
Abstract
Highly active antiretroviral therapy (HAART) is recognized as the most effective treatment method for AIDS, and protease inhibitors play a very important role in HAART. However, poor bioavailability and unbearable toxicity are their common disadvantages. Thus, the development of safer and potentially promising protease inhibitors is eagerly needed. In this review, we introduced the chemical characteristics and associated side effects of HIV protease inhibitors, as well as the possible off-target mechanisms causing the side effects. From the chemical structures of HIV protease inhibitors and their possible off-target molecules, we could obtain hints for optimizing the molecular selectivity of the inhibitors, to provide help in the design of new compounds with enhanced bioavailability and reduced side effects.
Introduction
Since the discovery of HIV, 26 anti-HIV compounds have been approved by the US Food and Drug Administration (FDA). Among these compounds, ten are HIV protease inhibitors. The combination therapy of HIV protease inhibitors, reverse transcriptase inhibitors, and/or an integrase inhibitor, referred to as highly active antiretroviral therapy (HAART), is the current most effective AIDS therapy. The AIDS-related mortality has dropped sharply, and AIDS has gradually become a controllable, chronic disease. Based on global AIDS response progress reporting, there are nearly 13 million people receiving antiretroviral therapy, and this number could reach 16 million by 2015.Citation1 HIV protease inhibitor is one of the most important components in the combination therapy. In the preferred antiretroviral combination regimens, protease inhibitor-based therapy has returned a lower level of resistance compared with non-nucleoside reverse-transcriptase inhibitor (NNRTI)-based therapy.Citation2 However, the need for lifelong treatment and the frequently associated side effects of HIV protease inhibitors severely hurt patient compliance, which is one of the obstacles in the treatment of HIV/AIDS patients. Although the toxic effects of HIV protease inhibitors could result from drug-drug interactions and overdose, the off-target adverse drug effects of therapeutic doses is a major concern in drug design.
In the HIV life cycle, protease is an essential element for viral maturation. The HIV protease is a homodimeric aspartyl protease, and each monomer is composed of 99 amino acid residues with a catalytic Asp at position 25 (). HIV-1 protease cleaves Gag and Gag-Pol polyprotein precursor encoded by the HIV-1 virus genome at nine processing sites to produce mature active proteins. The Pol polyproteins is first cleaved off from the Gag-Pol polyproteins and then further digested into protease, reverse transcriptase (p51), RNase H (p15), and integrase. The active site is not fully exposed, being covered by two flexible β-hairpin flaps. The flaps need to open to allow the substrates to access the active site. The HIV-1 protease enzyme activity can be inhibited by blocking the active site of the protease.
Figure 1 The HIV-1 protease structure in complex with an inhibitor.
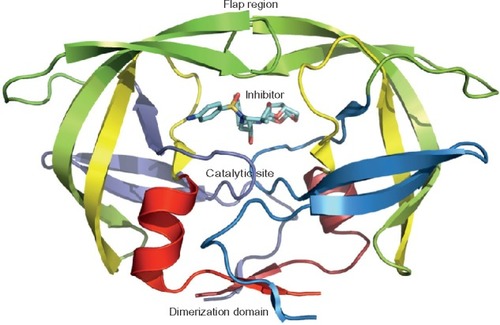
The indispensable role of HIV protease in viral maturation makes it a popular target for drug design. A large number of solved HIV protease protein structures have greatly facilitated the design of new and improved inhibitors. There are ten HIV protease inhibitors approved by the FDA; those inhibitors include: saquinavir, indinavir, ritonavir, nelfinavir, amprenavir, fosamprenavir, lopinavir, atazanavir, tipranavir, and darunavir (). Unfortunately, most of the inhibitors are accompanied by side effects in long-term treatment. The most common side effects are HIV protease inhibitor-induced metabolic syndromes, such as dyslipidemia, insulin-resistance, and lipodystrophy/lipoatrophy, as well as cardiovascular and cerebrovascular diseases.Citation3–Citation6 Protease inhibitor monotherapy is associated with a mild improvement in body fat distribution.Citation7,Citation8 However, regarding the serious adverse events of antiretroviral treatments, no significant between-group differences were found between HIV protease inhibitor monotherapy and the combination of protease inhibitors with the HIV integrase inhibitor raltegravir or nucleoside reverse transcriptase inhibitors (NRTIs),Citation9 indicating that HIV protease inhibitors may be responsible for the most serious adverse effects.
Figure 2 Chemical structures of the HIV protease inhibitors.
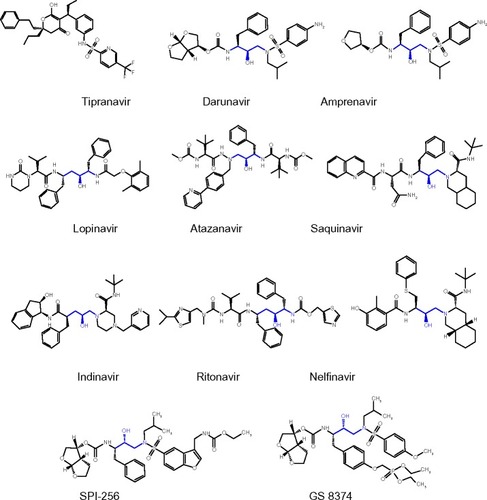
The FDA-approved HIV protease inhibitors share same structural similarities and a similar binding pattern, which may cause some of the common side effects of the protease inhibitor-containing regimens.
Saquinavir
Saquinavir (brand name: Invirase), developed by F. Hoffmann-La Roche Ltd (Basel, Switzerland), was the first FDA-approved HIV protease inhibitor used in the treatment of patients with AIDS (in 1995). The original design for the precursor of saquinavir comprised a proline at the P1′ site and a phenylalanine at the P1 site. The rationale is that HIV-1 protease cleaves the substrate between a phenylalanine and a proline, while mammalian proteases do not cleave substrates containing proline at the P1′ site. In the final structure of saquinavir, the proline was replaced by a z(S,S,S)-decahydro-isoquinoline- 3-carbonyl (DIQ) group to enhance the inhibitory potency. The carbonyl of the DIQ group contacts the bridging water molecule, which interacts with the inhibitor and the flaps of HIV-1 protease.Citation10 The mean 50% effective concentration (EC50) of saquinavir against HIV-1 in MT4 cells is 37.7 nM.Citation11 The adult dose is twice daily saquinavir 1,000 mg in combination with ritonavir 1 00 mg. Few side effects related to saquinavir have been reported.Citation12 However, saquinavir is not a preferred protease inhibitor regimen due to its low bioavailability.Citation13 The most common clinically significant saquinavir resistance mutations are 48VM, 54VTALM, 82AT, 84V, 88S, and 90M.
Indinavir
Indinavir (brand name: Crixivan) was developed by Merck & Co, Inc., (Whitehouse Station, NJ, USA) and approved in 1996. The EC50 of indinavir is ~5.5 nM. An advantage of indinavir is its effective inhibition of both HIV-1 and HIV-2, while the disadvantage is the quick decrease in the concentration of circulating indinavir. The low plasma concentration of indinavir usually leads to treatment failures.Citation14 In addition, the low solubility of indinavir may result in the development of kidney stones. Furthermore, indinavir could act as a competitive inhibitor of the cytoplasmic glucose binding site of GLUT4,Citation15 and lipodystrophy syndrome is strongly associated with indinavir.Citation16 Further, indinavir has a short acting time and requires a dosage of 800 mg every 8 hours. For these reasons, indinavir has been replaced by second-generation protease inhibitors. The most common clinically significant indinavir resistance mutations are 32I, 46IL, 47V, 54VTALM, 76V, 82ATFS, 84V, 88S, and 90M.
Ritonavir
Ritonavir (brand name: Norvir), developed by Abbott Laboratories (Abbott Park, IL, USA) and approved by the FDA in 1996, was originally designed as an HIV protease inhibitor (EC50 ~25 nM), but it was found later that ritonavir boosts the circulating concentration of other HIV protease inhibitors by inhibiting cytochrome P450 3A4.Citation17 Regarding the molecule structure, the isopropyl thiazolyl P3 group in ritonavir is longer than that in other FDA-approved HIV protease inhibitors. As a remarkably potent inhibitor of P450 3A4, a subtherapeutic dose of ritonavir has been used to boost the plasma concentration of the second generation of HIV protease inhibitors, since HIV protease inhibitors are extensively metabolized by cytochrome P450 3A4.Citation18 Ritonavir inhibits cytochrome P450 3A4 isoenzyme and prevents the metabolism of other protease inhibitors. Normally, boosted HIV protease inhibitors improve the side-effect and toxicity profile of HAART regimens. However, cytochrome P450 3A4 polymorphism, which encodes a nonfunctional protein, affects the metabolism of boosted HIV protease inhibitors, causing a higher plasma concentration of HIV protease inhibitors and increased toxicity.Citation19 Ritonavir-boosted protease inhibitor regimens require less frequent dosing, which benefits patients. However, higher doses could lead to hyperlipidemia in AIDS patients or healthy volunteers taking ritonavir.Citation20
Nelfinavir
Nelfinavir (brand name: Viracept) was developed by Agouron Pharmaceuticals (Pfizer, Inc., New York, NY, USA) and approved in 1997. One terminus of the nelfinavir molecule has the same DIQ group as saquinavir. The other terminus of nelfinavir contains a 2-methyl-3-hydroxybenzamide group. The S-phenyl group at the P1 site was designed to magnify the potency of this inhibitor. The EC50 of nelfinavir is 30–60 nM. The preferred regimen of nelfinavir is 1,250 mg orally, twice a day. The most common side effects related to nelfinavir are diarrhea and nausea.Citation21 The clinically significant nelfinavir resistance mutations are 30N, 33F, 46IL, 47V, 48VM, 54VTALM, 82ATFS, 84V, 88DS, and 90M.
Lopinavir
Lopinavir (brand name: Kaletra), containing lopinavir and ritonavir, from Abbott Laboratories, was approved by the FDA in 2000 and was developed as a ritonavir-based agent. The core region of lopinavir, a hydroxyethylene dipeptide isostere, is the same as that of ritonavir. The P2 and P2′ group are altered in lopinavir relative to ritonavir. The 5-thiazolyl P2 group of ritonavir is replaced by a phenoxyacetyl group, and the 2 -isopropylthiazolyl P2′ group of ritonavir is replaced by a six-member cyclic urea. In general, the new P2 and P2′ groups are smaller in order to decrease the contact with highly variable residues at the 8 2 site of HIV-1 protease.Citation22 The substitution of the P2 and P2′ groups improves the inhibitory potency of lopinavir against the drug-resistant variants of HIV-1 protease.Citation22 Lopinavir inhibits HIV protease activity, with the EC50 of ~17 nM. The dosage for adult patients is lopinavir 400 mg plus ritonavir 100 mg orally, twice a day. Lopinavir induces insulin resistance and could cause systemic hypersensitivity syndromes and Achilles tendinopathy.Citation23–Citation25 The most common clinically significant lopinavir resistance mutations are 32I, 33F, 46IL, 47VA, 48VM, 50V, 54VTALM, 76V, 82ATFS, 84V, and 90M.
Amprenavir
Amprenavir (brand name: Agenerase), developed by Vertex Pharmaceuticals Incorporated, (Boston, MA, USA) and approved in 1999, has a benzyl group at the P1 site and an isobutyl group at the P1′ site. The P1′ group and the phenyl amide P2′ group are connected by a sulfonamide. The asymmetry of the P1 and P1′ groups may favor the internal pseudosymmetry of HIV-1 protease. Amprenavir contains fewer chiral centers than do previous HIV protease inhibitors. This improvement simplifies the chemical synthesis and increases the oral availability.Citation26 The dosage of amprenavir is 1,200 mg orally, twice a day. Amprenavir is less effective on HIV-2 protease than on HIV-1 protease.Citation27 The EC50 of amprenavir has ranged from 12–80 nM. Amprenavir and its prodrug, fosamprenavir, cause the side effect of benign skin rash.
Fosamprenavir
Fosamprenavir (brand name: Lexiva), developed by GlaxoSmithKline plc, (Brentford, UK) and Vertex Pharmaceuticals Incorporated, was approved by the FDA in 2003. Fosamprenavir is the phosphate ester prodrug of amprenavir. The body metabolizes fosamprenavir to form the active ingredient, amprenavir. That metabolization increases the duration that amprenavir is available, making fosamprenavir a slow-release version of amprenavir and, thus, reducing the number of pills required versus standard amprenavir.Citation28 The recommended dose is fosamprenavir 1,400 mg in combination with ritonavir 100 mg orally, twice a day. Clinical studies demonstrated that fosamprenavir has a safer profile compared with amprenavir.Citation29,Citation30 The most common clinically significant fosamprenavir resistance mutations are 32I, 33F, 46IL, 47VA, 50V, 54VTALM, 76V, 82ATFS, 84V, and 90M.
Atazanavir
Atazanavir (brand name: Reyataz) developed by Bristol-Myers Squibb Co. (Princeton, NJ, USA) and approved in 2003, is an aza-dipeptide analog, which exhibits potent anti-HIV activity. The EC50 of atazanavir in cell culture is 2.6–5.3 nM. A unique characteristic of atazanavir is the presence of a large phenylpyridyl P1 group that is asymmetric relative to its benzyl P1′ group. Atazanavir shows good oral bioavailability.Citation31 Thus, the benefit of this is once-a-day dosing with atazanavir 300 mg plus ritonavir 100 mg. Fewer side effects are associated with atazanavir than with other protease inhibitors.Citation32,Citation33 Atazanavir has no effect on insulin sensitivity and serum lipid concentration.Citation23,Citation34 However, a significantly higher incidence of proximal tubulopathy was observed among atazanavir-treated patients.Citation35,Citation36 The most common clinically significant atazanavir resistance mutations are 32I, 33F, 46IL, 47V, 48VM, 50L, 54VTALM, 82ATFS, 84V, 88S, and 90M.
Tipranavir
Tipranavir (brand name: Aptivus) developed by Boehringer Ingelheim GmbH (Ingelheim, Germany), is the only nonpeptidomimetic HIV protease inhibitor and received approval from the FDA in 2005. Due to dissimilarities with other HIV protease inhibitors, tipranavir encounters a relatively different HIV-1 protease resistance profile.Citation37,Citation38 An innovation in the tipranavir design is the functional substitution of the bridging water molecule connecting the inhibitor and protease flaps. The lactone oxygen atom of the dihydropyrone ring of tipranavir interacts directly with the Ile50 residues in the flap region of the HIV-1 protease. The direct interaction stabilizes the protease-inhibitor complex. Tipranavir inhibits the HIV-1 protease that has developed resistance to other protease inhibitors. Though the structure of tipranavir is different from previous inhibitors, its contact residues for HIV-1 protease share many similarities with other HIV protease inhibitors. Tipranavir inhibits the replication of HIV-1 isolates, with EC50 ranging from 30–70 nM. The dosage is tipranavir 5 00 mg plus ritonavir 200 mg orally, twice a day. However, the adverse effects of tipranavir are more severe than those of other protease inhibitors. Intracranial hemorrhage and decompensated hepatitis have been reported to associate with tipranavir.Citation39 The most common clinically significant tipranavir resistance mutations are 32I, 33F, 46IL, 47VA 54VAM, 82TL, and 84V.
Darunavir
Darunavir (brand name: Prezista), approved in 2006, is the latest HIV protease inhibitor on the market. The hydrogen bonds that darunavir forms with the backbone of the HIV protease slow down the development of drug resistance.Citation40 The structure of darunavir is very similar to that of amprenavir, the only difference being that in darunavir, the P2 group bis-tetrahydrofuran replaces the tetrahydrofuran group of amprenavir. This change allows darunavir to have more hydrogen bonds with the Asp 29 residues of HIV protease.Citation41 The EC50 of darunavir is as low as 1–2 nM, and darunavir can inhibit both HIV-1 and HIV-2 with high potency.Citation27,Citation42 Furthermore, darunavir or atazanavir coadministrated with ritonavir is a current first-line antiretroviral regimen.Citation43 The dosage is darunavir 800 mg plus ritonavir 100 mg orally, once a day. Only minor darunavir-associated toxicities, such as rash, diarrhea, nasopharyngitis, and nausea, have been observed in some patients.Citation44 The most common clinically significant darunavir resistance mutations are 32I, 33F, 47VA, 50V, 54LM, 76V, 82F, and 84V.
The common mechanism of inhibitor binding
Most current HIV protease inhibitors were designed to mimic the substrate transition state. The hydroxyl group of the inhibitor interacts with the carboxyl group of the protease active site residues, Asp 25 and Asp 25′, by hydrogen bonds. The inhibitor-contacting residues of HIV protease are relatively conserved, including Gly 27, Asp 29, Asp 30, and Gly 48, but the accumulation of drug-resistance mutations alters the structure of HIV protease and causes treatment failure.
The major effort in inhibitor development is to modify the available HIV protease inhibitors, which inevitably causes the new inhibitors to inherit structural similarity with previous inhibitors. There have been many attempts to further modify current inhibitors; some have succeeded, while others have not shown significant benefits. A series of compounds has been tested to optimize the P1 group of lopinavir, but none of them has shown significant benefits over the original lopinavir.Citation45 The successful examples of modified inhibitors are darunavir and lopinavir, which were modified from amprenavir and ritonavir, respectively.
New designs of HIV protease inhibitors
It is possible to optimize the chemical structure of HIV protease inhibitors to avoid side effects. For example, not all HIV-1 proteases cause dyslipidemia, such as atazanavir.Citation34 Similarly, atazanavir does not inhibit glucose uptake as amprenavir, lopinavir, and ritonavir do.Citation46 GS-8374, developed by Gilead Sciences, Inc., (Foster City, CA, USA), is a modification of TMC-126 (darunavir analog).Citation47 This new inhibitor does not affect insulin-stimulated glucose uptake or peripheral glucose disposal, which is similar to atazanavir.Citation48 A possible explanation may be that the diethylphosphonate moiety modification on the P1 benzyl group of darunavir functions similarly to the large phenylpyridyl P1 group of atazanavir, to repel off-target molecules.
Designing inhibitors based on the substrate envelope of HIV protease is a recent strategy for developing HIV protease inhibitors. The substrate envelope is the overlapping volume occupied by multiple substrates. Based on this theory, a substrate-based HIV protease inhibitor, RO1, has been designed and synthesized.Citation49 The results demonstrated that RO1 has five- to tenfold higher inhibitory efficacy than current HIV protease inhibitors.Citation49 Among the current FDA-approved HIV protease inhibitors, only tipranavir forms hydrogen bonds with the flap residues of the protease. In contrast, RO1 forms more hydrogen bonds with HIV protease, which enhances the binding energy and is also a step toward a greater diversity of binding patterns. However, the contact residues of RO1 follow the old pattern, and no novel contact has been established. The difficulty of complete binding pattern alteration might be due to the inhibitor design concept of substrate mimics. To develop new scaffolds of HIV protease inhibitors, it is necessary to adopt drug discovery strategies other than substrate mimics.
New scaffolds of HIV protease inhibitors might be a possible solution to eliminate drug side effects. After extensive modifications and tests, the leading new scaffolds might become the next generation of HIV protease inhibitors possessing novel chemical structures and alternative binding patterns to HIV protease. However, it is very risky to explore new structures of inhibitors. One reason is that the new scaffolds are less likely to be superior to the well-established older scaffolds. The structure of mozenavir, which was developed based on the cyclic urea scaffold, is highly dissimilar from other HIV protease inhibitors.Citation50 However, the clinical studies showed that mozenavir did not show significant advantages compared with other protease inhibitors on the market, and therefore Gilead Sciences, Inc., stopped the development of mozenavir.Citation51 The other reason is that new scaffolds may cause severe adverse effects without thorough clinical studies. The relatively different structure of tipranavir, not only generates altered binding profiles but also, causes the unpredictable side effects, such as hemorrhage. Based on the above summary, the nonpeptidomimetic HIV protease inhibitors seem to have a higher tendency to cause severe adverse effects.
Several new HIV protease inhibitors are undergoing clinical trials. GlaxoSmithKline’s new inhibitor, CTP-518, was developed by replacing some hydrogen atoms of atazanavir with deuterium, which enables CTP-518 to inherit the once-daily dosage of atazanavir but slows down the drug metabolism.Citation52 Another new inhibitor is SPI-256 (), developed by Sequoia Pharmaceuticals (Gaithersburg, MD, USA), which is based on the darunavir scaffold.Citation53 Rather than coadministration with ritonavir, SPI-256 may be boosted using with Sequoia Pharmaceuticals’ own pharmacokinetic enhancer, SPI-452.Citation54 This pharmacokinetic enhancer targets cytochrome P450 3 A and boosts the circulating concentration of other protease inhibitors, as ritonavir does, but avoids the side effects caused by ritonavir.Citation55
Off-target molecules
The major metabolic side effects associated with HIV protease inhibitor therapy are lipodystrophy syndrome and insulin resistance.Citation56 The possible mechanisms of the metabolic alternations caused by HIV protease inhibitors have been studied. In the following paragraphs, the possible off-target effects of HIV protease inhibitors are discussed. Avoidance of interactions with off-target molecules may be considerations in the future development of new HIV protease inhibitors.
Proteases functioning in SREBP maturation
It has been demonstrated that most HIV protease inhibitors induce the accumulation of intracellular free cholesterol and lipid.Citation57 SREBP-1 is an endoplasmic reticulum (ER)-derived transcription factor that regulates the gene expression involved in lipogenesis. SREBP-1 mediates the effect of insulin on gene expression in adipocytesCitation58 and is associated with adipocyte differentiation and metabolism.Citation59 In the ER and nuclear envelope, SREBP exists as a full-length membrane-bound precursor.Citation60 A two-step sequential proteolytic cleavage activates the membrane-bound SREBP precursors. The first step is a sterol-regulated intramembrane proteolytic step to generate the N-terminal SREBP intermediate, which is processed by a membrane-anchored serine protease.Citation61 At the second step, the mature SREBP can be cleaved off by cysteine protease CPP32/SREBP cleavage activity-1 (SCA-1)Citation62 or Mch3/SCA-2.Citation63 The mature active SREBPs translocate to the nucleus. The inactive SREBP-1 precursor, rather than active SREBP-1, is processed by the ubiquitin-proteasome system.Citation64 Studies showed that HIV protease inhibitor therapy caused the sequestration of inactive SREBP-1 at the nuclear membrane level.Citation65,Citation66 Nelfinavir and indinavir have demonstrated effects in decreasing active SREBP-1 in the nucleus.Citation65,Citation67 The SREBP-1 sequestration prevents activation of the expression of downstream transcription factors for adipocyte differentiation.Citation68 Furthermore, SREBP-1 associates with the transcriptional regulation of a fat-derived hormone gene, adiponectin.Citation69 The effect of HIV protease inhibitors on SREBPs consequently leads to the deficiency of adiponectin. Therefore, adiponectin replacement therapy or adiponectin receptor agonist could ameliorate dyslipidemia, stabilize metabolic function, and prevent brain injury caused by HIV protease inhibitor-based treatment.Citation70,Citation71 Therefore, the off-target inhibition of the proteases required for SREBP maturation could cause inactive SREBP sequestration and affect adipocyte differentiation ().
Figure 3 Potential target molecules of the HIV protease inhibitors.
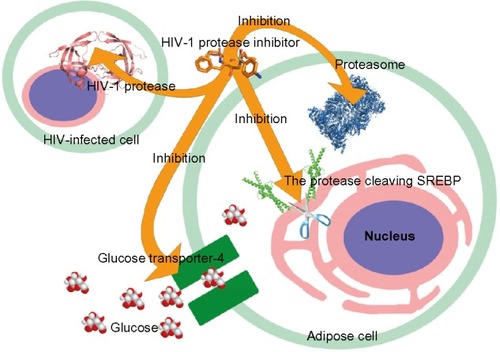
Glucose transporter-4
The insulin resistance observed in patients with HIV protease inhibitor treatment is associated with the blockage of a glucose transporter (). Glucose transporter-4 is an insulin-regulated transporter that can be directly inhibited by many HIV protease inhibitors, especially indinavir.Citation72,Citation73 The protease inhibitors specifically block the isoform of glucose transporter-4 and show no significant inhibition of glucose transporter-1 and glucose transporter-2.Citation72,Citation73 Therefore, the glucose uptake in adipocytes is blocked.
In vitro testing indicated that peptides with an aromatic core flanked by hydrophobic ends acutely inhibited glucose transporter-4 and weakly inhibited glucose transporter-2 but did not inhibit glucose transporter-1 and 3.Citation74 Those peptides represent the structure characteristic of currently approved HIV protease inhibitors and are also substrates of aspartyl proteases. Therefore, the aromatic core of HIV protease inhibitors could be the cause of a cross-interaction with glucose transporter-4. Interestingly, atazanavir, carrying an aromatic core, is known for having no effect on glucose uptake.Citation46 It is possible that the large phenylpyridyl P1 group of atazanavir, or its azapeptide backbone, disrupts the interaction with glucose transporter-4. Furthermore, atazanavir also differs from other HIV protease inhibitors in the hydrophobicity of the flanked molecule ends.Citation74
Proteasome
The proteasome is, not only a cell apparatus that degrades waste proteins but also, an important regulator in various signal pathways. But proteasome activity can be directly inhibited by some HIV protease inhibitors.Citation75 Ritonavir is a very potent proteasome inhibitor at clinically relevant levels.Citation73 Ritonavir, as well as nelfinavir and saquinavir, inhibits the proteasomal 20S subunit.Citation76,Citation77
Proteasome inhibition could cause metabolic complications (). HIV protease inhibitors prevent the proteasomal degradation of apolipoprotein B, and this may cause dyslipidemia,Citation78 as apolipoprotein B has a strong effect on the plasma lipid levels.Citation79 Saquinavir inhibits proteasome and therefore prevents the activation of NF-κB.Citation76 The inactivation of NF-κB inevitably leads to cell apoptosis. Furthermore, the inhibition of the proteasome complex that regulates NF-κB leads to an adipogenesis reduction.Citation80 Furthermore, the proteasome is also responsible for SREBP degradation in the nucleus.Citation64 The accumulation of SREBPs in the nucleus constitutively induces the expression of lipogenic and cholesterogenic genes.Citation81 HIV protease inhibitor treatment significantly elevates the total cholesterol, low-density lipoprotein cholesterol, and triglycerides in patients.Citation82
The toxicity of protease inhibitors also open up a method of drug repositioning. Study has demonstrated that nelfinavir, ritonavir, and saquinavir inhibited protein kinase B (Akt/PKB) and induced caspase-dependent apoptosis.Citation83 Darunavir had no significant effect on Akt/PKB, while atazanavir and lopinavir exerted a mild effect.Citation84 Among the HIV protease inhibitors tested, nelfinavir showed the great inhibition of Akt activity.Citation83 Currently, nelfinavir is undergoing a clinical trial in solid tumors.Citation85 Another study showed that the proteasome inhibitors downregulated the activation of Akt.Citation86 Thus, inhibition of Akt activity by nelfinavir could be a downstream event of proteasome inhibition. Due to proteasome inhibition, the accumulated unfolded proteins eventually result in ER stress and trigger autophagy.Citation87 The ER stress caused by protease inhibitors is one of the rationales for using HIV protease inhibitors as anticancer agents.
Prodrugs in improving bioavailability and reducing side effects
On one hand, side effects are the consequence of off-target binding. On the other hand, the physiological concentration of drugs is another detrimental factor causing side effects. To optimize the pharmacokinetics and decrease side effects, the development of protease inhibitor prodrugs could improve water solubility and bioavailability. One example is fosamprenavir, the prodrug of amprenavir. Study also showed that conjugating a valine residue to the protease inhibitor, through a hydrolyzable ester bond, improved cell absorption and decreased efflux.Citation88 In short, the design of HIV protease inhibitors that maintain potency and eliminate side effects is a long-term exploring process. Before the off-target effects could be eliminated by novel HIV protease inhibitor scaffolds, the development of HIV protease inhibitor prodrugs could decrease dosages and meliorate the side effects.
Future direction in preventing drug side effects
The identification of off-target mechanisms did not receive sufficient attentions in the past. Determining the potential toxic or benign chemical groups of HIV protease inhibitors helps in the design of newer inhibitors. For example, the P1 group and the two terminal groups of atazanavir may be benign chemical groups and can be considered for incorporation into new HIV protease inhibitors. With the accumulation of knowledge on the toxic chemical groups of HIV protease inhibitors, a “benign chemical group library” for HIV protease inhibitors can be compiled and benefit future drug design studies.
Disclosure
YW received research supports from the Hunan Provincial Science and Technology Project of China (grant number 2014SK3009) and the Scientific Research Foundation of Hunan Provincial Department of Health (grant number B2013-002). The authors report no other conflicts of interest in this work.
References
- UNAIDSGlobal Report: UNAIDS Report on the Global AIDS Epidemic 2010GenevaUNAIDS2010
- RiddlerSAHaubrichRDiRienzoAGAIDS Clinical Trials Group Study A5142 TeamClass-sparing regimens for initial treatment of HIV-1 infectionN Engl J Med2008358202095210618480202
- BozzetteSAAkeCFTamHKChangSWLouisTACardiovascular and cerebrovascular events in patients treated for human immunodeficiency virus infectionN Engl J Med2003348870271012594314
- HruzPWHIV protease inhibitors and insulin resistance: lessons from in-vitro, rodent and healthy human volunteer modelsCurr Opin HIV AIDS20083666066519373039
- KotlerDPHIV and antiretroviral therapy: lipid abnormalities and associated cardiovascular risk in HIV-infected patientsJ Acquir Immune Defic Syndr200849Suppl 2S79S8518725816
- SoontornniyomkijVUmlaufAChungSAHIV protease inhibitor exposure predicts cerebral small vessel diseaseAIDS20142891297130624637542
- CameronDWda SilvaBAArribasJRA 96-week comparison of lopinavir-ritonavir combination therapy followed by lopinavir-ritonavir monotherapy versus efavirenz combination therapyJ Infect Dis2008198223424018540803
- KoltaSFlandrePVanPNFat tissue distribution changes in HIV-infected patients treated with lopinavir/ritonavir. Results of the MONARK trialCurr HIV Res201191313921198431
- PatonNIKityoCHoppeAEARNEST Trial TeamAssessment of second-line antiretroviral regimens for HIV therapy in AfricaN Engl J Med2014371323424725014688
- KrohnARedshawSRitchieJCGravesBJHatadaMHNovel binding mode of highly potent HIV-proteinase inhibitors incorporating the (R)-hydroxyethylamine isostereJ Med Chem19913411334033421956054
- US Food and Drug AdministrationRoche, Fortovase® (saquinavir)Silver Spring, MDUS Food and Drug Administration2003 Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2003/20828s015ppi.pdfAccessed March 25, 2015
- WellerIVWilliamsIGABC of AIDS. Antiretroviral drugsBMJ200132272991410141211397751
- CameronDWJapourAJXuYRitonavir and saquinavir combination therapy for the treatment of HIV infectionAIDS199913221322410202827
- González de RequenaDGallegoOde MendozaCCorralAJiménez-NácherISorianoVIndinavir plasma concentrations and resistance mutations in patients experiencing early virological failureAIDS Res Hum Retroviruses200319645745912882654
- HreskoRCHruzPWHIV protease inhibitors act as competitive inhibitors of the cytoplasmic glucose binding site of GLUTs with differing affinities for GLUT1 and GLUT4PLoS One201169e2523721966466
- VirabenRAquilinaCIndinavir-associated lipodystrophyAIDS1998126F37F399583592
- KempfDJMarshKCKumarGPharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavirAntimicrob Agents Chemother19974136546609056009
- ZeldinRKPetruschkeRAPharmacological and therapeutic properties of ritonavir-boosted protease inhibitor therapy in HIV-infected patientsJ Antimicrob Chemother20045314914657084
- López AspirozECabrera FigueroaSEIglesias GómezAValverde MerinoMPDomínguez-Gil HurléACYP3A4 polymorphism and lopinavir toxicity in an HIV-infected pregnant womanClin Drug Investig20153516166
- PurnellJQZambonAKnoppRHEffect of ritonavir on lipids and post-heparin lipase activities in normal subjectsAIDS2000141515710714567
- MaxBShererRManagement of the adverse effects of antiretroviral therapy and medication adherenceClin Infect Dis200030Suppl 2S96S11610860894
- ShamHLKempfDJMollaAABT-378, a highly potent inhibitor of the human immunodeficiency virus proteaseAntimicrob Agents Chemother19984212321832249835517
- NoorMAParkerRAO’MaraEThe effects of HIV protease inhibitors atazanavir and lopinavir/ritonavir on insulin sensitivity in HIV-seronegative healthy adultsAIDS200418162137214415577646
- ManfrediRSabbataniSSerious, multi-organ hypersensitivity to lopinavir alone, involving cutaneous-mucous rash, and myeloid, liver, and kidney functionAIDS200620182399240017117031
- CresswellFVTomlinsJChurchillDRWalker-BoneKRichardsonDAchilles tendinopathy following Kaletra (lopinavir/ritonavir) useInt J STD AIDS2014251183383524516081
- St ClairMHMillardJRooneyJIn vitro antiviral activity of 141W94 (VX-478) in combination with other antiretroviral agentsAntiviral Res199629153568721545
- TieYWangYFBorossPICritical differences in HIV-1 and HIV-2 protease specificity for clinical inhibitorsProtein Sci201221333935022238126
- EronJJrYeniPGatheJJrKLEAN study teamThe KLEAN study of fosamprenavir-ritonavir versus lopinavir-ritonavir, each in combination with abacavir-lamivudine, for initial treatment of HIV infection over 48 weeks: a randomised non-inferiority trialLancet2006368953447648216890834
- GatheJCWoodRSanneILong-term (120-Week) antiviral efficacy and tolerability of fosamprenavir/ritonavir once daily in therapy-naive patients with HIV-1 infection: an uncontrolled, open-label, single-arm follow-on studyClin Ther200628574575416861096
- JuddADuongTGalliLEuropean Pregnancy and Paediatric HIV Cohort Collaboration (EPPICC) study group in EuroCoordPost-licensing safety of fosamprenavir in HIV-infected children in EuropePharmacoepidemiol Drug Saf201423332132524741696
- BeckerSAAtazanavir: improving the HIV protease inhibitor classExpert Rev Anti Infect Ther20031340341315482137
- SquiresKLazzarinAGatellJMComparison of once-daily atazanavir with efavirenz, each in combination with fixed-dose zidovudine and lamivudine, as initial therapy for patients infected with HIVJ Acquir Immune Defic Syndr20043651011101915247553
- MurphyRLSanneICahnPDose-ranging, randomized, clinical trial of atazanavir with lamivudine and stavudine in antiretroviral-naive subjects: 48-week resultsAIDS200317182603261414685054
- CahnPEGatellJMSquiresKAtazanavir – a once-daily HIV protease inhibitor that does not cause dyslipidemia in newly treated patients: results from two randomized clinical trialsJ Int Assoc Physicians AIDS Care (Chic)200433929815573713
- DauchyFALawson-AyayiSde La FailleRIncreased risk of abnormal proximal renal tubular function with HIV infection and antiretroviral therapyKidney Int201180330230921544066
- CalzaLTrapaniFSalvadoriCIncidence of renal toxicity in HIV-infected, antiretroviral-naïve patients starting tenofovir/emtricitabine associated with efavirenz, atazanavir/ritonavir, or lopinavir/ritonavirScand J Infect Dis201345214715422991923
- RusconiSLa Seta CatamancioSCitterioPSusceptibility to PNU-140690 (Tipranavir) of human immunodeficiency virus type 1 isolates derived from patients with multidrug resistance to other protease inhibitorsAntimicrob Agents Chemother20004451328133210770770
- LarderBAHertogsKBloorSTipranavir inhibits broadly protease inhibitor-resistant HIV-1 clinical samplesAIDS200014131943194810997398
- OrmanJSPerryCMTipranavir: a review of its use in the management of HIV infectionDrugs200868101435146318578560
- LefebvreESchifferCAResilience to resistance of HIV-1 protease inhibitors: profile of darunavirAIDS Rev200810313114218820715
- TieYBorossPIWangYFHigh resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strainsJ Mol Biol2004338234135215066436
- BrowerETBachaUMKawasakiYFreireEInhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical useChem Biol Drug Des200871429830518312292
- Panel on Antiretroviral Guidelines for Adults and AdolescentsGuidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and AdolescentsWashington, DCDepartment of Health and Human Services2011
- MolinaJMCohenCKatlamaCTMC114-C208 Study Group; TMC114-C215 Study GroupSafety and efficacy of darunavir (TMC114) with low-dose ritonavir in treatment-experienced patients: 2 4-week results of POWER 3J Acquir Immune Defic Syndr2007461243117621237
- ShamHLZhaoCLiLNovel lopinavir analogues incorporating non-Aromatic P-1 side chains – synthesis and structure – activity relationshipsBioorg Med Chem Lett200212213101310312372511
- Ya nQHruzPWDirect comparison of the acute in vivo effects of HIV protease inhibitors on peripheral glucose disposalJ Acquir Immune Defic Syndr200540439840316280693
- HeGXYangZYWilliamsMDiscovery of GS-8374, a potent human immunodeficiency virus type 1 protease inhibitor with a superior resistance profileMed Chem Commun201121110931098
- HruzPWYanQTsaiLGS-8374, a novel HIV protease inhibitor, does not alter glucose homeostasis in cultured adipocytes or in a healthy-rodent model systemAntimicrob Agents Chemother20115541377138221245443
- Prabu-JeyabalanMKingNMNalivaikaEAHeilek-SnyderGCammackNSchifferCASubstrate envelope and drug resistance: crystal structure of RO1 in complex with wild-type human immunodeficiency virus type 1 proteaseAntimicrob Agents Chemother20065041518152116569872
- AlaPJDeLoskeyRJHustonEEMolecular recognition of cyclic urea HIV-1 protease inhibitorsJ Biol Chem19982732012325123319575185
- HoffmannCART 2 010/2011: The horizon and beyondHoffmannCRockstrohJKHIV 2010 – A TextbookHamburgMedizin Fokus Verlag2010127159
- Concert PharmaceuticalsPharmacokinetics, safety and tolerability of isotopologs of atazanavir (ATV), with pharmacokinetic comparison to reyataz Available from: http://clinicaltrials.gov/ct2/show/NCT01458769?term=CTP-518&rank=1. NLM identifier: NCT01458769Accessed March 5, 2015
- HollandAWynneBRuffDGuttendorfRA first in human study evaluating the safety, tolerability, and pharmacokinetics (PK) of SPI-256, a novel HIV protease inhibitor (PI), administered alone and in combination with ritonavir (RTV) in healthy adult subjectsPoster presented at the 48th Annual ICAAC/46th Annual IDSA MeetingOctober 25–28, 2008Washington, DC
- GulnikSVEissenstatMApproaches to the design of HIV protease inhibitors with improved resistance profilesCurr Opin HIV AIDS20083663364119373035
- GulnikSEissenstatMAfoninaEPreclinical and early clinical evaluation of SPI-452, a new pharmacokinetic enhancer (PKE)Presented at the 16th CROI Conference on Retroviruses and Opportunistic InfectionsFebruary 8–12, 2009Montreal, Qc
- NolanDMetabolic complications associated with HIV protease inhibitor therapyDrugs200363232555257414636077
- ZhouHGurleyECJarujaronSHIV protease inhibitors activate the unfolded protein response and disrupt lipid metabolism in primary hepatocytesAm J Physiol Gastrointest Liver Physiol20062916G1071G108016861219
- KimJBSarrafPWrightMNutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1J Clin Invest19981011199421459
- FajasLSchoonjansKGelmanLRegulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: implications for adipocyte differentiation and metabolismMol Cell Biol19991985495550310409739
- SatoRYangJWangXAssignment of the membrane attachment, DNA binding, and transcriptional activation domains of sterol regulatory element-binding protein-1 (SREBP-1)J Biol Chem19942692517267172738006035
- SakaiJRawsonRBEspenshadePJMolecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cellsMol Cell1998245055149809072
- WangXPaiJTWiedenfeldEAPurification of an interleukin-1 beta converting enzyme-related cysteine protease that cleaves sterol regulatory element-binding proteins between the leucine zipper and transmembrane domainsJ Biol Chem19952703018044180507629113
- PaiJTBrownMSGoldsteinJLPurification and cDNA cloning of a second apoptosis-related cysteine protease that cleaves and activates sterol regulatory element binding proteinsProc Natl Acad Sci U S A19969311543754428643593
- HiranoYYoshidaMShimizuMSatoRDirect demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathwayJ Biol Chem200127639364313643711477106
- CaronMAuclairMVigourouxCGlorianMForestCCapeauJThe HIV protease inhibitor indinavir impairs sterol regulatory element-binding protein-1 intranuclear localization, inhibits preadipocyte differentiation, and induces insulin resistanceDiabetes20015061378138811375339
- GuanMFousekKChowWANelfinavir inhibits regulated intramembrane proteolysis of sterol regulatory element binding protein-1 and activating transcription factor 6 in castration-resistant prostate cancerFEBS J2012279132399241122540830
- TranHRobinsonSMikhailenkoIStricklandDKModulation of the LDL receptor and LRP levels by HIV protease inhibitorsJ Lipid Res200344101859186912837856
- BastardJPCaronMVidalHAssociation between altered expression of adipogenic factor SREBP1 in lipoatrophic adipose tissue from HIV-1-infected patients and abnormal adipocyte differentiation and insulin resistanceLancet200235993111026103111937183
- RahmouniKSigmundCDId3, E47, and SREBP-1c: fat factors controlling adiponectin expressionCirc Res2008103656556718796641
- XuAYinSWongLChanKWLamKSAdiponectin ameliorates dyslipidemia induced by the human immunodeficiency virus protease inhibitor ritonavir in miceEndocrinology2004145248749414592951
- PeppingJKOtvosLSurmaczEGuptaSKellerJNBruce-KellerAJDesigner adiponectin receptor agonist stabilizes metabolic function and prevents brain injury caused by HIV protease inhibitorsJ Neuroimmune Pharmacol20149338839824562631
- MurataHHruzPWMuecklerMThe mechanism of insulin resistance caused by HIV protease inhibitor therapyJ Biol Chem200027527202512025410806189
- MurataHHruzPWMuecklerMIndinavir inhibits the glucose transporter isoform Glut4 at physiologic concentrationsAIDS200216685986311919487
- HertelJStruthersHHorjCBHruzPWA structural basis for the acute effects of HIV protease inhibitors on GLUT4 intrinsic activityJ Biol Chem200427953551475515215496402
- AndréPGroettrupMKlenermanPAn inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation, and T cell responsesProc Natl Acad Sci U S A1998952213120131249789051
- PajonkFHimmelsbachJRiessKSommerAMcBrideWHThe human immunodeficiency virus (HIV)-1 protease inhibitor saquinavir inhibits proteasome function and causes apoptosis and radiosensitization in non-HIV-associated human cancer cellsCancer Res200262185230523512234989
- HamelFGFawcettJTsuiBTBennettRGDuckworthWCEffect of nelfinavir on insulin metabolism, proteasome activity and protein degradation in HepG2 cellsDiabetes Obes Metab20068666166817026490
- LiangJSDistlerOCooperDAHIV protease inhibitors protect apolipoprotein B from degradation by the proteasome: a potential mechanism for protease inhibitor-induced hyperlipidemiaNat Med20017121327133111726973
- SteinbergDCarewTEFieldingCLipoproteins and the pathogenesis of atherosclerosisCirculation19898037197232670321
- De BarrosSZakaroff-GirardALafontanMGalitzkyJBourlierVInhibition of human preadipocyte proteasomal activity by HIV protease inhibitors or specific inhibitor lactacystin leads to a defect in adipogenesis, which involves matrix metalloproteinase-9J Pharmacol Exp Ther2007320129129917038510
- ParkerRAFlintOPMulveyREndoplasmic reticulum stress links dyslipidemia to inhibition of proteasome activity and glucose transport by HIV protease inhibitorsMol Pharmacol20056761909191915755908
- MulliganKGrunfeldCTaiVWHyperlipidemia and insulin resistance are induced by protease inhibitors independent of changes in body composition in patients with HIV infectionJ Acquir Immune Defic Syndr2000231354310708054
- GillsJJLopiccoloJTsurutaniJNelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivoClin Cancer Res200713175183519417785575
- CapelEAuclairMCaron-DebarleMCapeauJEffects of ritonavir-boosted darunavir, atazanavir and lopinavir on adipose functions and insulin sensitivity in murine and human adipocytesAntivir Ther201217354955622293506
- National Cancer Institute (NCI)Nelfinavir in treating patients with metastatic, refractory, or recurrent solid tumors Available from: https://clinicaltrials.gov/ct2/show/NCT00436735. NLM identifier: NCT00436735Accessed March 25, 2015
- InoueTShirakiKFukeHProteasome inhibition sensitizes hepatocellular carcinoma cells to TRAIL by suppressing caspase inhibitors and AKT pathwayAnticancer Drugs200617326126816520654
- YorimitsuTNairUYangZKlionskyDJEndoplasmic reticulum stress triggers autophagyJ Biol Chem200628140302993030416901900
- RocheDGreinerJAubertinAMVierlingPSynthesis and in vitro biological evaluation of valine-containing prodrugs derived from clinically used HIV-protease inhibitorsEur J Med Chem20084371506151817950955