
Abstract
Maturity-onset diabetes of the young (MODY) is characterized by autosomal dominant inheritance, onset before 25 years of age, absence of β-cell autoimmunity, and sustained pancreatic β-cell function. To date, mutations have been identified in at least 14 different genes, including six genes encoding proteins that, respectively, correspond to MODY subtypes 1–6: hepatocyte nuclear factor (HNF) 4α (HNF4α), glucokinase (GCK), HNF1α (HNF1 α), pancreatic and duodenal homeobox 1 (PDX1), HNF1β (HNF1 β), and neurogenic differentiation 1 (NEUROD1). Diagnostic tools based on currently available genetic tests can facilitate the correct diagnosis and appropriate treatment of patients with MODY. Candidates for genetic testing include nonobese subjects with hyperglycemia, no evidence of β-cell autoimmunity, sustained β-cell function, and a strong family history of similar-type diabetes among first-degree relatives. Moreover, identification of the MODY subtype is important, given the subtype-related differences in the age of onset, clinical course and progression, type of hyperglycemia, and response to treatment. This review discusses the current perspectives on the diagnosis and treatment of MODY, particularly with regard to the six major subtypes (MODY 1–6).
Introduction
Maturity-onset diabetes of the young (MODY) was first reported in 1974 as mild familial diabetes with dominant inheritance.Citation1 Classically, MODY was characterized by autosomal dominant inheritance, onset before 45 years of age, the absence of β-cell autoimmunity,Citation2 absence of insulin resistance,Citation3 and sustained β-cell function. However, the new diagnostic criteria set forth in the Practice Guideline for MODY in 2008Citation4 include onset before 25 years of age in one family member, presence of diabetes in two consecutive generations, absence of β-cell autoantibodies, and sustained endogenous insulin secretion. Preserved β-cell function is indicated by the lack of need for insulin treatment or a serum C-peptide level of >200 pmol/L even after 3 years of insulin treatment.Citation4
Molecular methods for the diagnosis of MODY were first introduced after the 1990s. To date, mutations associated with MODY have been reported in least 14 different genes,Citation5–Citation14 including the following six genes encoding major factors: hepatocyte nuclear factor (HNF) 4α (HNF4α), glucokinase (GCK), HNF1α (HNF1α), pancreatic and duodenal homeobox 1 (PDX1), HNF1β (HNF1 β), and neurogenic differentiation 1 (NEUROD1), which correspond to MODY subtypes 1–6, respectively. The following eight genes have been identified as possibly causative in MODY subtypes 7–14, respectively: Kruppel-like factor 11 (KLF11); carboxyl ester lipase; paired-box-containing gene 4 (PAX4); insulin (INS); B-lymphocyte kinase; adenosine triphosphate (ATP)-binding cassette, sub-family C (CFTR/MRP) member 8 (ABCC8); potassium channel, inwardly rectifying subfamily J, member 11 (KCNJ11); and adaptor protein, phosphotyrosine interaction, PH domain, and leucine zipper containing 1 (APPL1).Citation11,Citation12 The causative genes for MODY and their medical conditions are shown in .
Table 1 The causative genes for maturity-onset diabetes of the young (MODY) and medical conditions associated with each MODY subtype
MODY is a rare condition, accounting for 1–5% of all cases of diabetesCitation12,Citation13 and 1–6% of pediatric cases of diabetes.Citation14 Mutations in GCK, HNF1α, HNF4α, and HNF1β are the most frequently identified etiologies of MODY and, respectively, account for 32%, 52%, 10%, and 6% of all affected patients in the United Kingdom.Citation11 However, the frequencies of these etiologies may differ among Asian and Caucasian populations. In Japan, 48.1% of pediatric cases of clinical MODY harbored already known MODY-related gene defects (GCK, 22.8%; HNF1A, 13.9%; HNF4A, 3.8%, and HNF1B, 7.6%), whereas in 51.9% cases, the causative mutations were not identifiedCitation15 In Korea, only 10% of clinical MODY or childhood-onset type 2 diabetes cases harbored known MODY-related gene defects (GCK, 2.5%; HNF1A, 5%, and HNF1B, 2.5%)Citation16 These data suggest that currently unidentified genes may cause MODY in Asian populations. The causative genes for MODY and their medical conditions are shown in .
To improve the prognosis of MODY, it is important to identify the affected subjects as early as possible. To this end, specific molecular analyses are available to predict the clinical disease course and offer the most appropriate treatment.Citation14 However, approximately 80% of patients with MODY may be misdiagnosed with type 1 or type 2 diabetes mellitus at diagnosis,Citation17 and current calculations indicate a delay of approximately 15 years from the diagnosis of diabetes to the genetic diagnosis of MODY.Citation17
This review discusses the current perspectives on the diagnosis and treatment of MODY, in particular, the six major subtypes (MODY 1–6).
Diagnosis of MODY
At diagnosis, MODY cannot be distinguished easily from type 1 and type 2 diabetes mellitus based on clinical characteristics.Citation13,Citation14 Rather, type 1 diabetes mostly differs from MODY in terms of disease etiology, as the pathogenesis of the latter does not involve pancreatic β-cell autoimmunity. Patients with MODY usually maintain β-cell function, exhibit a stimulated serum C-peptide level exceeding 200 pmol/L, and their diabetes is well-controlled with no or low-dose insulin for at least 5 years after diagnosis.Citation14 Although the clinical manifestations of youth-onset type 2 diabetes substantially resemble those of MODY, patients with the former condition are generally obese, whereas the latter condition is not associated with overweight. Still, approximately 10–15% of Japanese school children with type 2 diabetes are non-obese.Citation18,Citation19 Additionally, some patients with MODY, particularly those belonging to ethnic groups with a higher prevalence of obesity (eg, Hispanic), may become overweight or obese due to poor dietary habits and sedentary lifestyles. Furthermore, both MODY and type 2 diabetes are associated with a strong family history of diabetes. For example, studies revealed that approximately 70% of Japanese school children with type 2 diabetes had a family history of type 2 diabetes among first- and second-degree relatives. Those with nonobese type 2 diabetes had a particularly strong family background of the disease.Citation18,Citation19 Therefore, the correct diagnosis of MODY and determination of subtype should be based on genetic testing. Candidates for genetic testing include nonobese subjects with hyperglycemia, no evidence of β-cell autoimmunity, preserved β-cell function, and a strong family history of a similar type of diabetes among the first-degree relatives.Citation14
Currently, genetic testing is performed worldwide to facilitate predictions of the clinical course and prognosis of MODY. Known MODY-related genes can be identified by direct sequencing with sensitivity rates as high as 100%,Citation14 and next-generation sequencing methods (eg, gene-targeted and whole-exome sequencing) have been successfully used to identify mutations in MODY genes.Citation16 However, genetic testing remains expensive and is necessarily limited to cases of strongly suspected MODY. These patients should be offered the most suitable treatment from among various pharmacological therapies, including oral antidiabetic drugs (OADs) and insulin.
Diagnosis of the subtypes of MODY
sAs shown in , at least 14 MODY subtypes have been reported, of which 1–6 are the major subtypes. As noted above, eight other subtypes have been identified, including MODY 14, which was recently associated with the causative gene APPL1 in large families.Citation20 Most MODY-causative genes, except GCK, encode transcription factors expressed in pancreatic β-cells (). MODY subtype determination is important, as the subtypes differ in terms of the age of onset, clinical course and progression, type of hyperglycemia, and response to treatment.Citation14 Most patients with MODY exhibit the clinical characteristics of isolated diabetes or mild fasting hyperglycemia. However, some MODY subtypes are associated with additional manifestations, such as renal abnormalities (MODY5) or pancreatic exocrine dysfunction (MODY8). This review mainly describes the clinical characteristics of the six major subtypes of MODY.
Figure 1 Expression of maturity-onset diabetes of the young (MODY)-causative genes in pancreatic β-cells and mechanism of insulin secretion.
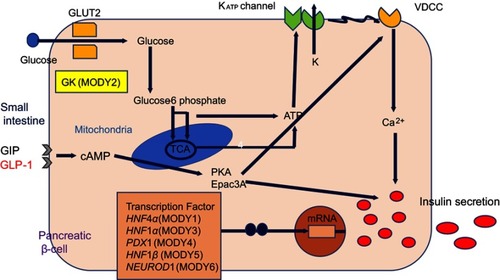
MODY2 (GCK-MODY)
GCK, a glucose sensor expressed in pancreatic β-cells, is a key enzyme in glucose metabolism that catalyzes the conversion of glucose to glucose-6-phosphate and thus controls glucose-mediated insulin secretion. More than 600 mutations in GCK have been identified in >1,000 families, and these alterations lead to both hyperglycemia and hypoglycemia.Citation21 Heterozygous inactive mutations are associated with mild and often subclinical hyperglycemia from birth, and this condition gradually deteriorates with ageCitation11 Essentially, these mutations elevate the glucose threshold for insulin secretion, resulting in an elevated fasting plasma glucose level (5.5–8 mmol/L). Such patients exhibit a slight increase in the 2-hr plasma glucose level during oral glucose tolerance testing (OGTT; increase in 4.5 mmol/L = 90th percentile).Citation4,Citation22 These patients usually have a HbA1c level of <64 mmol/mol.Citation23
Patients with MODY2 are usually asymptomatic. The majority are discovered through routine examinations during pregnancy or school-based urine glucose screening tests. MODY2 presents in approximately 2–6% of patients with gestational diabetes and can be distinguished based on clinical manifestations and the fasting glucose level.Citation24,Citation25 The birth weights of babies in MODY2 families depend on the mutation statuses of both the fetus and mother. If both harbor a GCK mutation, a maternal increase in plasma glucose will induce an appropriate insulin release in the fetus and a normal birth weight. However, if only the mother harbors the GCK mutation, maternal hyperglycemia will elicit increased fetal insulin secretion and a birth weight approximately 500 g higher than that of a baby who harbors the mutation.Citation26 In Japan, Yorifuji et al reported that 22.8% of patients in whom MODY was detected through school urine glucose screening programs were finally diagnosed with MODY2 through genetic testing. Accordingly, MODY2 is most frequent subtype in Japanese children.Citation15 The clinical course of this subtype may be mild and nonprogressive, and microvascular and macrovascular complications rarely occur despite long-term exposure to mild hyperglycemia.Citation26,Citation27
MODY 3 (HNF1α-MODY)
The transcription factor HNF1α is expressed in the liver, kidney, intestine and pancreatic β-cells. HNF1a knockout mice develop diabetes consequent to defective glucose-induced insulin secretion.Citation28,Citation29 Mutations in HNF1a are the most frequent causes of MODY in Europe, North America, and some Asian countries.Citation11,Citation14,Citation16 In Japan, mutations in HNF1 a were detected in 13.9% of patients with clinical MODY.Citation15 Studies of approximately 1200 families have identified >400 different HNF1a mutations,Citation30 of which a mutation (P291fsinsC) in exon 4 is most common.Citation31 Heterozygous mutations of HNF1a cause a progressive insulin deficiency that manifests as mild hyperglycemia in childhood and as diabetes during early adulthood.Citation15,Citation32 MODY3 is rarely discovered in children younger than 8 years of age.Citation15
Hyperglycemia associated with MODY3 may be progressive and deteriorating. In these patients, the risks of microvascular and macrovascular complications are similar to those observed in patients with type 1 and type 2 diabetes.Citation33 Interestingly, HNF1a mutation carriers develop postprandial glycosuria before the onset of diabetes due to renal tubular dysfunction and a consequently low renal threshold for glucose absorption.Citation34 In the fetus, HNF1a mutational heterozygosity does not influence the birth weight because insulin secretion in utero remains normal.Citation35
MODY1 (HNF4α-MODY)
The transcription factor HNF4α is expressed primarily in the liver but also in the pancreas and kidney,Citation36 where it regulates the expression of genes required for glucose transport and metabolism. Mutations in HNF4α are relatively uncommon, accounting for approximately 5% of all MODY cases. Phenotypically, MODY1 due to heterozygous HNF4α mutation manifests as progressive insulin deficiency, similar to that observed in MODY3. Fetal HNF4α heterozygosity results in macrosomia due to hyperinsulinemia in utero and subsequent neonatal transient or persistent hypoglycemia, which is responsive to diazoxide.Citation37 Glycosuria is not observed in MODY1, in contrast to MODY3. Hyperinsulinism associated with MODY1 generally remits during infancy, followed by a gradual decrease in endogenous insulin production and the emergence of diabetes in adolescence.Citation14 Moreover, HNF4α has been associated with triglyceride metabolism, and mutation carriers may exhibit reduced levels of apolipoproteins (apoAII, apoCIII, and apoB).Citation38
MODY5 (HNF1β-MODY)
The transcription factor HNF1β is involved in the organogenesis of the kidney, genito-urinary tract, liver, lungs, gut, and pancreas.Citation39 Accordingly, renal malformations, including renal cysts, renal dysplasia, urinary tract malformation, and hypoplastic glomerulonephritic kidney disease,Citation40 are seen in the majority of HNF1β mutation carriers and constitute the main presentation of MODY5 in children, regardless of the hyperglycemia statusCitation14HNF1β mutation also causes renal cysts and diabetes syndrome, and these renal abnormalities are evident from the 17th week of gestation.Citation41 Affected patients will develop renal dysfunction by 45 years of age, and half of these patients will progress to end-stage renal disease without developing diabetic kidney disease.Citation42 Accordingly, MODY5 should be suspected in patients with diabetes and nondiabetic renal disease. Genito-urinary tract malformations (especially uterine abnormalities), liver dysfunction, and pancreatic hypoplasia have also been reported.Citation43 Although diabetes associated with MODY5 develops in early adulthood, carriers of HNF1β mutations have significantly reduced birth weights.Citation44 Phenotypically, MODY5 differs from MODY3. Patients with MODY5 present with dyslipidemia, including a low high-density lipoprotein level and elevated triglyceride level. Diabetes develops during adolescence or early adulthood and usually progresses to an insulin-dependent state due to pancreatic hypoplasia, with hepatic insulin resistance, relatively earlier period of the disease.Citation11,Citation14 Patients harboring HNF1β mutations exhibit highly variable phenotype and clinical manifestations even within affected families. Accordingly, the diagnosis of MODY5 should be made in consultation with not only diabetes specialists but also other specialists, such as nephrologists, gynecologists, and urologists.Citation14
MODY4 (PDX1-MODY)
PDX1 is a homeodomain-containing transcription factor that acts in both the exocrine and endocrine pancreatic developmental programs and affects pancreatic development and INS expression.Citation45 Homozygous mutations in PDX1 cause pancreas agenesis and hypoplasia and permanent neonatal diabetes,Citation46 whereas heterozygous mutations lead to β-cell impairment and hyperglycemia, including permanent neonatal diabetes. MODY4 is a very rare subtype.
MODY6 (NEUROD1-MODY)
NEUROD1 is a basic-loop-helix transcription factor involved in pancreatic and neuronal development. It plays an important role in pancreatic β-cell maturation and maintenance. Islets lacking NEUROD1 respond poorly to glucose and show a glucose metabolic profile similar to immature β-cells. Heterozygous mutations in NEUROD1 induce childhood- or adult-onset diabetes, while homozygous mutations can cause neonatal diabetes, neurological abnormalities, and learning difficulties.Citation47–Citation49
The ages of onset, degrees of hyperglycemia, involvement of special tissues, and clinical features of other subtypes of MODY are shown in .
How can be MODY diagnosed correctly?
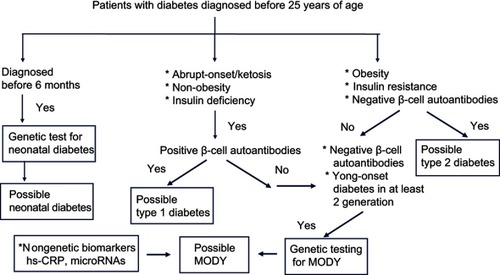
Cases of MODY are often misdiagnosed as type 1 or type 2 diabetes at presentation.Citation13,Citation14,Citation19 Accordingly, an improvement in diagnostic significance will require targeted selection of subjects for genetic testing, particularly in scenarios with limited resources. Various algorithms have been proposed to select candidates for genetic testing.Citation13,Citation50 According to a recent diagnostic criteria,Citation4 MODY is characterized by onset before 25 years of age, presence of diabetes in two consecutive family generations, absence of β-cell autoantibodies, and preserved endogenous insulin secretion with a serum C-peptide level of >200 pmol/L. These diagnostic criteria have been well accepted for distinguishing MODY from type 1 and type 2 diabetes. Shields et al.Citation50 further proposed a model in which an age younger than 30 years at diagnosis was the most useful discriminator between MODY and type 2 diabetes. In that model, a family background of diabetes increased the probability of diagnosis of MODY by 23 times among patients who were first diagnosed with type 1 diabetes.Citation51
The costs and limitations of genetic testing have encouraged the discovery of nongenetic biomarkers that might be used to identify appropriate subjects for molecular diagnosis. For example, high-sensitivity C-reactive protein (hs-CRP) may be a useful biomarker in the differential diagnosis of MODY3, as affected patients have a significantly lower level of hs-CRP than those with type 1 or type 2 diabetes or MODY2.Citation52–Citation54 A recent study suggested that microRNAs may be useful biomarkers in carriers of MODY3-related gene mutations.Citation55 However, some causative genes for MODY subtypes have also been associated with other forms of diabetes. KCNJ11, ABCC8, PDX1, PAX4, and NEUROD1 have been associated with permanent neonatal diabetes and type 2 diabetes, while INS is associated with both type 1 and type 2 diabetes.Citation56,Citation57 Moreover, variants of HNF1α, HNF4α, HNF1β, and GCK might be associated with an increased risk of type 2 diabetes.Citation57 It is often difficult to differentiate between an incompletely penetrant MODY mutation and a rare variant of type 2 diabetes, and such distinction may be a matter of semantics.Citation58,Citation59 Therefore, genetic testing is not sufficient to confirm a diagnosis of MODY, which should also include clinical observation, laboratory testing (serum C-peptide, β-cell autoantibodies, and specific biomarkers). presents a diagnostic algorithm used to identify candidates with diabetes who should undergo genetic testing to confirm the diagnosis of MODY.
Figure 2 Diagnostic algorithm for maturity-onset diabetes of the young (MODY).
Treatment of MODY
Correct determination of the MODY subtype is important, as this informs decisions regarding appropriate treatment and prognosis. Children and adolescents diagnosed with diabetes may initially be treated with insulin, and this regimen often continues even after the stabilization of glycemia. However, in some patients with MODY, hyperglycemia can be controlled by prescribing OADs (eg, sulfonylureas, without using insulin. Additionally, the selection of appropriate treatments for these patients is important to improve their quality of life.
MODY2 (GCK-MODY)
The majority of patients with MODY2 exhibit mildly elevated fasting plasma glucose levels but do not exhibit postprandial hyperglycemia; in other words, they secrete sufficient insulin in response to an increase in plasma glucose after consuming a meal. Therefore, dietary intervention alone is usually advised for these patients, as pharmacological intervention is not required to control hyperglycemia and prevent diabetic complications.Citation11,Citation14 However, some Japanese patients with MODY2 eventually require pharmacological treatment. In a study of 55 patients, Kawakita et al.Citation60 reported that seven were treated with OADs, two with sulfonylureas, one with metformin, and two with α-glucosidase inhibitors. Asians face a risk of insulin resistance, characterized by a higher Homeostatic Model Assessment (HOMA-IR) at a relatively low body mass index (BMI),Citation18,Citation61 possibly due to a greater amount of visceral fat than that observed in Caucasians.Citation62,Citation63 Accordingly, some Japanese patients who consume carbohydrate-rich foods and have sedentary lifestyles require OADs. Asians also exhibit a relatively lower level of insulin release in response to an increase in glucose, as well as a lower homeostasis model assessment for β-cell function (HOMA-β).Citation62,Citation63 This pattern may also lead to an increased requirement for OADs. During pregnancy, insulin might be offered to patients with MODY2 to prevent fetal overweight.
MODY3 (HNF1α-MODY) and MODY1 (HNF4α-MODY)
Patients with MODY3 and MODY1 can initially control glycemia with diet alone, although both tend to exhibit elevated postprandial glucose levels after consuming carbohydrate-rich foods.Citation22 Over time, however, most patients will experience deteriorating β-cell function and require pharmacological treatment. Patients with MODY3 and MODY1 are sensitive to sulfonylureas and can maintain optimal glycemic control with these drugs rather than insulin. One randomized clinical trial of gliclazide therapy demonstrated a 3.9-fold improvement in the fasting glucose levels of patients with MODY3 relative to BMI-matched patients with type 2 diabetes; this effect was particularly pronounced in children and young adults. That study also demonstrated the safety and efficacy of a switch from insulin to gliclazide despite using the former for a long duration.Citation64 The initial sulfonylurea dose should be low to avoid hypoglycemia. Reports suggest that optimal glycemic control without problematic hypoglycemia can be maintained for decades at gliclazide doses of 20–40 mg.Citation65,Citation66 If hypoglycemia occurs despite dose titration, a short-acting agent (eg, meglitinide) may be considered.Citation67 Another sulfonylurea, glimepiride, might offer a similar glucose-reducing effect with fewer episodes of hypoglycemia. It also exerts extrapancreatic effects such as decreased glucose output from the liver and enhanced sensitivity of peripheral tissues to insulin.Citation68
Another therapeutic option involving a glucagon-like peptide (GLP-1) agonist with a similar glucose-lowering effect as sulfonylureas and a low frequency of hypoglycemia has been proposed for patients with MODY3.Citation69,Citation70 A double-blind, randomized crossover trial that compared a GLP-1 agonist (liraglutide) with a sulfonylurea (glimepiride) found no difference between the two drugs in terms of controlling the fasting plasma glucose and responsive postprandial plasma glucose relative to baseline. However, glimepiride was clearly associated with a higher incidence of hypoglycemia.Citation70 We experienced similarly good glucose-reducing effects and a lack of hypoglycemia in children with MODY3 who were treated with liraglutide.Citation71 Moreover, we previously reported that a switch from glimepiride to a dipeptidyl-peptidase-4 (DPP-4) inhibitor, alogliptin, yielded similar glycemic control without hypoglycemia in a girl with MODY1.Citation72
During treatment, patients with MODY3 and MODY1 maintain substantial β-cell function for at least 2–4 years after diagnosis. As noted above, sulfonylureas are generally the first-line of treatment for patients with MODY3 and MODY1, despite the risk of hypoglycemia. Long-term treatment with sulfonylureas is also associated with body weight gain and the deterioration of endogenous insulin, which eventually induces insulin dependency in some patients with type 2 diabetes. In contrast, GLP-1 receptor agonists and DPP-4 inhibitors lower glucose levels and increase endogenous insulin secretion in a glucose-dependent manner. This effect occurs via a subpathway of insulin secretion in response to increased cyclic adenosine 3ʹ, 5ʹ-monophosphate (c-AMP) production, rather than by ATP production, which thus overcomes the impaired mitochondrial ATP production in response to glucose in patients with MODY3 and MODY1.Citation28 Therefore, the mode of insulin secretion induced by GLP-1 receptor agonists and DPP-4 inhibitors may more effectively increase insulin secretion in these patients. Additionally, GLP-1 receptor agonists and DPP-4 inhibitors might prevent β-cell apoptosis and promote β-cell generation to counter the progressive impairment of β-cell function during the courses of MODY3 and MODY1.Citation73–Citation75 These findings suggest that GLP-1 receptor agonists and DPP-4 inhibitors may be suitable alternatives to sulfonylureas.
MODY5 (HNF1β-MODY)
Unlike patients with MODY3 and MODY1, those with MODY5 do not respond adequately to treatment with sulfonylureas, possibly due to comorbid pancreatic hypoplasia and some degree of hepatic insulin resistance.Citation76 These patients may require intensive insulin treatment to control hyperglycemia. Additionally, microvascular complications have been described in these subjects,Citation6 and treatment for renal disease, liver dysfunction, and dyslipidemia is necessary. Renal management is a particularly important aspect of treatment in patients harboring HNF1β mutations, as these individuals will develop renal dysfunction by 45 years of age and half will progress to end-stage renal disease.Citation42
The current treatments of other MODY subtypes according to the molecular causes and clinical characteristics are listed in . However, standard treatments have not been established for most subtypes because of low numbers of cases and a lack of data confirming treatment efficacy. Furthermore, only metformin and insulin are approved for use in youth in the majority of countries. Sulfonylureas are approved for use in adolescents in some countries. Other OADs are not approved for use in those <18 years of age.Citation77
Conclusion
Advanced molecular genetic analyses have led to the identification of genes associated with clinically diagnosed subtypes of MODY. Accordingly, genetic testing is useful for the correct diagnosis and appropriate treatment of patients with MODY.Citation14 To confirm a correct diagnosis and improve prognosis, genetic testing is recommended at diagnosis or during early-stage disease, despite limitations associated with costs.
Patients with type 1 diabetes require insulin treatment for survival and metabolic control, whereas those with MODY do not usually require long-term insulin treatment. Therefore, misdiagnosis can lead to inappropriate treatment. MODY should be suspected in the presence of mild to moderate, nonketosis-prone hyperglycemia in usually nonobese patients with a strong family history of diabetes. The importance of genetic testing is emphasized by the effect of a general increase in the prevalence of obesity, which may complicate a differential diagnosis including both type 2 diabetes and MODY. Correct diagnosis of MODY is also important with respect to genetic counseling and the prevention of developing the disease.Citation78 Rapid advances in the fields of molecular analysis and laboratory technology are expected to improve the correct diagnosis of MODY and the provision of appropriate treatment based on the individual medical condition.
Abbreviation list
MODY, maturity-onset diabetes of the young; HNF, hepatocyte nuclear factor; GCK, glucokinase; PDX1, pancreatic and duodenal homeobox 1; NEUROD1, neurogenic differentiation 1; KLF11, Kruppel-like factor 11; CEL, carboxyl ester lipase; PAX4, paired-box-containing gene 4; INS, insulin; BLK, B-lymphocyte kinase; ABCC8, adenosine triphosphate (ATP)-binding cassette, sub-family C (CFTR/MRP) member 8; KCNJ 11, potassium channel, inwardly rectifying subfamily J, member 11; APPL1, adaptor protein, phosphotyrosine interaction, PH domain, and leucine zipper containing 1; OAD, oral antidiabetic drug; OGTT, oral glucose tolerance test; HOMA-IR, homeostasis model assessment for insulin resistance; BMI, body mass index; HOMA-β, homeostasis model assessment for β-cell function; hs-CRP, high-sensitivity C-reactive protein; GLP-1, glucagon-like peptide; DPP-4, dipeptidyl-peptidase-4; c-AMP, cyclic adenosine 3ʹ, 5ʹ-monophosphate.
Disclosure
The author declares no conflicts of interest in this work.
References
- Tattersall RB . Mild familial diabetes with dominant inheritance. Q J Med . 1974;43:339–357.4212169
- McDonald TJ , Colclough K , Brown R , et al. Islet autoantibodies can discriminate maturity-onset diabetes of the young (MODY) from type 1 diabetes. Diabet Med . 2011;20:1028–1033. doi:10.1111/j.1464-5491.2011.03287.x
- Owen KR , Roland J , Smith K , Hattersley AT . Adolescent onset type 2 diabetes in a non-obese Caucasian patient with an unbalanced translocation. Diabet Med . 2011;20:483–485. doi:10.1046/j.1464-5491.2003.00961.x
- Ellard S , Ballanné-Chantelot C , Hattersley AT . Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia . 2008;51:546–553. doi:10.1007/s00125-008-0942-y 18297260
- Froguel PM , Vallaire F , Sun G , et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin dependent diabetes mellitus. Nature . 1992;356:162–164. doi:10.1038/356162a0 1545870
- Horikawa Y , Iwasaki N , Hara M , et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) association with MODY. Nat Genet . 1997;17:384–385. doi:10.1038/ng1297-384 9398836
- Soffers DA , Ferrer J , Clarke WL , Habener JF . Early-onset type-II diabetes mellitus (MODY 4) linked to IPF1. Nat Genet . 1997;17:138–139. doi:10.1038/ng1097-138 9326926
- Vionnet N , Stoffel M , Takeda J , et al. Nonsense mutation in the glucokinase causes early-onset non-insulin-dependent diabetes mellitus. Nature . 1992;356:721–722. doi:10.1038/356721a0 1570017
- Yamagata K , Furuta H , Oda N , et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature . 1996;384:458–460. doi:10.1038/384458a0 8945471
- Yamagata K , Oda N , Kaisaki PJ , et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature . 1996;384:455–458. doi:10.1038/384455a0 8945470
- Kavvoura FK , Owen KR . Maturity onset diabetes of the young: clinical characteristics, diagnosis and management. Pediatr Endocrinol Rev . 2013;10:234–242.
- Kim SH . Maturity-onset diabetes of the young: what do clinicians need to know? Diabetes Metab J . 2015;39:468–477. doi:10.4093/dmj.2015.39.6.468 26706916
- Thanabalansingham G , Pal A , Selwood MP , et al. Systematic assessment of etiology in adults with a clinical diagnosis of young-onset type 2 diabetes is a successful strategy for identifying maturity-onset diabetes of the young. Diabetes Care . 2012;35:1206–1212. doi:10.2337/dc11-1243 22432108
- Hattersley AT , Greeley SA , Polak M , et al. ISPAD clinical practice consensus guidelines 2018: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes . 2018;19(Suppl.27):47–63. doi:10.1111/pedi.12772 30225972
- Yorifuji T , Fujimaru R , Hosokawa Y , et al. Comprehensive molecular analysis of Japanese patients with pediatric-onset MODY-type diabetes mellitus. Pediatr Diabetes . 2012;13:26–32. doi:10.1111/j.1399-5448.2011.00827.x 22060211
- Hwang JS , Shin CH , Yang SW , Jung SY , Huh N . Genetic and clinical characteristics of Korean maturity-onset diabetes of the young (MODY) patients. Diabetes Res Clin Pract . 2006;74:75–81. doi:10.1016/j.diabres.2006.03.002 16632067
- Shields BM , Hicks S , Shepherd MH , Colclough K , Hattersley AT , Ellard S . Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia . 2010;53:2504–2508. doi:10.1007/s00125-010-1799-4 20499044
- Urakami T , Kuwabara R , Habu M , et al. Clinical characteristics of non-obese children with type 2 diabetes mellitus without involvement of β-cell autoimmunity. Diabetes Res Clin Pract . 2013;99:105–111. doi:10.1016/j.diabres.2012.11.021 23260852
- Urakami T , Miyata M , Yoshida K , et al. Changes in annual incidence of school children with type 2 diabetes in the Tokyo metropolitan area during 1975–2015. Pediatr Diabetes . 2018;19:1385–1392. doi:10.1111/pedi.12750 30101568
- Prudente S , Jungtrakoon P , Marucci A , et al. Loss-of-function mutations in APPL1 in familial diabetes mellitus. Am J Hum Genet . 2018;97:177–185. doi:10.1016/j.ajhg.2015.05.011
- Osbak KK , Colclough K , Saint-Martin C , et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat . 2009;30:1512–1526. doi:10.1002/humu.21110 19790256
- Stride A , Vaxillaire M , Tuomi T , et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia . 2002;45:427–435. doi:10.1007/s00125-001-0770-9 11914749
- Martin D , Bellarnne-Chantelot C , Deschamps I , Froguel P , Robert JJ , Velho G . Long-term follow-up of oral glucose tolerance test-derived glucose tolerance and insulin secretion and insulin sensitivity indexes in subjects with glucokinase mutations (MODY2). Diabetes Care . 2008;31:1321–1323. doi:10.2337/dc07-2017 18411240
- Chakera AJ , Spyer G , Vincent N , Ellard S , Hattersley AT , Dunne FP . The 0.1% of the population with glucokinase monogenic diabetes can be recognized by clinical characteristics in pregnancy: the Atlantic diabetes in pregnancy cohort. Diabetes Care . 2014;37:1230–1236. doi:10.2337/dc13-2248 24550216
- Rudland VL , Hinchcliffe M , Pinner J , et al. Identifying glucokinase monogenic diabetes mellitus in a multiethnic gestational diabetes mellitus cohort: new pregnancy screening criteria and utility of HbA1c. Diabetes Care . 2016;39:50–52. doi:10.2337/dc15-1001 26109503
- Steele AM , Shields BM , Shepherd M , Ellard S , Colclough K , Hattersley AT . Microvascular complication risk in patients with 50 years of moderate hyperglycemia: are target ranges for glycemia control appropriate? Abstract A77. Diabet Med . 2011;28(S1):2. doi:10.1111/j.1464-5491.2011.03281.x 21166840
- Velho G , Blanche H , Vaxillaire M , et al. I dentification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia . 1997;40:217–224. doi:10.1007/s001250050666 9049484
- Dukes ID , Sreenan S , Roe M , et al. Defective pancreatic beta-cell glycolytic signaling in hepatocyte nuclear factor 1alpha-deficit mouse. J Biol Chem . 1998;273:24457–24464. doi:10.1074/jbc.273.38.24457 9733737
- Pontoglio M , Sreenan S , Roe M , et al. Defective insulin secretion in hepatocyte nuclear factor 1alpha-deficit mouse. J Clin Invest . 1998;101:2215–2222. doi:10.1172/JCI2548 9593777
- Colclough K , Bellanne-Chantelot C , Saint-Martin C , Flanagan SE , Ellard S . Mutations in the genes encoding the hepatocyte nuclear factor 1alpha and 4alpha in maturity-onset diabetes in the young and hyperinsulinemic hypoglycemia. Hum Mutat . 2013;34:669–685. doi:10.1002/humu.22279 23348805
- Ellard S , Colclough K . Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1alpha (HNF1A) and 4alpha (HNF4A) in maturity-onset diabetes in the young. Hum Mutat . 2006;27:854–869. doi:10.1002/humu.20357 16917892
- Frayling TM , Bulamn MP , Ellard S , et al. Mutations in the hepatocyte nuclear factor-1alpha gene are a common cause of maturity-onset diabetes of the young in the U.K. Diabetes . 1997;46:720–725. doi:10.2337/diab.46.4.720 9075818
- Steele AM , Shields BM , Shepherd M , Ellard S , Hattersley AT , Pearson ER . Increased all-cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabet Med . 2010;27:157–161. doi:10.1111/j.1464-5491.2009.02913.x 20546258
- Pontoglio M , Prie D , Cheret C , et al. HNF1alpha controls renal glucose reabsorption in mouse and man. EMBO Rep . 2000;1:359–365. doi:10.1093/embo-reports/kvd071 11269503
- Pearson ER , Boj SF , Steele AM , et al. Macrosomia and hyperinsulinaemic hypoglycemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med . 2007;4:e118. doi:10.1371/journal.pmed.0040118 17407387
- Stoffel M , Duncan SA . The maturity-onset diabetes of the young (MODY) transcription factor HNF4alpha regulates expression of genes required for glucose transport and metabolism. Proc Natl Acad Sci U S A . 1997;94:13209–13214. doi:10.1073/pnas.94.24.13209 9371825
- Pearson ER , Pruhova S , Tack CJ , et al. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large Europea collection. Diabeteologia . 2005;48:878–885. doi:10.1007/s00125-005-1738-y
- Lehto M , Bitzen PO , Isoma B , et al. Mutations in the HNF-4alpha gene affects insulin secretion and triglyceride metabolism. Diabetes . 1999;48:423–425. doi:10.2337/diabetes.48.2.423 10334325
- Barbacci E , Reber M , Ott MO , Breilat C , Huetz F , Cereghini S . Variant hepatocyte nuclear factor 1 is required for visceral endoderm specification. Development . 1999;126:4795–4805.10518496
- Edghill EL , Bingham C , Ellard S , Hattersley AT . Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet . 2006;43:84–90. doi:10.1136/jmg.2005.032854 15930087
- Bingham C , Ellard S , Allen L , et al. Abnormal nephron development associated with a frameshift mutation in the transcription factor hepatocyte nuclear factor-1beta. Kidney Int . 2000;57:898–907. doi:10.1046/j.1523-1755.2000.057003898.x 10720943
- Bingham C , Bulman MP , Ellard S , et al. Mutation in the transcription factor hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet . 2001;68:219–224. doi:10.1086/316945 11085914
- Bellanne-Chantelot C , Chauveau D , Gautier JF , et al. Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med . 2004;140:510–517.15068978
- Edghill EL , Bingham C , Singerland AS , et al. Hepatocyte nuclear factor-1beta mutations cause neonatal diabetes and intrauterine growth reduction: support for a critical role of HNF-1beta in human pancreatic development. Diabet Med . 2006;23:1301–1306. doi:10.1111/j.1464-5491.2006.01999.x 17116179
- Stoffers DA , Thomas MK , Habener JF . Homeodomain protein IDX-1: a master regulator of pancreas development and insulin gene expression. Trends Endocrinol Metab . 1997;8:145–151. doi:10.1016/S1043-2760(97)00008-8 18406800
- Schwitzgebel VM , Mamin A , Brun T , et al. Agenesis of human pancreas due to decreased half-life of insulin promoter factor 1. J Clin Endocrinol Metab . 2003;88:4398–4406. doi:10.1210/jc.2003-030046 12970316
- Malecki MT , Jhala US , Antonellis A , et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet . 1999;23:323–328. doi:10.1038/70539 10545951
- Gonsorcikova L , Pruhova S , Cinek O , et al. Autosomal inheritance of diabetes in two families characterized by obesity and a novel H241Q mutation in NEUROD1. Pediatr Diabetes . 2008;9(4 Pt 2):367–372. doi:10.1111/j.1399-5448.2008.00379.x 18331410
- Rubio-Cabezas O , Minton JA , Kantor I , Williams D , Ellard S , Hattersley AT . Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes . 2010;59:2326–2331. doi:10.2337/db10-0011 20573748
- Shields BM , McDonald TJ , Ellard S , Campbell MJ , Hyde C , Hattersley AT . The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia . 2012;55:1265–1272. doi:10.1007/s00125-011-2418-8 22218698
- DiabetesGenes. MODY Probability Calculator. Available from: http://www.diabetesgenes.org/content/mody-probability-calcualtor. Accessed June 27, 2019.
- Owen KR , Thanabalasingham G , James TJ , et al. Assessment of high-sensitivity C-reactive protein levels as diagnostic discriminator of maturity-onset diabetes of the young due to HNF1A mutations. Diabetes Care . 2010;33:1919–1924. doi:10.2337/dc10-0288 20724646
- McDonald TJ , Shields BM , Lawry J , et al. High-sensitivity CRP discriminates HNF1A-MODY from other subtypes of diabetes. Diabetes Care . 2011;34:1860–1862. doi:10.2337/dc11-0323 21700917
- Thanabalasingham G , Shah N , Vaxillaire M , et al. A large multi-centre European study validates high-sensitivity C-reactive protein (hsCRP) as a clinical biomarker for the diagnosis of diabetes subtypes. Diabetologia . 2011;54:2801–2810. doi:10.1007/s00125-010-2040-1 21814873
- Bonner C , Nyhan KC , Bacon S , et al. Identification of circulating microRNAs in HNF1A-MODY carriers. Diabetologia . 2013;56:1743–1751. doi:10.1007/s00125-013-2939-4 23674172
- Bakay M , Pandey R , Hakonarson H . Genes involved in type 1 daiabets: an update. Genes . 2013;4:499–521. doi:10.3390/genes4030499 24705215
- Yang Y , Chan L . Monogenic diabetes: what it teaches us on the common forms of type 1 and type 2 diabetes. Endocr Rev . 2016;37:190–222. doi:10.1210/er.2015-1116 27035557
- Flannick J , Ber NL , Bick AG , et al. Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes. Nat Genet . 2013;45:1380–1385. doi:10.1038/ng.2794 24097065
- Thomsen SK , Gloyn AL . The pancreatic β cell: recent insights from human genetics. Trends Endocrinol Metab . 2014;25:425–434. doi:10.1016/j.tem.2014.05.001 24986330
- Kawakita R , Hosokawa Y , Fujimaru R , et al. Molecular and clinical characterization of glucokinase maturity-onset diabetes of the young (GCK-MODY) in Japanese patients. Diabet Med . 2014;31:1357–1362. doi:10.1111/dme.12487 24804978
- Yoon KH , Lee JH , Kim JW , et al. Epidemic obesity and type 2 diabetes in Asia. Lancet . 2006;368:1681–1688. doi:10.1016/S0140-6736(06)69703-1 17098087
- Chan JCN , Malik V , Jin WP , et al. Diabetes in Asia. Epidemiology, risk factors and pathology. Jama . 2009;301:2129–2140. doi:10.1001/jama.2009.726 19470990
- Tfayli H , Bacha F , Gungor N , Arslanian A . Phenotypic type 2 diabetes in obese youth; insulin sensitivity and secretion in islet-cell antibody negative vs. antibody positive patients. Diabetes . 2009;58:738–744. doi:10.2337/db08-1372 19073767
- Pearson RB , Starkey BJ , Poweli RJ , Gribble FM , Clark PM , Hattersley AT . Genetic cause of hyperglycemia and response to treatment in diabetes. Lancet . 2003;362:1275–1281. doi:10.1016/S0140-6736(03)14571-0 14575972
- Fajans SS , Brown MB . Administration of sulfonylureas can increase glucose-induced insulin secretion for decades in patients with maturity-onset diabetes of the young. Diabetes Care . 1993;16:1254–1261. doi:10.2337/diacare.16.9.1254 8404429
- Shepherd M , Shields B , Ellard S , Rubio-Cabezas O , Hattersley AT . A genetic diagnosis of HNF-1A diabetes alters treatment and improves glycemic control in the majority of insulin-treated patients. Diabet Med . 2009;26:437–441. doi:10.1111/j.1464-5491.2009.02690.x 19388975
- Raile K , Schober K , Konrad K , et al. Treatment of young patients with HNF1A mutations (HNF1A-MODY). Diabet Med . 2015;32:526–530. doi:10.1111/dme.12662 25483937
- Campbell PK . Glimepiride: role of a new sulfonylurea in the treatment of type 2 diabetes mellitus. Ann Pharmacother . 1998;32:1044–1052. doi:10.1345/aph.17360 9793597
- Urakami T . New insights into the pharmacological treatment of pediatric patients with type 2 diabetes. Clin Pediatr Endocrinol . 2018;27:1–8. doi:10.1297/cpe.27.1 29403151
- Østoft SH , Babber JI , Hausen T , et al. Glucose-lowering effect and low risk of hypoglycemia in patients with maturity-onset diabetes of the young when treated with a GLP-1 receptor agonist: a double blind, randomized, crossover trial. Diabetes Care . 2014;37:1797–1805. doi:10.2337/dc13-3007 24929431
- Urakami T , Habu M , Okuno M , Suzuki J , Takahashi S , Yorifuji T . Three years of liraglutide treatment offers continuously optimal glycemic control in a pediatric patient with maturity-onset diabetes of the young type 3. J Pediatr Endocrinol Metab . 2015;28:327–331. doi:10.1515/jpem-2014-0211 25332292
- Tonouchi R , Mine Y , Aoki M , Okuno M , Suzuki J , Urakami T . Efficacy and safety of alogliptin in a pediatric patient with maturity-onset diabetes of the young type 1. Clin Pediatr Endocrinol . 2017;26:183–188. doi:10.1297/cpe.26.183 28804210
- DeFronzo RA , Fleck PR , Wilson CA , Mekki Q . Efficacy and safety of the dipeptidyl peptidase-4 inhibitor alogliptin in patients with type 2 diabetes and inadequate glycemic control: a randomized, double-blind, placebo-controlled study. Diabetes Care . 2008;31(12):2315–2317. doi:10.2337/dc08-1035 18809631
- Pratley RE , Kipnes MS , Fleck PR , Wilson C , Mekki Q . Efficacy and safety of the dipeptidyl peptidase-4inhibitor alogliptin in patients with type 2 diabetes inadequately controlled by glyburide mono therapy. Diabetes Obes Metab . 2009;11(2):167–176. doi:10.1111/j.1463-1326.2008.01016.x 19125778
- Nauck MA , Ellis GC , Fleck PR , Wilson CA , Mekki Q . Efficacy and safety of adding the dipeptidyl peptidase-4 inhibitor alogliptin to metformin therapy in patients with type 2 diabetes inadequately controlled with metformin monotherapy: a multicenter, randomized, double-blind, placebo-controlled study. Int J Clin Pract . 2009;63:46–55. doi:10.1111/j.1742-1241.2008.01933.x 19125992
- Murphy R , Ellard S , Hattersley AT . Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab . 1974;4:200–213. doi:10.1038/ncpendmet0778
- Zeitler P , Arslanian S , Fu J , et al. ISPAD clinical practice consensus guidelines 2018: type 2 diabetes mellitus in youth. Pediatr Diabetes . 2018;19(Suppl. 27):28–46. doi:10.1111/pedi.12719 29999228
- Gat-Yablonski G , Shalitin S , Phillip M . Maturity onset diabetes of the young-review. Pediatr Endocrinol Rev . 2006;3(Suppl 3):514–520.17551475