016/j.ejmech.2018.04.035
- Stephenson Clarke JR, Douglas LR, Duriez PJ, et al. Discovery of nanomolar-affinity pharmacological chaperones stabilizing the oncogenic p53 Mutant Y220C. ACS Pharmacol Transl Sci. 2022;5(11):1169–1180. doi:10.1021/acsptsci.2c00164
- Liang Y, Besch-Williford C, Hyder SM. PRIMA-1 inhibits growth of breast cancer cells by re-activating mutant p53 protein. Int J Oncol. 2009;35(5):1015–1023. doi:10.3892/ijo_00000416
- Degtjarik O, Golovenko D, Diskin-Posner Y, Abrahmsén L, Rozenberg H, Shakked Z. Structural basis of reactivation of oncogenic p53 mutants by a small molecule: methylene quinuclidinone (MQ). Nat Commun. 2021;12(1):7057. doi:10.1038/s41467-021-27142-6
- Wassman CD, Baronio R, Demir Ö, et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat Commun. 2013;4(1):1407. doi:10.1038/ncomms2361
- Lambert JMR, Gorzov P, Veprintsev DB, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15(5):376–388. doi:10.1016/j.ccr.2009.03.003
- Guiley KZ, Shokat KM. A small molecule reacts with the p53 somatic mutant Y220C to rescue wild-type thermal stability. Cancer Discov. 2023;13(1):56–69. doi:10.1158/2159-8290.Cd-22-0381
- Scotcher J, Clarke DJ, Weidt SK, et al. Identification of two reactive cysteine residues in the tumor suppressor protein p53 using top-down FTICR mass spectrometry. J Am Soc Mass Spectrom. 2011;22(5):888–897. doi:10.1007/s13361-011-0088-x
- Pichon MM, Drelinkiewicz D, Lozano D, et al. Structure–reactivity studies of 2-sulfonylpyrimidines allow selective protein arylation. Bioconjug Chem. 2023;34(9):1679–1687. doi:10.1021/acs.bioconjchem.3c00322
- Zhang Q, Bykov VJN, Wiman KG, Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018;9(5):439. doi:10.1038/s41419-018-0463-7
- Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2(9):2212–2221. doi:10.1038/nprot.2007.321
- Gao K, Oerlemans R, Groves MR. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys Rev. 2020;12(1):85–104. doi:10.1007/s12551-020-00619-2
- Huynh K, Partch CL. Analysis of protein stability and ligand interactions by thermal shift assay. Curr Protoc Protein Sci. 2015;79:28.29.21–28.29.14. doi:10.1002/0471140864.ps2809s79
- Huber K, Brault L, Fedorov O, et al. 7,8-Dichloro-1-oxo-β-carbolines as a versatile scaffold for the development of potent and selective kinase inhibitors with unusual binding modes. J Med Chem. 2012;55(1):403–413. doi:10.1021/jm201286z
- Bullock AN, Debreczeni JÉ, Fedorov OY, Nelson A, Marsden BD, Knapp S. Structural basis of inhibitor specificity of the human protooncogene proviral insertion site in Moloney Murine Leukemia Virus (PIM-1) Kinase. J Med Chem. 2005;48(24):7604–7614. doi:10.1021/jm0504858
- Fedorov O, Huber K, Eisenreich A, et al. Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem Biol. 2011;18(1):67–76. doi:10.1016/j.chembiol.2010.11.009
- Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene. 2000;19(10):1245–1256. doi:10.1038/sj.onc.1203434
- Baell JB, Nissink JWM. Seven Year Itch: pan-Assay Interference Compounds (PAINS) in 2017-Utility and Limitations. ACS Chem Biol. 2018;13(1):36–44. doi:10.1021/acschembio.7b00903
- Sun C, Li Y, Yates EA, Fernig DG. SimpleDSFviewer: a tool to analyze and view differential scanning fluorimetry data for characterizing protein thermal stability and interactions. Protein Sci. 2020;29(1):19–27. doi:10.1002/pro3703
- Kharenko OA, Patel RG, Brown SD, et al. Design and Characterization of Novel Covalent Bromodomain and Extra-Terminal Domain (BET) Inhibitors Targeting a Methionine. J Med Chem. 2018;61(18):8202–8211. doi:10.1021/acs.jmedchem.8b00666
- Cimmperman P, Baranauskiene L, Jachimoviciūte S, et al. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys J. 2008;95(7):3222–3231. doi:10.1529/biophysj.108.134973
- Dai R, Wilson DJ, Geders TW, Aldrich CC, Finzel BC. Inhibition of Mycobacterium tuberculosis transaminase BioA by aryl hydrazines and hydrazides. ChemBioChem. 2014;15(4):575–586. doi:10.1002/cbic.201300748
- Backus KM, Correia BE, Lum KM, et al. Proteome-wide covalent ligand discovery in native biological systems. Nature. 2016;534(7608):570–574. doi:10.1038/nature18002
- Johansson H, Isabella Tsai Y-C, Fantom K, et al. Fragment-based covalent ligand screening enables rapid discovery of inhibitors for the RBR E3 Ubiquitin Ligase HOIP. J Am Chem Soc. 2019;141(6):2703–2712. doi:10.1021/jacs.8b13193
- Tan L, Akahane K, McNally R, et al. Development of selective covalent janus kinase 3 inhibitors. J Med Chem. 2015;58(16):6589–6606. doi:10.1021/acs.jmedchem.5b00710
- Serafimova IM, Pufall MA, Krishnan S, et al. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat Chem Biol. 2012;8(5):471–476. doi:10.1038/nchembio.925
- Pettinger J, Jones K, Cheeseman MD. Lysine-Targeting Covalent Inhibitors. Angew Chem Int Ed. 2017;56(48):15200–15209. doi:10.1002/anie.201707630