 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Introduction
The present study was conducted to compare the bioavailability of two candesartan cilexetil 16 mg tablet formulations (test and reference formulations).
Materials and methods
This study was a randomized, single- blind, two-period, cross-over study which included 24 healthy adult male and female subjects under fasting conditions. The pharmacokinetic parameters were determined based on the concentrations of candesartan (CAS 139481-59-7), using ultra-pressure high-performance liquid chromatography with a tandem mass spectrometer detector. In each of the two study periods (separated by a washout period of 1 week), a single dose of test or reference product was administered. The pharmacokinetic parameters assessed were area under the plasma concentration time curve (AUC) from time 0 hours to 24 hours, AUC from time zero to infinity, the peak plasma concentration of the drug (Cmax), time to achieve the Cmax, and the elimination half-life.
Results
The geometric mean ratios (90% confidence interval) of the test drug/reference drug for candesartan were 100.92% (92.15%–110.52%) for the AUC from 0 hours to 24 hours, 100.24% (92.24%–108.95%) for the AUC from time zero to infinity, and 106.71% (93.20%–122.18%) for the Cmax. The differences between the test and reference product in the time to achieve Cmax values and elimination half-life values were not statistically significant (P > 0.05). The 90% confidence intervals of the test/reference AUC ratio and Cmax ratio of candesartan were within the acceptance range for bioequivalence. There was no adverse event encountered during this bioequivalence study.
Conclusion
It was concluded that the two candesartan tablet formulations (the test and reference product) were bioequivalent.
Introduction
Candesartan (CAS 139481-59-7) is an antihypertensive agent exerting its action as a selective AT1-subtype angiotensin-2 receptor antagonist.Citation1,Citation2 In oral formulations, the drug is available in an ester form, candesartan cilexetil (C33H34N6O6, molecular weight of 610.67), with its chemical structure described in .Citation3
Figure 1 Chemical structure of candesartan cilexetil (CAS 139481-59-7).
Plasma concentrations of angiotensin-1, angiotensin-2, and plasma renin activity increased in a dose-dependent manner after single and repeated dosing of candesartan to healthy subjects, as well as to hypertensive and heart failure patients. The antihypertensive action of the drug results from decreased systemic peripheral resistance, without a reflex increase in heart rate. There is no indication of serious or exaggerated first-dose hypotension or rebound effect after cessation of treatment. Blood pressure response is dose-dependent over the range of 2 mg to 32 mg; the usual recommended starting dose is 16 mg once daily. In healthy subjects, once-daily regimen of up to 16 mg of candesartan Cilexetil did not influence the levels of plasma aldosterone; yet administration of the drug at a dose of 32 mg to hypertensive patients was found to decrease the levels. Most of the antihypertensive effect of candesartan is seen within 2 weeks, and the full-effect generally occurs within 4 to 6 weeks of treatment. In patients with moderate hepatic and/or renal dysfunction, or intravascular volume depletion, candesartan should be administered at a lower dose and initiated under close medical supervision.Citation2 In heart failure, treatment with candesartan reduces mortality, hospitalization due to heart failure, and improves symptoms in patients with left ventricular systolic dysfunction.Citation2
Following oral administration, candesartan cilexetil, which is a prodrug, is converted to the active substance, candesartan, by ester hydrolysis during absorption from the gastrointestinal tract.Citation2 Food with a high fat content has no effect on the bioavailability of the drug. After single and repeated administration, the pharmacokinetics of candesartan are linear for oral doses of up to 32 mg of candesartan cilexetil. Candesartan and its inactive metabolite do not accumulate in serum upon repeated once-daily dosing. The absolute bioavailability for candesartan is 15% following administration of candesartan cilexetil as a tablet.Citation2 Peak plasma concentrations (Cmax) were reached approximately within 4.64 hours (4–8 hours) after dosing.Citation1,Citation2 Candesartan is more than 99% bound to plasma proteins and the volume of distribution in healthy individuals is 0.13 L/kg. The total plasma clearance of candesartan is 0.37 mL/minute/kg, with a renal clearance of 0.19 mL/minute/kg. Candesartan is mainly excreted unchanged in urine and feces (via bile). It undergoes minor hepatic metabolism by o-de-ethylation to an inactive metabolite.Citation1,Citation2 In healthy subjects, 67% of an oral dose of candesartan is excreted in feces and only about 5% to 10% of the administered dose is excreted unchanged in the urine within 24 hours.Citation2 The terminal elimination half-life (t1/2) of candesartan is approximately 9.7 hours. No relevant pharmacokinetic drug–food or drug–drug interactions are known.Citation1
The present study evaluated the bioavailability of two different oral candesartan tablet formulations following single dosing in healthy adult subjects in order to prove the bioequivalence between both preparations. For a drug to be considered bioequivalent to the reference drug, the area under the plasma concentration time curve (AUC) and the Cmax of the drug should be within 80%–125% of the AUC and Cmax of the reference drug.Citation4,Citation5
Materials and methods
Subjects and study design
This randomized, single-blind, two-period, two-sequence, cross-over study involved 24 healthy subjects under fasting condition, with a 1-week washout period. Screening for the eligibility, which was based on physical examination; vital signs (blood pressure, heart rate, respiratory rate and body temperature); electrocardiography (ECG) and biochemical tests for liver function (alkaline phosphatase, Alanine transaminase [ALT], aspartate aminotransferase [AST] and bilirubin), renal function (serum creatinine and ureum), hematology (hemoglobin, leucocyte and platelet count), blood glucose, urinalysis (pH, glucose, protein, and urine sediment), and seroimmunology (hepatitis B surface antigen [HBsAg], anti-hepatitis C virus [anti-HCV], and antihuman immunodeficiency virus [anti-HIV]), was performed within 2 weeks prior to the subjects’ first dosing day; pregnancy test (for female subjects) was performed just before the dosing in each period.
Subjects aged between 18–55 years, with bodyweight within a normal range (body mass index between 18 kg/m2 and 25 kg/m2), and who had signed the informed consent, were enrolled in this study. Pregnant women, nursing mothers, women of childbearing potential without adequate contraception, subjects with known contraindications or hypersensitivities to candesartan, chronic gastrointestinal problems, liver dysfunction, clinically significant hematology, renal insufficiency, and positive test results for HbsAg, anti-hepatitis C virus, and/or antihuman immunodeficiency virus were excluded.
This study was carried out in accordance with the Declaration of Helsinki,Citation6 Good Clinical Practice,Citation7 and Good Laboratory Practice.Citation8 The protocol, the consent form, and the patient information sheet were reviewed and approved by an independent Ethics Committee of the Medical Faculty, University of Indonesia, prior to the study conduct. Written informed consent from every study subject was obtained prior to any trial-related activities, and the investigator retained the consent forms.
Study products
The test preparation (candesartan cilexetil 16 mg tablets, batch number K-10057-F-PSC-1) was manufactured by PT Dexa Medica (Tangerang, Indonesia). The reference preparation (candesartan cilexetil 16 mg tablets, batch number GBHF) was the innovator product (Blopress®; Takeda Indonesia, licensed under Takeda Pharmaceutical Company limited, Osaka, Japan), and was purchased from the local pharmacy.
Prior to the conduct of the bioequivalence study, the test product was subject to undergo a series of comparative dissolution tests with the innovator product used as the comparator. The dissolution test was performed in three different media of certain pH levels (ie, pH 1.2, pH 4.5, and pH 6.8), each of which represents the pH along the gastrointestinal tract where the absorption of the study drug may occur. The dissolution profiles of both the test and comparator product in each media showed similarity factors of more than 50 (ie, the similarity factors were 74, 95, and 54, in pH 1.2, pH 4.5, and pH 6.8, respectively).
Treatment phase and blood sampling
Subjects attended PT Equilab International (Jakarta, Indonesia) a night before drug administration, and they were requested to fast from any food and drink except mineral water from 9 pm. In the morning (approximately 6 am) of the dosing day (day 1), after an overnight fast, a predose pharmacokinetic blood sample was taken. The study drug (one tablet of the test drug or the reference drug) was given at 7 am with 200 mL of water.
The date and the time at which each sample was taken were recorded. Lunch and dinner were provided 4 hours and 10 hours after drug administration, respectively. The amounts of food and water intake and physical activity for each individual subject were standardized during the sampling days. Xanthine-containing food or beverages and fruit juices were not allowed for 24 hours before and during the entire sampling days.
From each subject, venous blood samples (10 mL) were drawn immediately before taking the drug (control), and 5 mL each at 0.5 hours, 1 hour, 1.5 hours, 2 hours, 2.5 hours, 3 hours, 3.5 hours, 4 hours, 5 hours, 7 hours, 9 hours, 12 hours, 16 hours, 24 hours, and 36 hours after dosing. One week after the first dosing (ie, the washout period), the same procedure was repeated with the alternate drug. Blood samples from each subject were collected in vacuum polypropylene tubes containing ethylenediaminetetraacetic acid by using a 22 G drawing needle during each period. The predose samples were collected within 1 hour prior to drug dosing. The postdose samples were collected within 1 minute of the scheduled time where the end time of collection to the nearest minute was recorded on case report forms. Blood samples were drawn and transferred into ethylenediaminetetraacetic acid tubes. Plasma was separated from the blood by centrifugation at 1,500 g (rotor’s diameter was 86 mm) for 15 minutes and the plasma was transferred into a clean tube. The separated plasma was divided in duplicate aliquots in pre-labeled polypropylene tubes during each period. These tubes were stored in a sample storage room (ie, in a deep freezer with temperatures maintained at −20°C) for temporary storage, and at the end of the day, these tubes were then transferred to a deep freezer with temperatures maintained below −20°C for long-term storage. All plasma samples were stored at temperatures equal to or lower than −20°C until assayed. The dates and times at which each sample was taken were recorded in the case report forms.
Analysis of drug concentration
Method of analysis
The plasma concentrations of candesartan were assayed using a thoroughly validated ultra-pressure, high-performance liquid chromatography with a tandem mass spectrometer detector method (UPLC®-MS/MS; ACQUITY® TQD; Waters™, Milford, MA, USA). This method is characterized by adequate sensitivity, specificity, linearity, recovery, accuracy, and precision, both within and between days. The validation data, as presented in , were taken from our validation report.
Table 1 The validation data of the analytical method used to determine candesartan in human plasma by UPLC-MS/MS using irbesartan as the internal standard
Assay procedure
The procedures described below were applied for the extraction of subject samples, calibration, and quality control standards. An aliquot of human plasma was transferred to an appropriate glass test tube, then ether was added in acid condition. The content of the tube was vortexed and centrifuged. The organic phase was transferred to a vial and an aliquot was evaporated until dryness. The residue was reconstituted with the mobile phase, vortexed and injected into the validated UPLC®-MS/MS system. Calibration standards, controls, and samples were processed in batches.
The analytical column was ACQUITY® UPLC® C18 1.7 μm, 2.1 × 50 mm. The mobile phase was a mixture of acetonitrile and formic acid in such a composition so as to obtain the mass transition ion pair value of 429.5 > 195.2 for candesartan and 441.4 > 263.3 for irbesartan (as an internal standard), with an isocratic mode and flow rate of 0.2 mL/minute. Irbesartan was selected as the internal standard based on the analytical method formerly developed by Prasad et al,Citation9 when they studied losartan potassium and irbesartan in rat plasma using the liquid chromatography tandem mass spectrometry system (LC-MS/MS).Citation9
All chromatograms in the same batch were processed automatically by a software program using the same processing parameters, such as integration, peak-to-peak amplitude, and peak detection. Manual integration was performed only when necessary.
Pharmacokinetic evaluation
The noncompartmental pharmacokinetic analysis method was employed to determine the pharmacokinetic parameters of candesartan. Cmax (ng/mL) and the time to reach Cmax (tmax, hours) were obtained directly from the observed data. The AUC from time zero to the last measurable concentration time (AUCt) was calculated by the trapezoidal method. The AUC from time zero extrapolated to infinite time (AUCinf) was calculated as
where Ct is the last quantifiable concentration, and ke is the terminal elimination rate constant, which was determined by least-squares regression analysis during the terminal log-linear phase of the concentration time curve. The t1/2 time (hours) was calculated as 0.693/ke.
Statistical analysis
EquivTest version 2.0 (Statistical Solutions, Ltd Saugus, MA, USA) was used to perform the statistical analyses of AUC, AUCinf, and Cmax using analysis of variance (ANOVA) after transformation of the data to their logarithmic (ln) values. Using the error variance (S 2) obtained from the ANOVA, the 90% confidence intervals (CIs) were calculated from the following equation:
where
,
are the means of the ln transformed values for the test product (T) and the reference product (R); S
Citation2 is the error variance obtained from the ANOVA; n is the number of subjects; t
0.1 is the t-value for the 90% CI; and v is the degree of freedom of the error variance from the ANOVA.
The anti-lns of the above CIs were the 90% CIs of the ratios of the test/reference geometric means.
The power of study would be 80% with 0.05-alpha. The acceptance criteria for bioequivalence were that the 90% CIs of the geometric mean ratios were 0.80–1.25 for the AUC and Cmax. The tmax difference was analyzed using a non-parametric statistical method, i.e. the Wilcoxon matched-pairs test on the original data. The t1/2 difference was analyzed using Student’s paired t-test.
Results
All of the subjects were healthy Indonesians, had normal values of all clinical and laboratory parameters measured, and were compliant with the inclusion/exclusion criteria of the study. Blood samples pertaining to a total of 24 subjects (16 males and eight females) with a body mass index between 18.82 kg/m2 and 24.72 kg/m2 were analyzed for pharmacokinetic evaluation of candesartan.
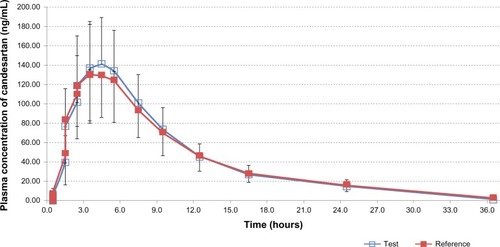
The Profiles of area under the curve (AUC) of mean plasma concentration versus time in subjects (n = 24) after oral administration of 16 mg of the candesartan cilexetil tablet (both the test drug and the reference drug) are displayed in . The geometric mean ratios (90% CI) of the test drug/reference drug for candesartan were 100.92% (92.15%–110.52%) for AUCt; 100.24% (92.24%–108.95%) for AUCinf; and 106.71% (93.20%–122.18%) for Cmax. The 90% CIs of the test/reference AUC ratio and Cmax ratio of candesartan were within the acceptance range for bioequivalence. The differences between the test and reference drug products for tmax and t1/2 values were not found to be statistically significant (P > 0.05). The values of the pharmacokinetic parameters (AUCt, AUCinf, Cmax, t1/2, and tmax) and the geometric mean ratios (90% CI) of AUCt, AUCinf, and Cmax of candesartan that resulted from the test drug as well as from the reference drug are presented in detail in .
Table 2 Pharmacokinetic parameters and statistical comparison of candesartan after single-dose oral administration of a 16 mg candesartan cilexetil tablet of the test and the reference drug
Figure 2 Mean plasma concentrations versus time profiles of candesartan in human subjects (n = 24) after single-dose oral administration of 16 mg of candesartan cilexetil tablets of the test drug and the reference drug.
There was no adverse event encountered during this bioequivalence study.
Discussion
The basic rationale behind conducting a bioequivalence study is that if two products are shown to be bioequivalent – with the same active substance, the same dosage form, but different formulations – they are therapeutically equivalent in terms of efficacy and safety; both products are therapeutically interchangeable.Citation10,Citation11 In bioequivalence studies, the reference products used should be those with proven efficacy and safety that have been demonstrated through robust clinical studies.Citation4,Citation5,Citation10,Citation11 Therefore, generic products that are bioequivalent to the reference products, which are usually the innovator products, do not require comparative clinical studies to prove their efficacy and safety.
The aim of the present randomized, single-blind, two-period, two-sequence, cross-over bioequivalence study, conducted under fasting conditions with a 1-week washout period, was to compare the bioavailability of the test formulation of candesartan cilexetil with that of the reference formulation (ie, the innovator or originator of the candesartan cilexetil preparation), Blopress® (Takeda Indonesia).
Similar studies on candesartan cilexetil preparations have been conducted previously in healthy Chinese and Korean subjects.Citation12–Citation14 However, those studies apparently did not evaluate the same preparation that was tested in the present study. The tested preparation in the current study had its own formulation, which was different from those of similar studies. In fact, the findings of a particular bioequivalence study were absolutely unique only to the tested formulation used in the study; thus, the findings cannot be generalized to other generic drug products, even though they contain the same active substance.
Further, unlike the findings from studies in China by Jin and HanCitation12 and Yu et al,Citation13 which used high-performance liquid chromatography as the analytical method, the present study employed a more sensitive and robust method, LC-MS/MS, to quantify the plasma concentration of candesartan.Citation9 A Korean study,Citation14 which was conducted after the Chinese ones, also employed the LC-MS/MS method. However, the study had a parallel design rather than the cross-over design we used for our present study. The cross-over design is preferable for a bioequivalence study since it can eliminate intersubject differences. In this cross-over designed study, the comparison between the two formulations was conducted within subjects rather than between subjects. Since food and water intake as well as the physical activity of all the subjects were standardized in the study, the cross-over design would minimize all the factors associated with subject differences, and would focus only on the differences between the formulations evaluated. The carry-over effect, which may interfere in a cross-over design study, was eliminated by providing each subject a 1-week washout period. A washout period of 1 week is adequate to eliminate candesartan from the body, considering that the t1/2 of candesartan is only about 9.7 hours.Citation1
In the present study, the 16 mg candesartan cilexetil tablet formulations were administered to overnight fasting subjects in order to eliminate the influence of food on drug absorption. The pharmacokinetic parameters of the 16 mg candesartan cilexetil tablet were assessed based on the plasma concentrations of candesartan.
In order to perform the two one-sided test procedures for bioequivalence on log-transformed plasma candesartan concentration data, with bioequivalence limits of 0.80 and 1.25 for AUC and Cmax, alpha = 0.05 and power = 80%, the number of subjects needed for the bioequivalence study was determined by means of CIs, as formerly presented by Diletti et al.Citation15 In the present study, the intrasubject coefficient of variance obtained from the ANOVA for the candesartan AUCt was 18.33% (). Hence, the number of subjects in this study (24 subjects) ensured that this study had adequate power to confirm the statistical conclusions.
Healthy subjects were selected under the eligibility criteria, which were set to ensure that only a subject population without accompanying diseases interfering with the conduct and scientific evaluation of the study were enrolled in the study. Additionally, involving healthy subjects alone would minimize risk to the subjects’ well-being.
In this study, the AUCt, AUCinf, and Cmax of candesartan were defined as the main parameters in order to assess possible bioequivalence between both preparations. Based on standard bioequivalence guidelines, the criteria for bioequivalence are the 90% CI of the test/reference geometric means ratio in the range of 80% to 125% for both the AUC and Cmax.Citation4,Citation5 The results of the present study showed that the geometric mean ratios (90% CIs) of the AUCt, AUCinf, and Cmax of candesartan were 100.92% (92.15%–110.52%), 100.24% (92.24%–108.95%), and 106.71% (93.20%–122.18%), respectively. The values were within the acceptance range for bioequivalence.
In each subject, the AUCt/AUCinf ratio of candesartan was more than 80% (81.45%–95.20% for the test drug and 82.07%–96.22% for the reference drug), indicating that the sampling time was sufficiently long to ensure an adequate description of the absorption phase.
The mean (standard deviation) t1/2 times of candesartan were 7.29 hours (1.72 hours) and 7.63 hours (1.85 hours) for the test drug and reference drug, respectively. These values were around the candesartan t1/2 value observed in the literature, which was approximately 9 hours.Citation2 Utilizing Student’s paired t-test, the t1/2 values of the test and reference drugs were not significantly different, demonstrating a comparable rate of drug elimination from the body.
The results obtained for the median (range) tmax of candesartan was 4.00 hours (2.00–7.00 hours) for the test drug and 4.00 hours (2.00–7.00 hours) for the reference drug. Using the Wilcoxon matched-pairs test on the original data, the difference between the tmax values of the two drugs (test and reference drug) was not statistically significant.
There were no adverse events encountered during this bioequivalence study.
Conclusion
Based on the pharmacokinetics and the results of this study, it was concluded that the two formulations of the candesartan cilexetil 16 mg tablets were bioequivalent.
Acknowledgments
We deeply thank the volunteers for their participation in this study.
Disclosure
The authors report no conflicts of interest in this work.
References
- Gleiter CH Mörike KE Clinical pharmacokinetics of candesartan Clin Pharmacokinet 2002 41 1 7 17 11825094
- Husain A Azim MS Mitra M Bhasin PS A review on candesartan: pharmacological and pharmaceutical profile Journal of Applied Pharmaceutical Science 2011 1 10 12 17
- Sweetman SC Martindale: The Complete Drug Reference 36th ed New York, NY Pharmaceutical Press 2009 1238 1239
- The European Agency for the Evaluation of Medicinal Products (EMA) Committee for Proprietary Medicinal Products (CPMP) Note for Guidance on the Investigation of Bioavailability and Bioequivalence. CPMP/EWP/QWP/1401/98 Rev.1 2010 Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039 Accessed August 7, 2013
- [Indonesian Guideline for Bioequivalence Studies] Badan Pengawas Obat dan Makanan Republik Indonesia (BPOM RI) Pedoman Uji Bioekuivalensi Jakarta Badan Pengawas Obat dan Makanan 2004 Bahasa
- World Medical Association Declaration of Helsinki Recommendations Guiding Physicians in Biomedical Research Involving Human Patients. Amended by the 52nd WMA General Assembly Edinburgh Scotland World Medical Association 2000
- International Conference on Harmonisation Expert Working Group ICH Harmonised Tripartite Guideline: Guideline for Good Clinical Practice E6 (R1) Geneva, Switzerland International Conference on Harmonisation 1996
- Organization for Economic Co-operation and Development OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring Number 1: OECD Principles on Good Laboratory Practice Paris, France Organization for Economic Co-operation and Development 1997
- Prasad SVSGB Shivakumar S Sudhir T Mital R Devala Rao G LC/MS/MS method for the simultaneous estimation of losartan potassium and irbesartian in rat plasma International Journal of Pharmacy and Pharmaceutical Sciences 2009 1 Suppl 1 206 215
- Cooper-Dehoff RM Elliott WJ Generic drugs for hypertension: are they really equivalent? Curr Hypertens Rep 2013 15 4 340 345 23700299
- Kesselheim AS Misono AS Lee JL Clinical equivalence of generic and brand-name drugs used in cardiovascular disease: a systematic review and meta-analysis JAMA 2008 300 21 2514 2526 19050195
- Jin X Han J [Studies on the bioequivalence of two candesartan preparations.] China Pharmacy 2007 11 Chinese
- Yu J Ma K Qi JW [Pharmacokinetics and bioequivalence of candesartan cilexetic tablet and capsule in healthy volunteers.] Chinese Journal of Clinical Pharmacology and Therapeutics 2009 7 Chinese
- Jeon JY Im YJ Kim Y Pharmacokinetic properties and bioequivalence of candesartan cilexetil in Korean healthy volunteers Drug Dev Ind Pharm Epub 10 3 2012
- Diletti E Hauschke D Steinijans VW Sample size determination for bioequivalence assessment by means of confidence intervals Int J Clin Pharmacol Ther Toxicol 1991 29 1 1 8 2004861