 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
The purpose of this study was to evaluate absorption and elimination from the gastrointestinal tract of glycyrrhizic acid diethyl ester (GZ-DE) which was prepared as a prodrug of glycyrrhizic acid (a poorly absorbed compound) in rats.
Methods
After the GZ-DE solution was administered via the intravenous, intraduodenal, intraileal, and stomach routes, GZ-DE and GZ concentrations in bile were determined by high-performance liquid chromatography. The stability of GZ-DE was estimated from residual GZ-DE and GZ produced in GZ-DE solutions prepared with distilled water, a pH 1.2 solution, 0.9% NaCl solution, and phosphate-buffered solution (pH 7.4) at 37°C.
Results
GZ-DE was eliminated into bile by the pharmacokinetic parameters of apparent distribution rate constant (4.56 ± 0.36 per hour) and apparent elimination rate constant (0.245 ± 0.042 per hour). After intravenous and intraduodenal administration of GZ-DE, the concentration ratio of GZ-DE to GZ in bile was approximately 4:1, and the bioavailability of GZ containing GZ-DE was three-fold higher compared with the bioavailability of GZ after intraduodenal administration. GZ-DE was immediately precipitated in pH 1.2 solution and was converted to GZ by hydrolysis in pH 7.4 solution.
Conclusion
Improvement of intestinal absorption of GZ was made possible by administration of GZ-DE into the intestine where absorption of GZ is lower than in the strong acidic environment of the stomach. However, because the elimination rate in bile simulated from kinetic parameters of GZ-DE was higher than the conversion rate from GZ-DE to GZ by hydrolysis, it is thought that the availability of GZ as a revolutionary prodrug was not high from the viewpoint of bioavailability of GZ in the liver by intestinal administration of GZ-DE.
Introduction
Glycyrrhizic acid (GZ, ), a main component of the plant Glycyrrhiza glabra L, has been used as a treatment for hepatitis for a long time. In particular, injections and oral formulations of GZ have been used with high safety and efficacy in patients with chronic hepatitis for over 60 years in Japan. The therapeutic efficacy of these injections is high in patients with chronic hepatitis,Citation1–Citation4 but there is a need to receive the injection while ambulatory, and some problems exist with regard to time restrictions, pain on injection, and hardening of the skin as a result of consecutive dosing. Therefore, an oral formulation with therapeutic efficacy comparable with that of the injection is needed. Absorption of the commercial oral formulation of GZ from the gastrointestinal tract is poor,Citation5,Citation6 and the membrane permeability of GZ in the intestinal tract is low,Citation7 because two glucuronic acids are included in the structure of GZ.
Figure 1 Structure of GZ (A) and GZ-DE (B).
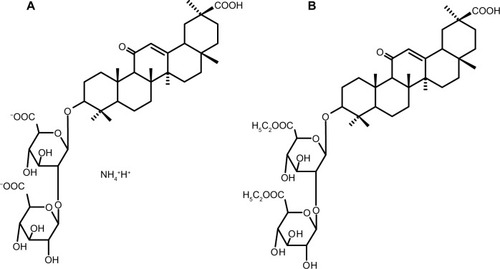
In order to enhance absorption of GZ from the intestinal tract, a diethyl ester prodrug form of GZ was synthesized (GZ-DE, ). GZ-DE (CAS registry number 1413393-38-0) is a new compound synthesized by Cokey Co, Ltd (Tokyo, Japan) in 2012. It was predicted that absorption of GZ-DE from the gastrointestinal tract would be improved compared with that of GZ, and that an enhanced therapeutic effect would be seen in the liver in patients with chronic hepatitis after conversion from GZ-DE to GZ by internal esterase and hydrolysis. Therefore, a study of the pharmacokinetic parameters of GZ-DE was needed. In this experiment, the pharmacokinetic parameters of GZ-DE and GZ were investigated after their administration via the intravenous, intraduodenal, intraileal, and oral routes in rats. The stability of GZ-DE in various types of solution was also studied. Here we present the early data from our in vitro and in vivo experiments with GZ-DE.
Materials and methods
Materials
Glycyrrhizic acid monoammonium (GZ-NH4) and GZ-DE (85% purity) were gifts from Cokey Systems Co, Ltd (Matsusaka, Japan). GZ as a standard for high-performance liquid chromatography (HPLC) assay (GLY0605, Japanese pharmacopoeia reference standard) was purchased from the Pharmaceutical and Medical Device Regulatory Science Society of Japan (Tokyo, Japan). Propylene glycol, L-arginine, 60% perchloric acid, and 25% ammonia solution were purchased from Wako Pure Chemicals Industries Ltd (Osaka, Japan). Other chemicals were of HPLC or reagent grade.
Animals
The protocol for this study was approved by the Committee of Animal Use at Hokuriku University. All animal experiments were conducted in accordance with the Institutional Guidelines for Care and Use of Laboratory Animals. Male Sprague Dawley rats (aged 7 weeks, body weight approximately 180 g) were purchased from Sankyo Laboratories Co, Ltd (Toyama, Japan), and housed for at least 7 days in a clean room. The rats were given free access to commercial chow and water, and were maintained according to the Hokuriku University animal guidelines. For in vivo experiments using the GZ-DE formulations, the rats (240–265 g) were randomly divided into treatment groups containing three to four rats per group.
Preparation of dosage formulations
GZ-DE is only slightly soluble in water, but is soluble in propylene glycol. Therefore, GZ-DE was dissolved in propylene glycol. The concentration of GZ-DE was adjusted to 23.85 mg/mL. This concentration is accepted as being equivalent to a GZ concentration of 20 mg/mL. Conversion from a GZ-DE concentration to a GZ concentration in the test solution was calculated as follows: GZ-DE bulk is composed of 85.0% GZ-DE, 4.4% GZ monoester (GZ-ME), and 10.6% other compounds. The molecular weights of GZ, GZ-DE, and GZ-ME are 822.9, 878.9, and 850.9, respectively. Based on the conversion to molar concentration, the GZ-DE concentration in a 23.85 mg/mL GZ-DE solution is 20.27 mg/mL (23.06 mmol/L). A GZ concentration of 23.06 mmol/L is equivalent to 18.98 mg/mL GZ, and the GZ-ME concentration in a 23.85 mg/mL GZ-DE solution is 1.05 mg/mL (1.23 mmol/L). A GZ concentration of 1.23 mmol/L is equivalent to 1.02 mg/mL GZ. The sum concentration of GZ which is converted completely from GZ-DE and GZ-ME in 23.85 mg/mL GZ-DE solution is 20.0 mg/mL. Namely, justification of the concentration of GZ-DE at 23.85 mg/mL is equivalent to GZ at 20.0 mg/mL. This GZ-DE propylene glycol solution was maintained at 20°C until in vivo experiments, ie, intravenous, intraduodenal, intraileal, and oral administration. GZ-NH4 was dissolved in 100 mM phosphate-buffered solution containing 4% L-arginine (pH 7.4), and the concentration of GZ-NH4 solution was adjusted to a GZ concentration of 20 mg/mL (for intraduodenal administration). L-arginine was added to prevent gelation of the GZ solution.Citation8
In vivo experiments in rats
The rats were given water only for 12 hours, and were then anesthetized with urethane saline solution via the intraperitoneal route (2.5 g/mL/kg body weight). The rats were fixed in a supine position on a surgical plate maintained at 32°C. After the abdomen was carefully opened (approximately 3 cm in length) with a surgical knife, the common bile duct was cannulated with polyethylene tubing (PE10, 20 cm in length, Becton Dickinson, Sparks, MD, USA). After the abdomen was closed using a surgical stapler, the opposite end of the tubing was placed into a 2 mL sampling microtube.
For intravenous administration, GZ-DE propylene glycol solution (GZ dose 2 mg/100 μL per rat) was injected via the right subclavian vein using a microsyringe (1710N 100 μL SYR, 22 s/s”/2, Hamilton Company, Reno, NV, USA). The injection speed was 100 μL over 30 seconds. Bile was collected at 30 minutes and 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 7, and 8 hours after injection of GZ-DE. The liver was carefully excised using a surgical knife at 8 hours after injection of GZ-DE. The bile and liver samples were stored at −30°C until assay.
For intraduodenal and intraileal administration, GZ-NH4 solution or GZ-DE propylene glycol solution (GZ dose 5 mg/250 μL per rat) was slowly administered into the duodenum (lower position, 1 cm beneath the stomach) or ileum (upper position, 20 cm from the cecum) after bile cannulation using a microsyringe (725RN 250 μL SYR, 22 s/s”/2, Hamilton Company) before the abdomen was closed using a surgical stapler. The intestinal part that pointed at a needle was blocked up by a surgical adhesion glue (Aron Alpha A Sankyo, Toagosei Co, Ltd Toyama, Japan). After administration of GZ-NH4 or GZ-DE, bile collection was immediately started. The continuous sampling periods were set to be 30 minutes, and 1, 1.5, 2, 4, 6, 8, and 10 hours. The liver was carefully excised using a surgical knife at 10 hours after administration of GZ-NH4 or GZ-DE. The bile and liver samples were stored at −30°C until assay.
For oral administration, GZ-DE propylene glycol solution (GZ dose 5 mg/mL per rat) was administered into the rat stomach using a gastric tube with a 1 mL syringe and without anesthesia. The rats were returned to the breeding cage for 90 minutes. Next, the rats were anesthetized with ethyl carbamate saline solution, and PE10 tubing was inserted into the bile duct as described above. Bile was collected for 2–10 hours after administration (at 4, 6, 8, and 10 hours). The liver was carefully excised using a surgical knife at 10 hours after administration of GZ-DE. The bile and liver samples were stored at −30°C until assay.
Extraction of GZ and GZ-DE from bile and liver
Bile (20 μL), 50 mM phosphate-buffered solution (pH 7.4, 180 μL), and acetonitrile/0.6% perchloric acid solution adjusted to pH 8.0 with 200 μL of 25% ammonia solution (2:8, v/v) were mixed. The mixed solution was then filtered using a membrane filter (13HP020 Dismic®, Toyo Roshi, Tokyo, Japan). Next, 10 μL of the filtered solution was injected into the HPLC system.
Liver (200 mg), 50 mM phosphate-buffered solution (pH 7.4, 200 μL), and stainless steel balls (two balls 3.2 mm in diameter and one ball 5.5 mm in diameter) were added to a 2 mL polyethylene screw vial. The liver tissue was crushed at 3,000 rpm for one minute using a cell crusher machine (MS-100R, Tomy, Tokyo, Japan). After addition of 1.0 mL of methanol to the screw vial, the vial was shaken for 20 minutes at 500 rpm using a vortex shaker (VR-36, Taitec, Saitama, Japan), and then centrifuged for 10 minutes at 14,000 × g. Next, 0.8 mL of the supernatant was transferred to a 10 mL glass tube and evaporated to dryness under a continuous stream of nitrogen gas on a heat block set to 80°C. After cooling the glass tube, 0.3 mL of acetonitrile/0.6% perchloric acid solution adjusted to pH 8.0 with 25% ammonia solution (2:8, v/v) was added to the glass tube, which was then vortexed for one minute using a touch mixer. The solution was filtered using a membrane filter (13HP020), and a10 μL aliquot of the solution was then injected into the HPLC system.
HPLC assay
The drug concentration in bile and liver in in vivo experiments was assayed by HPLC as the concentration of GZ or GZ-DE. A standard GZ solution (100 μg/mL) was prepared with 100 mM phosphate-buffered solution containing 4% L-arginine (pH 7.4). The concentration of GZ was determined according to a previous report.Citation9 Namely, the HPLC system was equipped with a LC-10 ADvp pump, an SIL-20A autosampler, a DGU-20As degassing apparatus, a SPD-20A ultraviolet detector, and a CR7A-plus data processor (Shimadzu, Kyoto, Japan). A Capcell Pak C18 column (1.5 mm inner diameter, 150 mm length) was used at 40°C during separation. Detection was performed at an ultraviolet wavelength of 254 nm. As the mobile phase, the ratios of acetonitrile and 0.6% perchloric acid solution adjusted to pH 8.0 with 25% ammonia solution were set to be 2:8 (v/v) for GZ assay. The flow rate of mobile phase was set to be 0.08 mL per minute. On the other hand, the GZ-DE standard solution (100 μg/mL) was prepared with 50% ethanol. The concentration of GZ-DE was determined according to a modification of the GZ assay. The different assay condition was only for mobile phase, ie, acetonitrile and 0.6% perchloric acid solution adjusted to pH 8.0 with 25% ammonia solution 2:3 (v/v) was used for the GZ-DE assay.
Calculation of pharmacokinetic parameters
The target organ for GZ is the liver. Therefore, it was important that the analysis was focused on its speed of excretion from the liver to bile. The pharmacokinetic parameters for GZ-DE after intravenous administration were analyzed using a nonlinear least-squares program (MULTICitation10) with a two-compartment model following the following equation:
where C is the bile GZ or GZ-DE concentration at time t, A and B are ordinate intercepts, and α and β are the corresponding first-order elimination rate constants in bile. Elimination half-life (t ½β) was calculated by dividing ln2 by β. CLtotal (total clearance) was calculated by dose (2 mg)/AUC (area under the concentration-time curve). AUC was calculated by the trapezoidal rule and extrapolated to infinity The bioavailability (BA) of GZ-DE or GZ was calculated from the AUCnon-iv for oral, intraduodenal, or intraileal administration and AUCiv by the following equation:
where mean AUC values for the intravenous, oral, intraduodenal, and intraileal dosing groups were used.
Stability of GZ-DE
Each solution (99 mL) of distilled water, 2 mg/mL sodium chloride (NaCl) solution adjusted to pH 1.2 with hydro chloric acid (pH 1.2 solution), and 50 mM phosphate-buffered solution (pH 7.4) was incubated at 37°C for 2 hours. One milliliter of GZ-DE propylene glycol solution (GZ concentration 20 mg/mL) was added to the three types of solution described above. Next, 1 mL of each mixed solution was put into a 1.5 mL microtube and then cooled to 4°C. The solutions were used as samples of the initial concentration of GZ-DE in the stability study. After incubation at 37°C, 1 mL of each mixture solution was put into a 1.5 mL microtube every 2 hours for up to 10 hours. The GZ-DE and GZ concentration in the samples was determined by HPLC.
Further, to estimate the long-term stability of GZ-DE in aqueous solution, a stability study similar to the above was carried out. Namely, 1 mL of GZ-DE propylene glycol solution (GZ concentration 0.3 mg/mL) and 99 mL of aqueous solution (distilled water, 0.9% NaCl solution, or pH 7.4 solution) were mixed and incubated at 37°C. After incubation, 1 mL of each mixed solution was taken at 1, 2, 5, 6, 7, 8, 9, 10, and 20 days. The GZ-DE and GZ concentrations in the mixed solutions were then measured by HPLC.
Results and discussion
Pharmacokinetic parameters
shows concentration versus time curves for GZ-DE and GZ in bile, and shows the pharmacokinetic parameters after intravenous administration of GZ-DE (GZ dose 2 mg per rat).
Table 1 Pharmacokinetic parameters for GZ-DE and GZ after intravenous administration of GZ-DE (GZ dose 2 mg per rat)
Figure 2 Bile GZ-DE (A) and GZ (B) concentration versus time profiles after intravenous administration of GZ-DE (GZ dose 2 mg per rat). The data represent the mean of three experiments. Standard deviation bars are shown with each symbol.
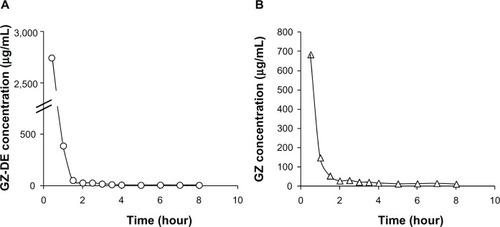
From the concentration-time profile of GZ-DE in bile, the biliary GZ-DE concentration was 2,744 μg/mL at 30 minutes after administration, and decreased rapidly to 390 μg/mL at one hour. However, the GZ-DE concentration at 1.5–8 hours decreased only moderately. These results suggest that this biphasic change was controlled by two types of elimination kinetics, ie, an elimination rate of GZ-DE to bile as first-pass after intravenous administration and an elimination rate of GZ-DE to bile after distribution to other organs via the systemic circulation. Elimination of GZ-DE into bile at one hour and 8 hours after administration was 68.5% and 72.6% of the administered dose, respectively. On the other hand, the presence of GZ became clear from determination of GZ in bile by HPLC. It was confirmed that GZ did not exist in the GZ-DE formulation before administration. Therefore, the result indicates that GZ-DE was changed to GZ in rats after administration. From the concentration-time profile for GZ in bile, rapid elimination (at 0.5–2 hours) and moderate elimination (2–8 hours) of GZ were observed. The GZ concentration in bile at 30 minutes after administration was 684 μg/mL, suggesting that conversion from GZ-DE to GZ occurred rapidly. Elimination of GZ to bile at 30 minutes and 2 and 8 hours was 12.3%, 16.6%, and 19.4% of the administered GZ dose (2 mg), respectively. The sum of GZ-DE and GZ elimination into bile at 8 hours was 92.0% of the administered dose. Namely, the ratio of GZ-DE to GZ in bile was approximately 4:1 after intravenous administration.
It has been reported that almost all GZ is rapidly excreted into bile via the liver after intravenous or subcutaneous administration. Specifically, 85% of the administered GZ dose (5 mg/kg) in rats was excreted into bile over 4 hours;Citation11 93.3% of the administered GZ dose (4 mg per rat) was excreted into bile until 8 hours;Citation8 and 80.6% of the administered GZ dose (100 mg/kg) in rats were excreted into bile until 48 hours.Citation12 Comparison of these reports indicates that approximately 20% of the GZ-DE administered is converted into GZ, which is then excreted rapidly into bile. Further, it is interesting that both apparent elimination rate constants (α and β) for GZ-DE shown in (4.56 per hour and 0.245 per hour) were higher than those for GZ (3.35 per hour and 0.148 per hour). From these results, it is suggested that the efflux rate of GZ-DE into bile by efflux transporters in the liver, such as multidrug resistance protein 2, may be high in comparison with the efflux rate of GZ into bile. GZ is one of the substrates of multidrug resistance protein 2 in the liver,Citation13,Citation14 and the initial elimination rate constant for GZ-DE (4.56 per hour) was remarkably high compared with that for GZ (1.99 per hour) after intravenous administration of GZ in rats.Citation15 Therefore, it was predicted that the efflux rate of GZ-DE from liver to bile would be rapid as compared with the conversion rate from GZ-DE to GZ after intravenous administration of GZ-DE.
Gastrointestinal absorption
shows the GZ-DE and GZ eliminated in bile versus the time curves after intraduodenal, intraileal, and oral administration of GZ-DE (GZ dose 5 mg per rat).
Figure 3 GZ-DE and GZ eliminated in bile after intraduodenal (A), intraileal (B), and oral (C) administration of GZ-DE (GZ dose 5 mg per rat). The data represent the mean ± standard deviation of four experiments.
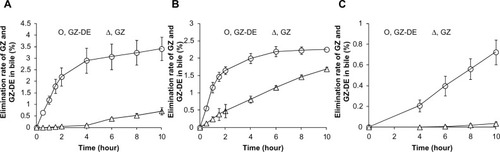
Initial elimination of GZ-DE into bile until one hour after intraduodenal administration was 54.9 μg/hour. GZ-DE until 2 hours after administration was excreted into bile at a constant level, but elimination over 4–10 hours changed slightly. In , the GZ-DE eliminated into bile after administration of the dose was 0.65% at 30 minutes, 1.19% at one hour, and 3.41% at 10 hours. On the other hand, elimination of GZ into bile was accepted as a slow curve at 4 hours after administration, but elimination of GZ was almost maintained in the range of 1.8–4.8 μg/hour. Elimination of GZ into bile increased in a time-dependent manner, ie, the amount of GZ eliminated after administration of a 5 mg GZ dose was 0.013% at one hour, 0.10% at 4 hours, and 0.71% at 10 hours. The sum of GZ-DE and GZ eliminated into bile at 10 hours was 4.12% of the administered dose. To clarify how this value of 4.12% for absorption of GZ-DE and GZ from the intestinal tract arose, an in vivo absorption study was carried out by administration of a GZ-NH4 solution (GZ dose 5 mg per rat). When the GZ-NH4 solution was administered via the duodenum in rats, elimination of GZ into bile was 1.39% of the administered dose after 10 hours. Therefore, intraduodenal administration of GZ-DE was improved approximately three-fold times when compared with GZ.
On the other hand, there are problems concerning conversion from GZ-DE to GZ before it was eliminated into bile. Specifically, the ratio of GZ-DE to GZ 10 hours after intraduodenal administration of GZ-DE was 83% and 17%, ie, approximately 4:1. This result indicates that the GZ-DE:GZ elimination ratio (4:1) after intraduodenal administration of GZ-DE was the same as that after intravenous administration of GZ-DE. On the basis of these results, it is suggested that conversion of GZ-DE to GZ might not be affected by administration route.
A wide range of esterases exist, with differences in their substrate specificity, organization such as in the liver and small intestine, and biological function.Citation16 Further, the distribution of esterase in the bowel is different from that in the duodenum and colon, and when metabolism due to intestinal bacteria participates in de-esterification, the lower portion of the intestinal tract becomes important. To confirm the conversion rate from GZ-DE to GZ in the intestinal tract, the ileum was the administration site for the in vivo experiments.
The initial elimination rate of GZ-DE into bile in the hour after intraileal administration of GZ-DE was 51.2 μg/hour, beyond which the elimination of GZ-DE gradually decreased. As shown in , elimination of GZ-DE into bile increased in a time-dependent manner. Elimination of the administered dose of GZ-DE was 1.65% at 2 hours and 2.25% at 10 hours. On the other hand, elimination of GZ after intraileal administration of GZ-DE remained constant in the range of 9.78 ± 0.99 μg/hour until 10 hours. Elimination of GZ into bile increased approximately linearly in a time-dependent manner. Elimination of GZ over 10 hours was 1.69% of the administered dose (GZ 5 mg). The sum of GZ-DE and GZ eliminated into bile until 10 hours was 3.94% of the administered dose. The bioavailability (3.94%) was also the same as that after intraduodenal administration of GZ-DE (4.12%). Interestingly, the ratio of GZ-DE and GZ was 57% and 43%, ie, approximately 3:2. The elimination ratio of GZ after intraileal administration of GZ-DE was higher than that after intraduodenal administration of GZ-DE. As a predicted reason, according to GZ-DE administered in the ileum, conversion from GZ-DE to GZ might enhance in the ileum. Because, in spite of initial elimination rate (51.2 μg/hours) of GZ-DE into bile after intraileal administration was similar to that (54.9 μg/hour) after intraduodenal administration, GZ-DE elimination decreased and GZ elimination increased into bile until 10 hours. Therefore, it is thought that there are two possibilities, ie, enhanced metabolism and/or hydrolysis from GZ-DE to GZ and improved absorption of GZ from the ileum. In support of the latter hypothesis, there is a report indicating that GZ is easily absorbed in the lower small intestine and in the large intestine.Citation17
Elimination of GZ-DE occurred at a constant rate (3.85 ± 0.41 μg/hour) during the 4–10 hours after oral administration of GZ-DE. Similarly, elimination of GZ-DE into bile increased linearly in a time-dependent manner (). Elimination of GZ-DE into bile over 10 hours was 0.72% of the administered dose (GZ 5 mg). On the other hand, GZ was not detected in bile until 4 hours after oral administration of GZ-DE. The GZ eliminated in bile by 10 hours was 0.03% of the administered dose. GZ-DE and GZ levels eliminated into bile were extremely low compared with those after intraduodenal and intraileal administration. The following are considered as problems: prolonged retention of GZ-DE propylene glycol solution in the stomach; degradation of GZ-DE in acidic conditions; and deposition of GZ-DE in acidic conditions.
GZ-DE and GZ concentrations in the liver at 10 hours after intraduodenal, intraileal, and oral administration of GZ-DE were not detected by HPLC. These results suggest that the concentrations of both compounds were less than the detection limit (0.5 μg/mg liver for both compounds) at 10 hours after administration.
Stability of GZ-DE
It has important meaning in pharmaceutics that the availability of GZ after the oral administration is extremely lower than that in an intraduodenal administration in a pharmacokinetic model. Therefore, a stability study of GZ-DE in gastric juice (pH 1.2) was necessary. Further, as one of several factors inducing conversion from GZ-DE to GZ, the influence of hydrolysis in an in vivo absorption study seemed to have a strong effect on conversion because the time curves for elimination of GZ into bile after intraduodenal, intraileal, and oral administration increased linearly.
shows the residual GZ-DE and produced GZ dissolved in the test solutions. Both compound profiles were markedly affected by the components in each test solution. When distilled water, 0.9% NaCl solution, and 50 mM phosphate-buffered solution (pH 7.4) were used as the test solutions, their appearance was maintained as a colorless clear state. On the other hand when GZ-DE propylene glycol solution was added to pH 1.2 solution, it rapidly became turbid with a white color and precipitation was observed. After centrifugation (14,000 × g, 10 minutes) of the mixture of GZ-DE propylene glycol solution and the pH 1.2 solution with precipitation, the GZ-DE in the supernatant was not detected by the HPLC system, ie, all of the GZ-DE in the pH 1.2 solution was precipitated under acidic conditions (pH 1.2). GZ was not detected in the pH 1.2 solution because the GZ-DE was precipitated (). This result suggests that almost all of the GZ-DE dissolved in propylene glycol may be solidified under acidic conditions after oral administration in rats. In contrast, distilled water, of the three test solutions tested was the most amenable for stability of GZ-DE. Residual GZ-DE levels in distilled water were 79.3% ± 3.0% at 10 hours () and 83.7% ± 9.8% by one day () after incubation at 37°C. GZ levels produced in distilled water were 0.05% ± 0.01% at 10 hours () and 13.2% ± 0.02% after 20 days (). When the pH 7.4 solution was used as the test solution, the residual GZ-DE levels were 52.7% ± 2.2% at 10 hours () and 12.5% ± 0.1% at one day (). GZ levels produced in pH 7.4 solution were 4.85% ± 0.47% at 10 hours () and 22.1% ± 0.5% at one day () after incubation. Further, the GZ level produced in pH 7.4 solution was maintained at approximately 90% after 5 days (). When 0.9% NaCl solution was used as the test solution, residual GZ-DE levels at one day and 20 days were 65.3% ± 0.4% and 0%, respectively (), and GZ levels produced at one day and 20 days were 0% and 81.1% ± 0.3%, respectively ().
Figure 4 Stability of GZ-DE in distilled water, pH 1.2 solution, 0.9% NaCl solution, and 50 mM phosphate-buffered solution (pH 7.4). The initial GZ concentration in each test solution at the start was set to 200 μg/mL (A and B) and 3 μg/mL (C and D). The incubation temperature was set to 37°C. The data represent the mean ± standard deviation of three experiments.
Abbreviations: GZ, glycyrrhizic acid; GZ-DE, glycyrrhizic acid diethyl ester.
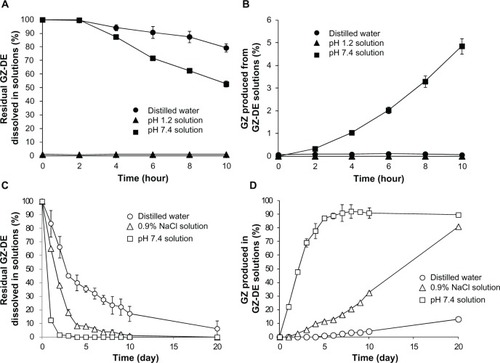
From the results in , it seemed that disappearance of GZ-DE was dependent on a one-order linear equation and production of GZ was dependent on a zero-order linear equation, because the disappearance profiles of GZ-DE decreased depending on half-life (4.46 days for distilled water, 1.97 days for 0.9% NaCl solution, and 0.43 days for pH 7.4 solution) and production of GZ in the three test solutions increased linearly in a time-dependent manner (). With the exception of the pH 1.2 solution, it was clear that there was a difference between the rate of decrease in GZ-DE concentration and rate of increase in GZ concentration in the test solutions. A question occurred in production rate of GZ being slow for disappearance rate of GZ-DE. It could be that the difference occurred between the disappearance rate of GZ-DE and the production rate of GZ. It is necessary to think carefully about the processes involved in conversion from GZ-DE to GZ. Because GZ-DE has two ester sites, it seems that a GZ monoester is probably involved during conversion from GZ-DE to GZ. Therefore, it guessed that the conversion speed from GZ-monoester to GZ might pose the difference between disappearance rate of GZ-DE and production rate of GZ. The existence of a GZ monoester was not addressed in this study, so will need to be investigated in future research. From these results, it is concluded that GZ-DE has very poor stability in pH 7.4 solution and converts to GZ in a time-dependent manner.
Conclusion
Improvement in bioavailability of GZ in the liver after oral administration of a GZ formulation is expected because of increased absorption of GZ-DE from the intestinal tract. Total bioavailability of GZ-DE and GZ after intraduodenal and intraileal administration of GZ-DE was three-fold higher compared with that after intraduodenal administration of GZ in rats. However, the conversion rates from GZ-DE to GZ were approximately 20% and 40% after intraduodenal and intraileal administration, respectively, from GZ-DE and GZ eliminated into bile until 10 hours after administration. As the reason of insufficient conversion from GZ-DE to GZ in rats, it was clear that GZ-DE was rapidly excreted into bile faster than GZ in pharmacokinetic parameters calculated from intravenous administration of GZ-DE. Further, our results strongly suggest that GZ-DE was converted to GZ mainly by hydrolysis in the pH 7.4 solution. From the pharmacokinetic characteristics of GZ-DE in rats, it is thought that the availability of GZ as a revolutionary prodrug was not high from the viewpoint of the bioavailability of GZ in the liver by intestinal administration of GZ-DE. Although the result with expected utility was not provided, as a future study, the synthesis of compound which has high absorption from the intestinal tract and can convert into GZ in the liver or before arriving at the liver is expected in a development of the prodrug of GZ.
Disclosure
The authors report no conflicts of interest in this work.
References
- Matsunaga K Suzuki M Matsumoto N Stricter control of alanine transaminase by long-term administration of glycyrrhizin counters the disease progression in patients with chronic hepatitis C J St Marianna Univ 2012 3 1 23 33
- Acharya SK Sreenivas V Datta GS Treatment of chronic hepatitis due to hepatitis C virus (CH-C) in India: a randomized controlled trial comparing daily interferon-alfa-2b and ribavirin with daily interferon-alfa-2b and glycyrrhizin – a multicenter study J Clin Exp Hepatol 2012 2 1 10 18
- Ashfaq UA Nawaz Z Riazuddin S Glycyrrhizin as antiviral agent against hepatitis C virus J Transl Med 2011 9 112 21762538
- Ikeda K Glycyrrhizin injection therapy prevents hepatocellular carcinogenesis in patients with interferon-resistant active chronic hepatitis C Hepatol Res 2007 37 Suppl 2 S287 S293 17877497
- Sasaki K Yonebayashi S Yoshida M Shimizu K Aotsuka T Takayama K Improvement in the bioavailability of poorly absorbed glycyrrhizin via various non-vascular administration routes in rats Int J Pharm 2003 265 1–2 95 102 14522122
- Shibata N Ohno T Shimokawa T Application of pressure-controlled colon delivery capsule to oral administration of glycyrrhizin in dogs J Pharm Pharmacol 2001 53 4 441 447 11341360
- Koga K Kawashima S Shibata N Takada K Murakami M Preparation and rectal absorption of highly concentrated glycyrrhizin solution Biol Pharm Bull 2003 26 9 1299 1305 12951475
- Koga K Kawashima S Shibata N Takada K Novel formulations of a liver protection drug glycyrrhizin Yakugaku Zasshi 2007 127 7 1103 1114 17603270
- Koga K Nishimon Y Ueta H Matsuno K Takada K Utility of nano-sized, water-in-oil emulsion as a sustained release formulation of glycyrrhizin Biol Pharm Bull 2011 34 2 300 305 21415545
- Yamaoka K Tanigawara Y Nakagawa T Uno T A pharmacokinetic analysis program (MULTI) for microcomputer J Pharmacobiodyn 1981 4 11 879 885 7328489
- Ichikawa T Ishida S Sakiya Y Sawada Y Hanano M Biliary excretion and enterohepatic cycling of glycyrrhizin in rats J Pharm Sci 1986 75 7 672 675 3761168
- Ishida S Sakiya Y Ichikawa T Taira Z Awazu S Prediction of glycyrrhizin disposition in rat and man by a physiologically based pharmacokinetic model Chem Pharm Bull 1990 38 1 212 218 2337943
- Horikawa M Kato Y Tyson CA Sugiyama Y The potential for an interaction between MRP2 (ABCC2) and various therapeutic agents: probenecid as a candidate inhibitor of the biliary excretion of irinotecan metabolites Drug Metab Pharmacokinet 2002 17 1 23 33 15618649
- Shimamura H Suzuki H Tagaya O Horie T Sugiyama Y Biliary excretion of glycyrrhizin in rats: kinetic basis for multiplicity in bile canalicular transport of organic anions Pharm Res 1996 13 12 1833 1837 8987080
- Koga K Tomoyama M Ohyanagi K Takada K Pharmacokinetics of glycyrrhizin in normal and albumin-deficient rats Biopharm Drug Dispos 2008 29 7 373 381 18548521
- Imai T Hydrolysis by carboxylesterase and disposition of prodrug with ester moiety Yakugaku Zasshi 2007 127 4 611 619 17409690
- Shibata N Shimokawa T Jiang Z Characteristics of intestinal absorption and disposition of glycyrrhizin in mice Biopharm Drug Dispos 2000 21 3 95 101 11113882