
Abstract
Patients with type-2 diabetes mellitus (T2DM) display chronic low-grade inflammation induced by activation of the innate immune system. Toll-like receptor (TLR)4 is a pattern recognition receptor that plays a vital part in activation of the innate immune system. Results from animal and computer-simulation studies have demonstrated that targeting TLR4 to block the TLR4-nuclear factor-kappa B (NF-κB) pathway reduces the inflammatory response and complications associated with T2DM. Therefore, TLR4-targeted therapy has broad prospects. Here, we reviewed the role of TLR4 in inflammation during chronic hyperglycemia in T2DM and its therapeutic prospects.
Keywords:
Introduction
The International Diabetes Federation showed that the number of people aged 20–79 years with diabetes mellitus (DM) worldwide was estimated to be 463 million in 2019. Almost 90% of these people will have type-2 diabetes mellitus (T2DM), and this number may increase to 700 million by 2045. However, 1 in 2 (~232 million) patients do not know that they are suffering from DM, and the number of people at risk for T2DM is ~374 million.Citation1 T2DM has become a major public-health problem worldwide.
T2DM is characterized by impaired insulin resistance and a dysregulated immune response. T2DM patients display chronic initiation of the innate immune system in pancreatic islets, which occurs in insulin-sensitive tissues and at the sites of diabetic complications. Several studies have shown that T2DM is associated significantly with activation of the innate immune system and that, in many aspects, T2DM is an inflammatory disease. Therefore, the proteins associated with components of the innate immune system could be potential targets for studying T2DM and its complications, including diabetic kidney disease (DKD), atherosclerosis and diabetic retinopathy.
Toll-like receptors (TLRs) are receptors that recognize pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs).Citation2 TLR4 has important roles in modulating innate immunity. TLR4 is expressed in macrophages, adipose tissue, pancreatic β-cells, vascular endothelial cells, skeletal muscle, airway epithelia and smooth muscle cells. TLR4 interacts with other endogenous and exogenous substances, such as heat-shock proteins, fibronectin, fibrinogen, free fatty acids (FFAs), and saturated fatty acids. This protein is also a crucial receptor for the lipopolysaccharide (LPS) component of Gram-negative bacteria. Most of the proinflammatory cytokines induced by activated TLR4 are involved in insulin resistance, which is associated significantly with T2DM risk.Citation3 Given this information, the relationship between T2DM and TLR4 will be highlighted in this review.
TLRs Family
The first identification of a Toll receptor was in the embryonic development of Drosophila. Results from subsequent studies showed that the Drosophila Toll receptor had a major role in the antifungal responses of the adult fly.Citation4 Since then, researchers have focused intensively on the role of Toll receptors in the innate immune response of insects and mammals. Consequently, the Toll receptor was identified in mammals, and revealed the important roles of Toll receptors in recognizing microorganisms.
Medzhitov and colleagues first identified that the mammalian homolog of the Drosophila Toll receptor was hToll (now termed TLR4) in 1997.Citation5 Consequently, many studies have suggested that several proteins that are structurally associated with TLR4 are called TLRs. To date, 10 TLRs in humans (TLR1–TLR10) and 12 TLRs in mice (TLR1 to TLR9 and TLR11 to TLR13) have been identified. It has been demonstrated that TLR-mediated recognition of microorganisms is complex. Different TLRs can recognize and bind with the specific patterns of microbial components.Citation6
Structure of TLRs
TLRs are evolutionarily conserved proteins that can detect PAMPs. TLRs are important for the innate immune response and are vital regulators of innate and adaptive immune responses.Citation2,Citation7 As type-I transmembrane glycoproteins, TLRs share three common domains: an intracellular Toll-interleukin 1 receptor (TIR) homology signaling domain; a single transmembrane domain; an extracellular recognition domain.Citation8 Since the first reported crystal structures of unliganded hTLR3 provided atomic detail,Citation9,Citation10 the hybrid long-range radar method has been established to crystallize TLRs.Citation11 Subsequently, eight structures of TLR complexes activated by ligand induction (TLR1–TLR2–triacyl lipopeptide,Citation12 TLR2–TLR6–diacylated lipopeptide,Citation13 TLR3–dsRNA,Citation10,Citation14 TLR4–MD-2–Eritoran (–LPS),Citation11,Citation15 TLR5–flagellin,Citation16 TLR8–imidazoquinoline derivatives,Citation17 TLR9–ssDNA,Citation18 and TLR13–ssRNA)Citation19 have been confirmed based on the hybrid long-range radar method.
Structures of the Intracellular TIR Domains and Signaling Adapters
The TIR domain is a pivotal module that adapts the pathways of the innate immune response. The TIR domain is mediated by TLRs and is contained in the C-terminal regions of all mammalian TLRs. Initiation of downstream signaling requires homo- or heterotypic dimerization of TIR domains. The latter and a five-stranded parallel β-sheet (βA–βE) in the center have a common fold, and there are five α-helices (αA–αE) on both sides.Citation20,Citation21 Including three highly conserved residues (arginine BB3, aspartic acid BB4 and glycine BB8), the BB loop connects helix αB and strand βB, and has a vital role in the dimerization of TIR and/or recruitment of adaptors.Citation21,Citation22 The BB loop can use different conformations to form TIR dimers.Citation22
Structures of Extracellular Domains
The extracellular recognition domains of TLRs belong to the leucine-rich repeat (LRR) family. The domains contain 16–28 LRR modules in the middle of the N-terminal and C-terminal modules.Citation23 Each of the LRR modules includes 20–30 amino acids that contain a mutable part and a conserved “LxxLxLxxN” motif.Citation24,Citation25 Evidence from crystal structures has shown that all LRR modules are horseshoe-like. The concave face and convex surface of the horseshoe shape consist of parallel β strands and short 310 helices and parallel loops, respectively.Citation26 There are two structural transitions in the central β-sheets of TLR4 so the LRR domains of TLR4 can be separated into three subdomains: C-terminal, central, and N-terminal.Citation11
TLR4 Signaling Pathways
The main pathways activated by TLR4 are the myeloid differentiation primary response protein (MyD)88-dependent and -independent pathways. The initiating step of each pathway is TLR4 identification of ligands. The complex combination of LPS and LPS binding protein (LBP) attaches to another protein known as cluster of differentiation (CD)14.Citation27 As a membrane-binding protein on innate immune cells, CD14 can also cycle in the cytoplasm in a soluble form. It has been suggested that the major role of CD14 is to strengthen TLR4 signaling by accelerating the transport of TLR4 to “lipid rafts” on the cellular membrane. Subsequently, the coreceptor MD-2 is recruited to facilitate TLR4 translocation to the cellular membrane.Citation28 Endocytosis of the TLR4/MD-2 complex occurs after translocation. The heterotrimer CD14/TLR4/MD-2 recognizes LPS and then induces the initiation of MyD88-dependent and MyD88-independent pathways. The general pattern of these pathways is shown in .
Figure 1 TLR4 signaling pathway (schematic). After LBP- and CD14-mediated LPS binding or FetA-mediated FFA binding, the TLR4/MD-2 complex recruits MyD88 or TRIF via TIRAP and TRAM.27 This action, in turn, induces (1) the MyD88-dependent pathway.29 MyD88 binds with the IRAK/TRAF6 complex (Complex I) and TAK1/TAB complex (Complex II), subsequently triggering the activation of IKK. IκBα is phosphorylated by activation of IKK and stimulates the translocation of NF-κB into the nucleus, where NF-κB leads to the production of ROS and proinflammatory cytokines. Complex II can also trigger the MAPK pathway and lead to the activation of p38, JNK and ERK1/2. This activation stimulates the translocation of AP-1 into the nucleus, and proinflammatory cytokines are subsequently transcribed. (2) In the MyD88-independent pathway,34 TLR4 is stimulated by LPS and associates with translocating chain-associated membrane protein (TRAM) and TIR-domain-containing adaptor-inducing interferon-β (TRIF). The dimerization of TRAM and TRIF activates the TRAF6-TAK1-IKK pathway. Alternatively, TBK1 kinase can activate interferon regulatory factor (IRF)3 by phosphorylating it. Activated IRF3 can migrate to the nucleus and act as a transcription factor. Interferon (IFN)-1β binds to activated IRF3 and subsequently activates the production proinflammatory cytokines. Furthermore, two potential targeting points blocking TLR4 signal-pathway have been found according to the literature: (a) FP7 blocks the TLR4 signaling pathway by targeting MD2 and CD14; (b) dioscin inhibits the TLR4 signaling pathway by decreasing NF-κB expression.
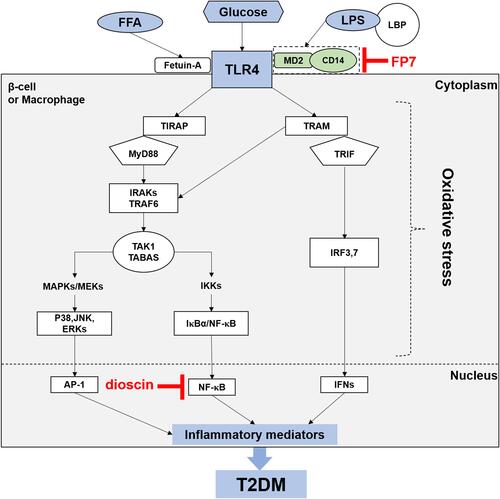
MyD88-Dependent Pathway
The MyD88-dependent pathway was the first to be discovered, and it is the most vital pathway. Evidence from several studies has suggested that MyD88 knockout mice do not respond to TLR ligands.Citation29 Through this mechanism, after stimulation, the binding of TLR4 and MyD88 at the cytoplasmic TIR domainCitation30 results in recruitment of the interleukin-1 receptor-associated kinase (IRAK)4, which is associated with the IL-1 receptor.Citation31 After binding to and activating tumor necrosis factor receptor-associated factor (TRAF)6, activated IRAK4 dissociates from MyD88, resulting in the formation of Complex-1. Subsequently, after dissociating from TLR4, this triple association stimulates the recruitment of numerous proteins, including TGFα-associated kinase (TAK)1 and TAK1 binding protein (TAB) 1–3, which form the core components of Complex-2. These processes result in the activation and initiation of two sub-pathways. In the first sub-pathway, TAK-1 phosphorylation activates the IKB kinase (IKK) complex, which in turn induces the phosphorylation of inhibitor of kappa B (IκB) protein. This action results in proteasomal degradation of IκB and stimulates the translocation of nuclear factor-kappa B (NF-κB) to the nucleus.Citation32 NF-κB then induces the activation of proinflammatory mediators, which are extremely effective in activating inducible nitric oxide synthase (iNOS) and inducible cyclo-oxygenase (COX)-2, and stimulates the release of many major inflammatory cytokines, such as tumor necrosis factor (TNF)-α, IL-1β, and IL-6.Citation33 In the second sub-pathway, TRAF6 induces the activation of the mitogen-activated protein kinases (MAPKs) c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK)1/2, and p38 via phosphorylation, subsequently modulating the activation of some transcription factors, including activator protein (AP)-1.Citation31
MyD88-Independent Pathway
Kawai and colleagues revealed that, following stimulation of TLRs, JNK and NF-κB were produced in MyD88-knockout mice. This result suggests that there are other MyD88-independent pathways.Citation34 After stimulation of TLR4 with its ligand, TLR4 recruits translocating chain-associated membrane protein (TRAM) and TIR-domain-containing adaptor-inducing interferon-β (TRIF). The dimerization of TRAM and TRIF activates the TRAF6-TAK1-IKK pathway. This interaction activates NF-κB and destroys IκBs.Citation35 Alternatively, TBK1 kinase activates interferon regulatory factor (IRF)3 by phosphorylating it. Activated IRF3 can translocate to the nucleus and act as a transcription factor. After binding to activated IRF3, interferon 1β (IFN-1β) subsequently activates the production of reactive oxygen species (ROS) and proinflammatory cytokines. Several endogenous inhibitors can regulate TLR4 overactivation, such as the ubiquitin ligase TRIAD3A, which strengthens ubiquitination and then degrades TLR4.Citation36 In addition, a homolog of the TLR4 protein RP105 can also antagonize TLR4 signaling on the surface of the cell membrane.Citation37 It has been shown that posttranslational modification of TLR4 signaling can be altered by the acetylation of lysine residues.Citation38
Role of TLR4 in T2DM Pathogenesis
Inflammation and T2DM
T2DM has become a growing issue as people become older, especially for those who have genetic and epigenetic predispositions. T2DM is associated mainly with obesity and physical inactivity. In people with predispositions, a defect in insulin secretion can be detected concomitantly with a reduced response to glucose uptake stimulated by insulin in the liver and adipose tissues, and this condition is called insulin resistance.Citation39–Citation42 In individuals, insulin resistance remains relatively constant over time and increases only slightly with age. However, an initial increase in insulin production was followed by a continuous deterioration in the insulin-secretory capacity of pancreatic β-cells, which causes the initiation and development of T2DM. Finally, the increased demand for peripheral insulin cannot be compensated for by insulin secretion. In T2DM, mechanisms associated with defective insulin secretion and responses include glucotoxicity, lipotoxicity, oxidative stress, endoplasmic reticulum (ER) stress, endocannabinoids, alterations in the gut microbiota and the formation of amyloid deposits in pancreatic islets.Citation43–Citation47 Interestingly, these factors are all associated with the inflammatory response.Citation48–Citation56 For example, increased glucose concentrations and LPS activate TLR4, which upregulates expression of caspase‑3 and poly ADP ribose polymerase (PARP), and modifies the ratio of expression of B‑cell lymphoma‑2 (Bcl‑2)/Bcl‑2-associated X protein, which results in enhanced oxidative stress, increased release of ROS and apoptosis.Citation57–Citation59 Armann et al showed that ROS generation in pancreatic islets is linked to the percentage of cells undergoing apoptosis and the functional potency of pancreatic islets in vivo.Citation60 Duprez et al revealed that oxidative stress led to the apoptosis of pancreatic-islet β‑cells and dysfunction of insulin secretion in rats.Citation61 ROS and the byproducts induced by ROS (degraded hyaluronic acid, oxidize phospholipids and activated high-mobility group protein B1) are recognized as DAMPs, which can activate the TLR4 pathway, thereby leading to increased production of ROS and inflammatory mediators. Consequently, there is a “vicious cycle” between TLR activation and ROS generation that contributes to the maintenance of chronic inflammation in pancreatic islets.Citation62 Furthermore, the increased ROS leads to activation of the NOD-, LRR- and pyrin domain-containing (NLRP)3 inflammasome and caspase-1, which promotes the production of mature interleukin-1β (IL-1β).
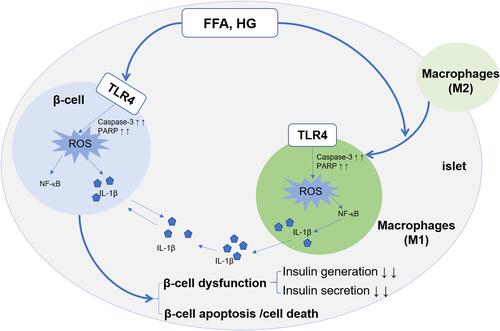
Initially, this inflammation can promote β-cell proliferation and the production of insulin to compensate for insulin resistance.Citation63 Various cytokines and chemokines are induced by IL-1β, such as monocyte chemoattractant protein (MCP)1; also called (CCL2) and TNF, which results in the infiltration of macrophages and other immune cells.Citation64–Citation66 Such infiltration of immune cells is increased due to the vicious cycle of IL-1β autostimulation.Citation67 Therefore, macrophage infiltration into resident pancreatic islets induces their dysfunction. In adipocytes, lipid overload can result in adipocyte death, further triggering an inflammatory response, which forms a vicious cycle. In addition, a review by Kitamura indicated the contribution of Forkhead box protein O1 (FOXO)1 (a multifunctional protein that regulates proliferation, apoptosis, senescence, differentiation, stress resistance, autophagy and metabolism) signaling to the initiation and development of loss of pancreatic β-cells in T2DM.Citation68 The inflammatory microenvironment in pancreatic islets stimulated by FFAs and high glucose includes macrophage polarization, and the conversion of M2 macrophages to M1 macrophages. Moreover, activation of TLR4 in M1 macrophages and β-cells mediates the inflammatory pathway, which leads to β-cell dysfunction (reduced production and secretion of insulin) and apoptosis/death. Eventually, these processes contribute to T2DM development (). Insulin resistance and β-cell dysfunction are hallmarks of T2DM.
Figure 2 Implications of TLR4 in inflammation in pancreatic islets. TLR4 on the surface of β-cells and macrophages is activated by FFA and high glucose, which induces oxidative stress and ROS generation by upregulating expression of caspase-3 and PARP. The cytokine IL-1β is released via the TLR4-NF-κB signaling pathway in pancreatic β-cells and macrophages. Eventually, the generation of IL-1β and ROS induces the pancreatic β-cell dysfunction (decreased production and secretion of insulin) and the apoptosis/cell death of β-cells. Moreover, M2 macrophages from bone marrow or adipose tissue enter pancreatic-islet tissue and translate into M1 macrophages. Activated M1 macrophages increase the level of ROS and IL-1β to aggravate inflammation and further deteriorate dysfunction of β-cell in pancreatic islets.62
Expression of TLR4 in T2DM
T2DM has become the most prevalent metabolic disease worldwide. The effects of T2DM have become the main cause of mortality in humans.Citation69 Moreover, the results obtained from several studies have suggested that proinflammatory cytokines in low-grade inflammation are involved in the pathological processes that lead to T2DM.Citation70,Citation71 TLR4 is expressed not only in various immune-related cells (eg, neurons, oligodendrocytes, astrocytes, microglia and the cerebral endothelium) but also functions in several types of nonimmune cells. Garay-Malpartida et al observed variable levels of TLR4 expression in pancreatic β-cells isolated from adult nondiabetic brain-dead donors. Their results showed that LPS could increase TLR4 expression and impair the viability of pancreatic β-cells, which led to a decrease in the synthesis and secretion of insulin.Citation72 Moreover, activation of TLRs induced by any stimuli in injured tissue plays a key part in the human inflammatory response: TLRs are important mediators of the human inflammatory response because they facilitate the production of inflammatory cytokines. Given the findings mentioned above, Gupta et al measured TLR1-10 expression in neutrophils and monocytes derived from patients with T2DM and healthy participants. They showed that in neutrophils, mRNA expression of most TLRs was enhanced in glycemic control (GC) patients compared with that in normal glucose tolerance (NGT) cases. Similarly, mRNA expression of TLRs was increased in monocytes derived from GC cases.Citation73 The data collected from T2DM patients with good GC were consistent, and studies have suggested that TLR4 expression was enhanced in monocytes isolated from patients with T2DM; the mean duration of such expression was 29 months.Citation74 Moreover, data from other studies indicated that mRNA expression of TLR4 was enhanced in peripheral blood mononuclear cells (PBMCs) derived from patients with T2DMCitation75 (though the mean duration of disease was not stated).Citation76 Mohan et al showed that high glucose could stimulate the generation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in human monocytes, and that TLR4 expression was induced by NADPH oxidase via protein kinase C (PKC)-δ.Citation77 Recently, Sindhu et al reported that expression of TLR10 (an immune marker of metabolic inflammation) was induced by oxidative stress, and that ROS-mediated oxidative stress induced TLR10 expression in monocytic cells, an ER stress response, as well as NF-κB and MAPK signaling pathways.Citation78
Besides macrophagesCitation79 or immunocompetent cells (such as dendritic cells, DCs),Citation80 TLR4 is also expressed in the cardiovascular system, including endothelial cells,Citation79 vascular smooth muscle cells (VSMCs)Citation81 and adventitial fibroblasts.Citation82 Several studies have identified the role of TLR4 in the development and progression of atherosclerotic disease. For instance, TLR4 on adventitial fibroblasts engaged in neointima formation in atherosclerosis and the TLR4 pathway lead to expression of apoptotic molecules in the Fas death pathway.Citation83 Moreover, we should also pay close attention to the expression and role of TLR4 in adipose tissue because it can communicate with multiple organs or tissues through many adipocytokines, which affects many physiologic and pathophysiologic processes. Several studies have shown that adipose tissue in obese people is infiltrated with macrophages; they may participate in the inflammatory process activated in adipose tissue.Citation84–Citation86 Notably, the infiltration of macrophages and expression of inflammation-associated genes in adipose tissue precede insulin resistance.Citation84,Citation85 Hence, infiltrating macrophages play an essential part in inflammation initiation in adipose tissue. Accumulated evidences have suggested that TLR4 has a vital role in inflammation of adipose tissue induced by obesity and systemic glucose and lipid metabolism.Citation87–Citation90 TLR4 is expressed in macrophages more abundantly than in adipocytes, so the chronic inflammatory responses induced by the interaction between macrophages and adipocytes could be mediated via TLR4 in macrophages.Citation91
Role of TLR4 as an Inflammatory Target in T2DM
The innate immune system is activated by TLRs, particularly TLR4, which plays a vital part in chronic, low-grade inflammation. A high-fat diet can lead to an increase in plasma LPS concentrations in T2DM cases and healthy individuals,Citation92,Citation93 whereas LPS induces insulin resistance rapidly in healthy people.Citation94 LPS is derived from a group of Gram-negative bacteriaCitation95 called Enterobacteriaceae (ie, Escherichia coli),Citation96 as well as fungal and viral productsCitation97 that are well-known extracellular pathogens and can induce TLR4 activation. Similarly, an excessive amount of glucose and FFAs leads to increased expression of TLR4 mRNA and protein in patients with untreated T2DM. FFAs induce activation of the inflammatory response and metabolic signaling in insulin resistance, primarily via the TLR4-NF-κB signaling pathway in pancreatic β-cells and macrophages.Citation98 However, TLR4 cannot bind to FFAs directly; thus, the interaction between TLR4 and FFAs is mediated by fetuin A (FetA), which may be regarded as a new therapeutic target.Citation99,Citation100
Therapeutic Prospects of TLR4 in T2DM
Application of TLR4 Antagonists in T2DM
Having shown the effects of TLR4 in T2DM development, one could hypothesize that blockade of TLR4 signaling would inhibit the progression of inflammation in pancreatic islets. Consistent with this hypothesis, Shi et al observed that a lack of TLR4 could improve obesity-induced insulin resistance.Citation98 Moreover, Wang et al established that pretreatment with TLR4-short hairpin (sh)RNA and a TLR4 antibody led to significantly reduced expression of TLR4 compared with that observed upon treatment with LPS. After pretreatment with a TLR4 antibody and TLR4-shRNA, insulin secretion was increased significantly in isolated pancreatic islets in rats.Citation101 This result showed that inhibition of TLR4 expression may protect pancreatic islet cells from the damage induced by ligands.
One study suggested, for the first time, that a TLR4 antagonist improved atherogenesis and vascular inflammation in diabetic apolipoprotein E (ApoE)−/− mice and lowered serum levels of triglyceride and cholesterol in nondiabetic ApoE−/− mice.Citation102 Furthermore, Perrin-Cocon et al examined the activity of FP7 (a small-molecule synthetic TLR4 antagonist) on human monocytes and monocyte-derived DCs in vitro and on mice with influenza-virus infection in vivo. They showed that FP7 inhibited activation of TLR4-mediated monocytes and DCs, thereby avoiding excessive TLR4 signaling in inflammatory disorders.Citation103 FP7 binds to MD-2 and CD14 and inhibits LPS-induced TLR4 signaling.Citation104 Inhibition was TLR4-specific, suggesting a more precise molecular interaction with MD-2.Citation103 Therefore, TLR4 antagonists such as FP7 could be developed as new drugs that target TLR4-related inflammatory diseases, such as T2DM.
Blockade of the TLR4 Signaling Pathway in T2DM Complications
Recently, several studies have revealed that blocking TLR4 expression is a strategy for treating T2DM complications. Diabetic retinopathy (DR), atherosclerosis and DKD are three of the most common complications of DM. Hu et al treated retinal ganglion cells (RGCs) isolated from retinas of Sprague–Dawley rats with a selective inhibitor of TLR4, TAK-242, under a high-glucose environment for 48 h. Subsequently, they showed that treatment with TAK-242 decreased expression of TLR4 downstream signaling molecules and significantly reduced levels of inflammatory cytokines, accompanied by a reduced rate of RGC apoptosis. Hu et al identified the potential effect of TAK-242 for treating the degeneration of primary RGCs in DR.Citation105
Moreover, low-density lipoprotein receptor-deficient (LDLR−/−) mice (which are atherosclerotic and can be induced to develop T2DM with a high-fat diet) were treated with Rhodobacter sphaeroides LPS (Rs-LPS), an established TLR4 antagonist, in a study by Lu et al They found that the number of monocytes and macrophages in atherosclerotic lesions was reduced, and that expression of IL-6 and matrix metallopeptidase-9 in atherosclerotic plaques was inhibited; they also showed that Rs-LPS inhibited atherosclerosis in diabetic LDLR−/− mice and reduced collagen levels in diabetic mice.Citation106
Emerging evidence has identified that the TLR4-NF-κB pathway in the kidney plays a vital part in DKD pathogenesis.Citation107,Citation108 Wu et al suggested that high glucose would increase expression of microRNA (miR)-199a-5p to decrease expression of Klotho, leading to inflammation and fibrosis by activating the TLR4/NF-κB p65/NGAL signaling pathway. In contrast, they showed that initiation of the TLR4/NF-κB p65/NGAL signaling pathway might be attenuated under high glucose conditions by inhibiting miR-199a-5p or adding Klotho exogenously, thereby preventing the progression of inflammation and fibrosis in the kidney.Citation109 Furthermore, Liu et al found that 1.25-dihydroxyvitamin D3 (1,25(OH)2D3) treatment reduced TLR4 expression, inhibited expression of NF-κB and MyD88 in cultured renal tubular epithelial cells, and reduced expression of collagen-associated proteins, resulting in a significant decrease in the inflammatory response in the renal tissues of rats with DKD. Immunohistochemistry results showed that 1.25(OH)2D3 treatment could suppress the immune response by reducing the infiltration of macrophages significantly. Therefore, 1.25(OH)2D3 could downregulate the innate immune TLR4-NF-κB pathway to protect against DKD.Citation110 Recently, Cai et al reported the potential role of dioscin in treating DKD in patients suffering from DM.Citation111
Conclusions and Future Perspectives
TLR4, which is expressed in pancreatic β-cells, adipocytes and macrophages, plays a key part in the activation of chronic low-grade inflammation. This action induces the initiation and progression of T2DM under high glucose concentrations in peripheral blood in obese people. Hence, TLR4 could be an ideal therapeutic target to inhibit the inflammatory pathway and improve T2DM and its complications. Moreover, recent studies have shown that treatments targeting TLR4 could inhibit the initiation and progression of inflammation effectively, thereby eliciting a potential hypoglycemic effect. Ullah et al used a computer-based study and found that curcumin analogs are natural antagonists of TLR4 that could inhibit the TLR4 pathway.Citation112 Lakhia et al reported that miR-214 could attenuate inflammation by inhibiting TLR4 expression.Citation113 Shah et al found that MAL/MyD88-inhibitory peptide 2, an inhibitor, has broad specificity for TLRs and attenuates the activation of TLRs (including TLR4) that is induced by microbial or environmental factors.Citation114 Yang et al demonstrated that D. officinale polysaccharide could inhibit TLR4 expression to achieve potent anti-inflammatory effects.Citation115 Therefore, TLR4 represents a promising therapeutic target in patients with T2DM, and researchers prefer to identify natural anti-inflammatory drugs. However, another important feature is the consequence of blocking the TLR4 pathway, which has a substantial role in initiating the immune response. Zheng et al suggested that TLR4 knockout influenced the growth, development, and reproduction of mice.Citation116 Furthermore, Coenen et al showed no differences in body composition or plasma lipids between MθTLR4+/+ mice (recipient mice with TLR4 expression in bone marrow) and MθTLR4−/− mice (recipient mice with TLR4 knock-down in bone marrow) on any of the diets. Moreover, similar insulin sensitivity between MθTLR4+/+ mice and MθTLR4−/− mice fed a high-fat diet with saturated fatty acids was found.Citation117 Similarly, Eritoran (E5564, TLR4 antagonist) has shown its safety in several studies.Citation118,Citation119 Therefore, further research and understanding of related mechanisms are necessary to explore the potential future clinical importance of these treatments.
Abbreviations
T2DM, type-2 diabetes mellitus; TLR4, Toll-like receptor 4; NF-κB, nuclear factor κB; TLRs, toll-like receptors; PAMPs, pathogen-associated molecular patterns; DAMPs, danger-associated molecular patterns; LPS, lipopolysaccharide; TIR, Toll-interleukin 1 receptor; MD2, myeloid differentiation protein 2; LRR, leucine-rich repeat; MyD88, myeloid differentiation primary response protein 88; LBP, LPS-binding protein; CD14, cluster of differentiation 14; IRAK4, interleukin-1 receptor-associated kinase 4; TRAF6, TNF receptor-associated factor 6; TAK-1, TGFα-associated kinase-1; TAB, TAK1 binding protein; IKK, IκB kinase; IκB, inhibitor of kappa B; iNOS, inducible nitric oxide synthase; AP-1, activator protein-1; TRAM, translocating chain-associated membrane protein; TRIF, TIR-domain-containing adaptor-inducing interferon-β; IRF3, interferon regulatory factor 3; IFN-1β, interferon 1β; ROS, reactive oxygen species; ER, endoplasmic reticulum; NLRP3, NOD-, LRR- and pyrin domain-containing 3; IL-1β, interleukin-1β; MCP1, monocyte chemoattractant protein 1; TNF, tumor necrosis factor; GC, glycemic control; NGT, normal glucose tolerance; PBMCs, peripheral blood mononuclear cells; MAPK, mitogen-activated protein kinase; FFAs, free fatty acids; DCs, dendritic cells; DR, diabetic retinopathy; DKD, diabetic kidney disease; RGCs, retinal ganglion cells; Rs-LPS, Rhodobacter sphaeroides LPS; NGAL, Neutrophil Gelatinase-Associated Lipocalin; MIP2, MAL/MyD88-inhibitory peptide 2; DOP, D. officinale polysaccharide; MθTLR4+/+, recipient mice with TLR4 expression in bone marrow; MθTLR4−/−, recipient mice with TLR4 knockdown in bone marrow; PARP, poly ADP ribose polymerase.
Acknowledgments
All authors made substantial contributions to conception and design, acquisition of data, or analyses and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to this journal; gave approval of the final version to be published; and agree to be accountable for all aspects of the work.
This work was supported by the Natural Science Foundation of China (81870552, 81400790, 81872096, 81571385, 91849118, and 91849132), National Key R&D Program of China (2018YFC2000400), Beijing Hospital Doctoral Scientific Research Foundation (BJ-2018-024), Beijing Hospital Nova Project (BJ-2018-139) and Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2018RC330003).
Disclosure
The authors report no conflicts of interest in this work.
References
- IDF Diabetes atlas, 9th edn. International Diabetes Federation. Available from: https://diabetesatlas.org/en/. Accessed June 4, 2020.
- West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. 2006;22:409–437. doi:10.1146/annurev.cellbio.21.122303.115827
- Garibotto G, Carta A, Picciotto D, Viazzi F, Verzola D. Toll-like receptor-4 signaling mediates inflammation and tissue injury in diabetic nephropathy. J Nephrol. 2017;30(6):719–727. doi:10.1007/s40620-017-0432-8
- Imler JL, Hoffmann JA. Toll receptors in innate immunity. Trends Cell Biol. 2001;11(7):304–311. doi:10.1016/s0962-8924(01)02004-9
- Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388(6640):394–397. doi:10.1038/41131
- Takeda K, Akira S. Toll-like receptors. Curr Protoc Immunol. 2015;109:1–10. doi:10.1002/0471142735.im1412s109
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi:10.1038/nri1391
- Brodsky I, Medzhitov R. Two modes of ligand recognition by TLRs. Cell. 2007;130(6):979–981. doi:10.1016/j.cell.2007.09.009
- Bell JK, Botos I, Hall PR, et al. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci U S A. 2005;102(31):10976–10980. doi:10.1073/pnas.0505077102
- Choe J, Kelker MS, Wilson IA. Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science. 2005;309(5734):581–585. doi:10.1126/science.1115253
- Kim HM, Park BS, Kim JI, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130(5):906–917. doi:10.1016/j.cell.2007.08.002
- Jin MS, Kim SE, Heo JY, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130(6):1071–1082. doi:10.1016/j.cell.2007.09.008
- Kang JY, Nan X, Jin MS, et al. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity. 2009;31(6):873–884. doi:10.1016/j.immuni.2009.09.018
- Liu L, Botos I, Wang Y, et al. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320(5874):379–381. doi:10.1126/science.1155406
- Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458(7242):1191–1195. doi:10.1038/nature07830
- Yoon SI, Kurnasov O, Natarajan V, et al. Structural basis of TLR5-flagellin recognition and signaling. Science. 2012;335(6070):859–864. doi:10.1126/science.1215584
- Tanji H, Ohto U, Shibata T, Miyake K, Shimizu T. Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science. 2013;339(6126):1426–1429. doi:10.1126/science.1229159
- Ohto U, Shibata T, Tanji H, et al. Structural basis of CpG and inhibitory DNA recognition by Toll-like receptor 9. Nature. 2015;520(7549):702–705. doi:10.1038/nature14138
- Song W, Wang J, Han Z, et al. Structural basis for specific recognition of single-stranded RNA by Toll-like receptor 13. Nat Struct Mol Biol. 2015;22(10):782–787. doi:10.1038/nsmb.3080
- Nyman T, Stenmark P, Flodin S, Johansson I, Hammarström M, Nordlund P. The crystal structure of the human toll-like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J Biol Chem. 2008;283(18):11861–11865. doi:10.1074/jbc.C800001200
- Xu Y, Tao X, Shen B, et al. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 2000;408(6808):111–115. doi:10.1038/35040600
- Tao X, Xu Y, Zheng Y, Beg AA, Tong L. An extensively associated dimer in the structure of the C713S mutant of the TIR domain of human TLR2. Biochem Biophys Res Commun. 2002;299(2):216–221. doi:10.1016/s0006-291x(02)02581-0
- Matsushima N, Tanaka T, Enkhbayar P, et al. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genom. 2007;8:124. doi:10.1186/1471-2164-8-124
- Kajava AV. Structural diversity of leucine-rich repeat proteins. J Mol Biol. 1998;277(3):519–527. doi:10.1006/jmbi.1998.1643
- Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol. 2001;11(6):725–732. doi:10.1016/s0959-440x(01)00266-4
- Gay NJ, Gangloff M. Structure and function of Toll receptors and their ligands. Annu Rev Biochem. 2007;76:141–165. doi:10.1146/annurev.biochem.76.060305.151318
- Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi:10.1126/science.282.5396.2085
- Shimazu R, Akashi S, Ogata H, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189(11):1777–1782. doi:10.1084/jem.189.11.1777
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11(1):115–122. doi:10.1016/s1074-7613(00)80086-2
- Laird MH, Rhee SH, Perkins DJ, et al. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J Leukoc Biol. 2009;85(6):966–977. doi:10.1189/jlb.1208763
- Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16(1):3–9. doi:10.1016/j.smim.2003.10.003
- Delaney JR, Mlodzik M. TGF-beta activated kinase-1: new insights into the diverse roles of TAK1 in development and immunity. Cell Cycle (Georgetown, Tex). 2006;5(24):2852–2855. doi:10.4161/cc.5.24.3558
- Saha RN, Pahan K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid Redox Signal. 2006;8(5–6):929–947. doi:10.1089/ars.2006.8.929
- Kawai T, Takeuchi O, Fujita T, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167(10):5887–5894. doi:10.4049/jimmunol.167.10.5887
- Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Rev. 2009;59(2):278–292. doi:10.1016/j.brainresrev.2008.09.001
- Chuang TH, Ulevitch RJ. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol. 2004;5(5):495–502. doi:10.1038/ni1066
- Divanovic S, Trompette A, Atabani SF, et al. Inhibition of TLR-4/MD-2 signaling by RP105/MD-1. J Endotoxin Res. 2005;11(6):363–368. doi:10.1179/096805105X67300
- Hu X, Yu Y, Eugene Chin Y, Xia Q. The role of acetylation in TLR4-mediated innate immune responses. Immunol Cell Biol. 2013;91(10):611–614. doi:10.1038/icb.2013.56
- Cerasi E. Insulin deficiency and insulin resistance in the pathogenesis of NIDDM: is a divorce possible? Diabetologia. 1995;38(8):992–997. doi:10.1007/BF00400591
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi:10.1038/nature05482
- Bonner-Weir S. Islet growth and development in the adult. J Mol Endocrinol. 2000;24(3):297–302. doi:10.1677/jme.0.0240297
- Kahn BB. Type 2 diabetes: when insulin secretion fails to compensate for insulin resistance. Cell. 1998;92(5):593–596. doi:10.1016/S0092-8674(00)81125-3
- Robertson RP, Harmon J, Tran POT, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53(Suppl 1):S119–S124. doi:10.2337/diabetes.53.2007.S119
- Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 2004;53(Suppl 3):S16–S21. doi:10.2337/diabetes.53.suppl_3.S16
- Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest. 2006;116(7):1802–1812. doi:10.1172/JCI29103
- Hull RL, Westermark GT, Westermark P, Kahn SE. Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab. 2004;89(8):3629–3643. doi:10.1210/jc.2004-0405
- Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes. 2002;51(Suppl 3):S455–S461. doi:10.2337/diabetes.51.2007.S455
- Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8(12):923–934. doi:10.1038/nri2449
- Donath MY, Størling J, Maedler K, Mandrup-Poulsen T. Inflammatory mediators and islet beta-cell failure: a link between type 1 and type 2 diabetes. J Mol Med. 2003;81(8):455–470. doi:10.1007/s00109-003-0450-y
- Ehses JA, Ellingsgaard H, Böni-Schnetzler M, Donath MY. Pancreatic islet inflammation in type 2 diabetes: from alpha and beta cell compensation to dysfunction. Arch Physiol Biochem. 2009;115(4):240–247. doi:10.1080/13813450903025879
- Westwell-Roper CY, Ehses JA, Verchere CB. Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1β production and β-cell dysfunction. Diabetes. 2014;63(5):1698–1711. doi:10.2337/db13-0863
- Westwell-Roper C, Dai DL, Soukhatcheva G, et al. IL-1 blockade attenuates islet amyloid polypeptide-induced proinflammatory cytokine release and pancreatic islet graft dysfunction. J Immunol 2011;187(5):2755–2765. doi:10.4049/jimmunol.1002854
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010;11(10):897–904. doi:10.1038/ni.1935
- Jourdan T, Godlewski G, Cinar R, et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med. 2013;19(9):1132–1140. doi:10.1038/nm.3265
- Oslowski CM, Hara T, O’Sullivan-Murphy B, et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012;16(2):265–273. doi:10.1016/j.cmet.2012.07.005
- Lerner AG, Upton J-P, Praveen PVK, et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16(2):250–264. doi:10.1016/j.cmet.2012.07.007
- Ling PR, Smith RJ, Bistrian BR. Acute effects of hyperglycemia and hyperinsulinemia on hepatic oxidative stress and the systemic inflammatory response in rats. Crit Care Med. 2007;35(2):555–560. doi:10.1097/01.Ccm.0000253310.02180.C2
- Apostolova N, Garcia-Bou R, Hernandez-Mijares A, Herance R, Rocha M, Victor VM. Mitochondrial antioxidants alleviate oxidative and nitrosative stress in a cellular model of sepsis. Pharm Res. 2011;28(11):2910–2919. doi:10.1007/s11095-011-0528-0
- Lu H, Koshkin V, Allister EM, Gyulkhandanyan AV, Wheeler MB. Molecular and metabolic evidence for mitochondrial defects associated with beta-cell dysfunction in a mouse model of type 2 diabetes. Diabetes. 2010;59(2):448–459. doi:10.2337/db09-0129
- Armann B, Hanson MS, Hatch E, Steffen A, Fernandez LA. Quantification of basal and stimulated ROS levels as predictors of islet potency and function. Am J Transpl. 2007;7(1):38–47. doi:10.1111/j.1600-6143.2006.01577.x
- Duprez J, Roma LP, Close AF, Jonas JC, Nadal A. Protective antioxidant and antiapoptotic effects of ZnCl2 in rat pancreatic islets cultured in low and high glucose concentrations. PLoS One. 2012;7(10):e46831. doi:10.1371/journal.pone.0046831
- Lucas K, Maes M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol Neurobiol. 2013;48(1):190–204. doi:10.1007/s12035-013-8425-7
- Maedler K, Fontana A, Ris F. FLIP switches Fas-mediated glucose signaling in human pancreatic β cells from apoptosis to cell replication. Proc Natl Acad Sci USA. 2002;99:8236–8241. doi:10.1073/pnas.122686299
- Ehses JA, Perren A, Eppler E, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56(9):2356–2370. doi:10.2337/db06-1650
- Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. Islet-associated macrophages in type 2 diabetes. Diabetologia. 2009;52(8):1686–1688. doi:10.1007/s00125-009-1410-z
- Butcher MJ, Hallinger D, Garcia E, et al. Association of proinflammatory cytokines and islet resident leucocytes with islet dysfunction in type 2 diabetes. Diabetologia. 2014;57(3):491–501. doi:10.1007/s00125-013-3116-5
- Böni-Schnetzler M, Thorne J, Parnaud G, et al. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J Clin Endocrinol Metab. 2008;93(10):4065–4074. doi:10.1210/jc.2008-0396
- Kitamura T. The role of FOXO1 in beta-cell failure and type 2 diabetes mellitus. Nat Rev Endocrinol. 2013;9(10):615–623. doi:10.1038/nrendo.2013.157
- Guest CB, Park MJ, Johnson DR, Freund GG. The implication of proinflammatory cytokines in type 2 diabetes. Front Biosci. 2008;13:5187–5194. doi:10.2741/3074
- Festa A, D’Agostino R, Howard G, Mykkänen L, Tracy RP, Haffner SM. Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation. 2000;102(1):42–47. doi:10.1161/01.cir.102.1.42
- Navarro JF, Mora C. Diabetes, inflammation, proinflammatory cytokines, and diabetic nephropathy. ScientificWorldJournal. 2006;6:908–917. doi:10.1100/tsw.2006.179
- Garay-Malpartida HM, Mourao RF, Mantovani M, Santos IA, Sogayar MC, Goldberg AC. Toll-like receptor 4 (TLR4) expression in human and murine pancreatic beta-cells affects cell viability and insulin homeostasis. BMC Immunol. 2011;12:18. doi:10.1186/1471-2172-12-18
- Gupta S, Maratha A, Siednienko J, et al. Analysis of inflammatory cytokine and TLR expression levels in Type 2 Diabetes with complications. Sci Rep. 2017;7(1):7633. doi:10.1038/s41598-017-07230-8
- Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care. 2010;33(4):861–868. doi:10.2337/dc09-1799
- Sepehri Z, Kiani Z, Nasiri AA, et al. Human Toll like receptor 4 gene expression of PBMCs in diabetes mellitus type 2 patients. Cell Mol Biol (Noisy-Le-Grand). 2015;61(3):92–95.
- Ahmad R, Al-Mass A, Atizado V, et al. Elevated expression of the toll like receptors 2 and 4 in obese individuals: its significance for obesity-induced inflammation. J Inflamm (Lond). 2012;9(1):48. doi:10.1186/1476-9255-9-48
- Dasu MR, Devaraj S, Zhao L, Hwang DH, Jialal I. High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes. 2008;57(11):3090–3098. doi:10.2337/db08-0564
- Sindhu S, Akhter N, Kochumon S, et al. Increased expression of the innate immune receptor TLR10 in obesity and type-2 diabetes: association with ROS-mediated oxidative stress. Cell Physiol Biochem. 2018;45(2):572–590. doi:10.1159/000487034
- Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002;105(10):1158–1161. doi:10.1161/circ.105.10.1158
- Bobryshev YV, Lord RS. S-100 positive cells in human arterial intima and in atherosclerotic lesions. Cardiovasc Res. 1995;29(5):689–696. doi:10.1016/S0008-6363(96)88642-1
- Stoll LL, Denning GM, Li WG, et al. Regulation of endotoxin-induced proinflammatory activation in human coronary artery cells: expression of functional membrane-bound CD14 by human coronary artery smooth muscle cells. J Immunol. 2004;173(2):1336–1343. doi:10.4049/jimmunol.173.2.1336
- Vink A, Schoneveld AH, van der Meer JJ, et al. In vivo evidence for a role of toll-like receptor 4 in the development of intimal lesions. Circulation. 2002;106(15):1985–1990. doi:10.1161/01.cir.0000032146.75113.ee
- Li H, Sun B. Toll-like receptor 4 in atherosclerosis. J Cell Mol Med. 2007;11(1):88–95. doi:10.1111/j.1582-4934.2007.00011.x
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi:10.1172/jci19246
- Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–1830. doi:10.1172/jci19451
- Clément K, Viguerie N, Poitou C, et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J. 2004;18(14):1657–1669. doi:10.1096/fj.04-2204com
- Suganami T, Mieda T, Itoh M, Shimoda Y, Kamei Y, Ogawa Y. Attenuation of obesity-induced adipose tissue inflammation in C3H/HeJ mice carrying a Toll-like receptor 4 mutation. Biochem Biophys Res Commun. 2007;354(1):45–49. doi:10.1016/j.bbrc.2006.12.190
- Shi L, Kishore R, McMullen MR, Nagy LE. Lipopolysaccharide stimulation of ERK1/2 increases TNF-alpha production via Egr-1. Am J Physiol Cell Physiol. 2002;282(6):C1205–C1211. doi:10.1152/ajpcell.00511.2001
- Poggi M, Bastelica D, Gual P, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia. 2007;50(6):1267–1276. doi:10.1007/s00125-007-0654-8
- Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56(8):1986–1998. doi:10.2337/db06-1595
- Saberi M, Woods NB, de Luca C, et al. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009;10(5):419–429. doi:10.1016/j.cmet.2009.09.006
- Ghanim H, Abuaysheh S, Sia CL, et al. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care. 2009;32(12):2281–2287. doi:10.2337/dc09-0979
- Harte AL, Varma MC, Tripathi G, et al. High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care. 2012;35(2):375–382. doi:10.2337/dc11-1593
- van der Crabben SN, Blümer RME, Stegenga ME, et al. Early endotoxemia increases peripheral and hepatic insulin sensitivity in healthy humans. J Clin Endocrinol Metab. 2009;94(2):463–468. doi:10.1210/jc.2008-0761
- Takeuchi O, Akira S. Toll-like receptors; their physiological role and signal transduction system. Int Immunopharmacol. 2001;1(4):625–635. doi:10.1016/S1567-5769(01)00010-8
- Garate I, Garcia-Bueno B, Madrigal JL, et al. Stress-induced neuroinflammation: role of the Toll-like receptor-4 pathway. Biol Psychiatry. 2013;73(1):32–43. doi:10.1016/j.biopsych.2012.07.005
- Schaefer TM, Desouza K, Fahey JV, Beagley KW, Wira CR. Toll-like receptor (TLR) expression and TLR-mediated cytokine/chemokine production by human uterine epithelial cells. Immunology. 2004;112(3):428–436. doi:10.1111/j.1365-2567.2004.01898.x
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi:10.1172/JCI28898
- Shen X, Yang L, Yan S, et al. Fetuin A promotes lipotoxicity in beta cells through the TLR4 signaling pathway and the role of pioglitazone in anti-lipotoxicity. Mol Cell Endocrinol. 2015;412:1–11. doi:10.1016/j.mce.2015.05.014
- Pal D, Dasgupta S, Kundu R, et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18(8):1279–1285. doi:10.1038/nm.2851
- Wang X, Ge QM, Bian F, Dong Y, Huang CM. Inhibition of TLR4 protects rat islets against lipopolysaccharide-induced dysfunction. Mol Med Rep. 2017;15(2):805–812. doi:10.3892/mmr.2016.6097
- Lu Z, Zhang X, Li Y, Jin J, Huang Y. TLR4 antagonist reduces early-stage atherosclerosis in diabetic apolipoprotein E-deficient mice. J Endocrinol. 2013;216(1):61–71. doi:10.1530/JOE-12-0338
- Perrin-Cocon L, Aublin-Gex A, Sestito SE, et al. TLR4 antagonist FP7 inhibits LPS-induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection. Sci Rep. 2017;7:40791. doi:10.1038/srep40791
- Cighetti R, Ciaramelli C, Sestito SE, et al. Modulation of CD14 and TLR4.MD-2 activities by a synthetic lipid A mimetic. Chembiochem. 2014;15(2):250–258. doi:10.1002/cbic.201300588
- Hu L, Yang H, Ai M, Jiang S. Inhibition of TLR4 alleviates the inflammation and apoptosis of retinal ganglion cells in high glucose. Graefes Arch Clin Exp Ophthalmol. 2017;255(11):2199–2210. doi:10.1007/s00417-017-3772-0
- Lu Z, Zhang X, Li Y, Lopes-Virella MF, Huang Y. TLR4 antagonist attenuates atherogenesis in LDL receptor-deficient mice with diet-induced type 2 diabetes. Immunobiology. 2015;220(11):1246–1254. doi:10.1016/j.imbio.2015.06.016
- Foresto-Neto O, Albino AH, Arias SCA, et al. NF-kappaB system is chronically activated and promotes glomerular injury in experimental type 1 diabetic kidney disease. Front Physiol. 2020;11:84. doi:10.3389/fphys.2020.00084
- Yuan S, Liu X, Zhu X, et al. The role of TLR4 on PGC-1alpha-mediated oxidative stress in tubular cell in diabetic kidney disease. Oxid Med Cell Longev. 2018;2018:6296802. doi:10.1155/2018/6296802
- Wu C, Lv C, Chen F, Ma X, Shao Y, Wang Q. The function of miR-199a-5p/Klotho regulating TLR4/NF-kappaB p65/NGAL pathways in rat mesangial cells cultured with high glucose and the mechanism. Mol Cell Endocrinol. 2015;417:84–93. doi:10.1016/j.mce.2015.09.024
- Liu P, Li F, Xu X, et al. 1,25(OH)2D3 provides protection against diabetic kidney disease by downregulating the TLR4-MyD88-NF-kappaB pathway. Exp Mol Pathol. 2020;114:104434. doi:10.1016/j.yexmp.2020.104434
- Cai S, Chen J, Li Y. Dioscin protects against diabetic nephropathy by inhibiting renal inflammation through TLR4/NF-κB pathway in mice. Immunobiology. 2020;225(3):151941. doi:10.1016/j.imbio.2020.151941
- Ullah MA, Johora FT, Sarkar B, Araf Y, Rahman MH. Curcumin analogs as the inhibitors of TLR4 pathway in inflammation and their drug like potentialities: a computer-based study. J Recept Signal Transduct Res. 2020;1–15. doi:10.1080/10799893.2020.1742741
- Lakhia R, Yheskel M, Flaten A, et al. Interstitial microRNA miR-214 attenuates inflammation and polycystic kidney disease progression. JCI Insight. 2020;5(7). doi:10.1172/jci.insight.133785
- Shah M, Kim GY, Achek A, et al. The alphaC helix of TIRAP holds therapeutic potential in TLR-mediated autoimmune diseases. Biomaterials. 2020;245:119974. doi:10.1016/j.biomaterials.2020.119974
- Yang K, Zhan L, Lu T, et al. Dendrobium officinale polysaccharides protected against ethanol-induced acute liver injury in vivo and in vitro via the TLR4/NF-kappaB signaling pathway. Cytokine. 2020;130:155058. doi:10.1016/j.cyto.2020.155058
- Zheng Y, Wang JH, Liu LL, Li GY, Peng Y, Zhao TJ. Experimental study on growth and reproduction of Tlr4 knockout mice. Chin J Compar Med. 2018;28(12):41–43+50. doi:10.3969/j.issn.1671-7856.2018.12.007
- Coenen KR, Gruen ML, Lee-Young RS, Puglisi MJ, Wasserman DH, Hasty AH. Impact of macrophage toll-like receptor 4 deficiency on macrophage infiltration into adipose tissue and the artery wall in mice. Diabetologia. 2009;52(2):318–328. doi:10.1007/s00125-008-1221-7
- Lynn M, Rossignol DP, Wheeler JL, et al. Blocking of responses to endotoxin by E5564 in healthy volunteers with experimental endotoxemia. J Infect Dis. 2003;187(4):631–639. doi:10.1086/367990
- Opal SM, Laterre PF, Francois B, et al. Effect of Eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis the ACCESS randomized trial article. JAMA. 2013;309(11):1154–1162. doi:10.1001/jama.2013.2194