
Abstract
Progressive familial intrahepatic cholestasis (PFIC) is a group of autosomal recessive cholestatic liver diseases which are subgrouped according to the genetic defect, clinical presentation, laboratory findings and liver histology. Progressive liver fibrosis, cirrhosis, and end stage liver disease (ESLD) may eventually develop. PFIC was first described in Amish descendants of Jacob Byler, therefore it was originally called Byler disease. But it can be seen anywhere on the globe. This review summarizes the main features of the subtypes of the disease and discusses the current available diagnosis, conservative and surgical therapeutic options.
Video abstract
Point your SmartPhone at the code above. If you have a QR code reader the video abstract will appear. Or use:
Introduction
Impaired production and excretion of bile results in cholestatic liver disease, where biliary substances cannot be eliminated from the liver and thus reenter the circulation. This results in the deposition of bilirubin pigments in the tissues as skin, sclerae, mucous membranes and so on, which overall is called jaundice.Citation1
Pruritus is the most obvious and the most unbearable symptom in cholestasis. In the patients, pruritus is probably induced by the stimulation of nonmyelinated subepidermal free nerve ends because of increased serum bile acids.Citation2
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of liver disorders of autosomal recessive inheritance, characterized by an early onset of cholestasis (usually during infancy) with pruritus and malabsorption, which rapidly progresses and ends up as liver failure.Citation3,Citation4 PFIC appears equally in both genders.
PFICs are responsible for about 10–15% of children with cholestatic liver diseases. PFIC-1 and 2 are rare diseases with an estimated incidence of 1:50–100,000. The total number of cases from the two subtypes reported in the literature is less than 200. PFIC-3 subtype is even rarer with a reported case number of less than 20.Citation5,Citation6
PFIC cases are more often observed in some cultures where consanguineous marriages are more popular. They were first described in Amish descendants of Jacob Byler, therefore it was originally called Byler disease. But they can be seen anywhere on the globe. So the name has been superseded by PFIC.Citation7
General approaches to PFIC
PFICs are subgrouped according to the genetic defect, clinical presentation, laboratory findings, and liver histology. The disease has been classified into three subgroups. All the three types of PFIC are caused by defects in bile secretion from hepatocyte to canaliculi. In PFIC-1 and PFIC-2, bile acid secretion is depleted, while in PFIC-3, bile phospholipid secretion is impaired.Citation3,Citation8,Citation9
All subtypes are shown in the –. The PFIC subgroups will be closely reviewed at the end of the article.
Table 1 Genetic features of PFIC subtypes
Table 2 Laboratory findings in PFIC subtypes. (N= normal, H= high/elevated, L= low/depleted)
Table 3 Clinical features of PFIC subtypes
Table 4 Liver biopsy, electron microscopy features and treatment modalities for PFIC subtypes
Diagnosis
Diagnosis is made through a detailed history, physical examination, laboratory tests, radiologic or histological evaluations if needed.
Biliary atresia, infections, hormones, drugs, parenteral nutrition, extrahepatic obstruction due to common bile duct stones or choledochal cysts, sclerosing cholangitis, hypothyroidism and panhypopituitarism, metabolic disorders as tyrosinemia type I, galactosemia, inborn errors of bile acid metabolism, alpha-1-antitrypsin deficiency, cystic fibrosis, benign recurrent intrahepatic cholestasis (BRIC), intrahepatic cholestasis of pregnancy (ICP), Alagille syndrome, and Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) may also cause neonatal cholestasis and should be included in differential diagnosis of PFICs.Citation5,Citation10–Citation13
History
In a patient with abnormal liver function tests, obtaining a detailed history is critical. It should include age of onset of jaundice, age of onset of abnormal liver function tests, pattern of abnormal liver function tests either hepatocellular or cholestatic, any infections, medication, feeding, bowel movements, stool color, urine color and the family history as well. Cholestasis should be considered in a baby with irritability, pruritus, cutaneous mutilation, scratching, jaundice, watery diarrhea, steatorrhea and failure to thrive.Citation1,Citation3,Citation9
Physical examination
Cholestatic liver diseases have a wide spectrum of presentation from isolated abnormalities in liver function tests to liver failure. Patients with PFIC can further present with jaundice, pruritus, splenomegaly, hepatomegaly, and altered anthropometrics ().Citation1,Citation3,Citation9
Figure 1 An infant with PFIC, jaundice and scratching marks.
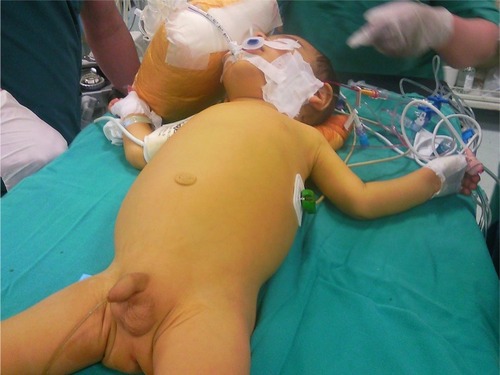
Pruritus is induced by the stimulation of nonmyelinated subepidermal free nerve ends related with the increased serum bile acids and is the most prominent and distressing symptom.Citation10
PFIC-1 patients may also present with short height, growth retardation, deafness, diarrhea, pancreatitis, increased sweat electrolyte concentration, hepatic steatosis and epistaxis despite bleeding diathesis. Secondary vitamin K deficiency related with fat malabsorption and inadequate dietary intake, may predispose to hemorrhagic disease of the newborn (HDN) and late HDN (seen in infants aged 1 week to 6 months) may be associated with serious and life-threatening intracranial hemorrhage.Citation14 PFIC-2 may directly refer with a malignancy. PFIC-3 patients are likely to present with cirrhosis in late childhood and young adulthood.
Clinical presentation, liver biochemistry, radiological and histological evaluations all help to diagnose PFIC and help to distinguish the subtype.
Laboratory evaluation
Liver function tests, serum bile acids and imaging studies help to rule out the cause of liver disease. Since PFIC is a rare disease, all other more common liver diseases and biliary diseases such as biliary atresia, Alagille syndrome, alpha-1 antitrypsin deficiency, cystic fibrosis, sclerosing cholangitis, and biliary obstruction shall be excluded first.
While evaluating a jaundiced infant, the initial step is measuring the serum total (TB) and direct bilirubin. In PFICs, laboratory abnormalities include increased serum alkaline phosphatase (ALP), variable elevation of bilirubin, decreased/increased gamma-glutamyltransferase (GGT) and increased bile acids, serum copper, ceruloplasmin, cholesterol and lipoprotein X. Accompanying tests shall include alanine aminotransferase (ALT), aspartate aminotransferase (AST), prothrombin time (PT), international normalized ratio (INR), serum 5′-Nucleatidase, glucose and albumin. When TB is elevated, a direct/conjugated ratio greater than 1.0 mg/dL (17 mmol/L) is considered abnormal.Citation1,Citation9
Patients with PFIC-1 and PFIC-2 have normal GGT levels, while patients with PFIC-3 have increased GGT levels. All 3 subtypes of PFIC have increased serum bile acid levels.
Radiologic studies
In cholestasis patients, ulltrasound (US) comes first in radio-logic studies. Magnetic Resonance Cholangiopancreticography (MRCP) contributes in excluding extrahepatic biliary obstruction and sclerosing cholangitis. The use of hepatic scintigraphy is under evaluation.Citation9,Citation15
Liver biopsy
A liver biopsy may help in the diagnosis of PFIC. But, assessment of liver biopsy specimens is not adequate in the differential diagnosis. Typical pattern of laboratory findings in serum contribute for diagnosis, especially low to normal level of gamma glutamyl transferase (GGT), absent lipoprotein X, low cholesterol level, and high levels of bile acids.
At a liver biopsy, canalicular cholestasis can be seen in PFIC-1 and PFIC-2 with more fibrosis and a giant cell hepatitis in PFIC-2 subtype. PFIC-3 specimens usually show proliferation of the bile ducts and fibrosis. The defective bile salt export pump protein (BSEP) in PFIC-2 and the multidrug resistance three protein (MDR3) in PFIC-3 can be stained and the subtype can be named.Citation7,Citation9
Immunohistochemistry studies of liver biopsy specimen may be useful in the diagnosis. BSEP antibodies for PFIC2 and MDR3 antibodies for PFIC3 are available. But, there is no standardized antibodies for PFIC-1. Absent or decreased hepatic canalicular expression of these antibodies may be diagnostic for PFIC2 and PFIC3.Citation16
Electronmicroscopy (EM)
Evaluation of the composition of bile may also support diagnosis of PFIC. Bile salt concentration is low in PFIC-1 and PFIC-2 and phospholipid level is low in the bile in PFIC-3.
On light microscopy and transmission electron microscopy of liver tissues of PFIC-1 patients have coarsely granular bile. While canalicular dilatation, microvilli loss, abnormal mitochondrial internal structure, and varying intracanalicular accumulation of finely granular, rather amorphous or finely filamentous bile is seen in PFIC-2 patients.
Genetic testing
Genetic testing can further confirm the diagnosis of PFIC in the vast of patients. Deletion/duplication analysis, sequence analysis of the coding region, targeted variant analysis can be performed. Targeted next-generation sequencing (NGS) has successfully been used for molecular genetic diagnosis in subjects with neonatal/infantile intrahepatic cholestasis.Citation17
Treatment
Treatment includes medical and surgical approaches. Diets, medications, and vitamins are used for medical treatment and external or internal biliary diversions are applied for surgical treatment.Citation17,Citation18 Surgical approaches have an important role in the relief of symptoms such as pruritus and prevention of development of cirrhosis of the liver. Details of the treatment approaches are discussed below.
Diet
Dietary fat is mainly provided as medium chain triglycerides. The fat-soluble vitamin supplements (A, D, E and K) are administered to ensure proper absorption. Calcium intake and adequate exposure to sunlight are also essential.
The patients can also benefit from cold tubs, local steroids and moisturizers, antihistaminics and sedatives as for pruritus. Phototherapy and plasmapheresis are other conservative options.Citation18,Citation19
Drug treatment
Drug therapy is the first line of treatment in all PFIC patients. The purpose of medical treatment in cholestasis and its complications can be summarized as follows; enhancing the bile flow and inhibiting the accumulation of metabolites in the liver (choleresis), treatment of toxic effects of bile reentering the systemic circulation, avoiding the malabsorption of fat and fat soluble vitamins, preventing acute and chronic malnutrition and ensuring continuity of growth.
In the treatment of PFIC, general treatment principles of cholestasis are followed and infant formulas enriched with medium-chain triglycerides, fat-soluble vitamins, antihistamines, etc, are used.
Drug treatment aims to relieve pruritus which is the most distressing symptom in PFIC, to slow the disease progression, to improve the nutritional status, to correct vitamin deficiencies and to treat the complications of advanced liver disease like ascites and variceal bleeding. However, medical treatment often fails and surgical alternatives and liver transplantation might be necessary.Citation20
Ursodeoxycholic acid (UDCA)
UDCA is the initial treatment for all PFIC subtypes. Some reports suppose, at a dose of 10–30 mg/kg per day, it dissolves cholestasis and is successful in the treatment. It is a nontoxic hydrophilic bile acid and thought to reverse the potential hepatotoxicity of the accumulating endogenous bile acids. It regulates bile acid distribution, reduces the amount of cholesterol in the bile, and provides mitochondrial integrity. It has choleretic, immunomodulatory, antioxidant, antiapoptotic and cytoprotective effects.
UDCA is effective in two thirds of patients with PFIC-3 with ABCB4 alterations. In one third, where mutations resulted in no expression of the MDR3 protein, patients are usually non-responders to UDCA therapy. Progression of disease and insufficient symptom relief may necessitate further intervention.
Cholestyramine
Cholestyramine is an oral bile acid binding resin used to resolve pruritus. It forms nonabsorbable micelles with the bile acids in the intestines and prevents bile acids from entering the enterohepatic cycle. Cholestramine should be taken at least 1 hour before or 4–6 hours after meals, 1–4 gr/day. This drug induces liver enzyme activity and increases bilirubin excretion. In patients with reduced serum bilirubin levels, pruritus also regresses.
Rifampicin
Rifampicin acts by upregulating detoxification enzymes and export pumps through farnesoid X receptor (FXR) dependent mechanisms. Rifampicin indirectly induces hydroxylation of bile salts which are further glucuronidated and excreted in urine. It also induces conjugation and excretion of bilirubin through uridine diphosphate (UDP)-glucuronosyl transferase.Citation19 It is used 5–10 mg/kg/day.
Phenobarbital
Phenobarbital, is used to induce CYP/CYP450 system in the treatment of newborn hyperbilirubinemia and chronic cholestasis with low bilirubin levels at a dose of 3–10 mg/kg/day.
4-phenylbutyrate
Hasegawa et alCitation21 evaluated the therapeutic potency of 4-phenylbutyrate in three patients with PFIC1 and observed that at a dosage of 350 or 500 mg/kg/day per orally significantly relieved the intractable itch. Naoi et alCitation22 evaluated the effect of 4-phenylbutyrate in one patient with PFIC-2 and concluded that in patients with decreased cell-surface expression of BSEP among PFIC-2s, 4-phenylbutyrate (4PB) therapy has partially restored BSEP expression at the canalicular membrane, significantly improved liver tests and pruritus at a dosage of 500 mg/kg/day.
Other drugs
Antihistaminic agents, opiate antagonists, ondansetron, steroids, propofol, and carbamazepine are part of the other medical therapy options.
Nasobiliary drainage
Nasobiliary drainage is the non-surgical, temporary diversion of bile through an endoscopically introduced nasobiliary drain. The risk of pancreatitis should not be ignored.
Surgical management
Intractable pruritus despite medical treatment, growth failure and nutritional deficiencies necessitate surgery. Pruritus is assessed according to the Whitington scale. The severity of pruritus is important in the decision to proceed with surgery. There is epidermal bleeding at grade four according to this scale.
Biliary diversion procedures aim to interrupt the enterohepatic recirculation of bile salts via an anastomosis of the biliary tract to the intestines (internal drainages) or to the skin (external diversions). Thus, accumulating excess serum bile salts decrease, biliary acid composition changes, pruritus regresses, progression to cirrhosis delays. Partial biliary diversions have been used successfully in many patients with PFIC-1 and 2, who do not respond to medical therapy and are as yet not candidates for liver transplant.Citation23
Diversions help to improve liver functions, growth, liver histology, reduce progression of fibrosis and extend the time interval before liver transplantation in the majority of patients with PFIC-1 and 2.
If the patients have not developed cirrhosis at the time of surgery, the results are even more satisfying, therefore biliary diversions should be offered early before development of cirrhosis.
Partial external biliary diversions (PEBD)
The PEBD procedure, once described by Whitington et al,Citation24 involves use of a 10–15 cm jejunal conduit between the fundus of the gallbladder and abdominal skin where a permanent stoma is created. Diversion of bile interrupts the enterohepatic circulation of bile salts, diminishes subsequent reuptake and decreases the pool of bile salts.Citation24–Citation29
PEBD has gained popularity over the last few years. So far other modifications of the conduit between gall bladder and skin have been defined as the use of a button of gall bladder wall (cholecystostomy), appendix (cholecystoappen-dicocutaneostomy) or ileum (cholecystoileocutaneostomy). Cholecystostomy may seem practical but maintaining an adequately watertight stoma is challenging and this technique has a high risk of cholangitis.
PEBD is used extensively as the first line surgery in PFIC-1 and 2 patients. Many retrospective review reports of individual centers state that pruritus has been relieved, liver chemistry and liver function tests, serum lipid levels and growth are improved in most patients.Citation29,Citation30
Many studies have outlined regression of histological abnormalities in the liver after PEBD and Arnell et alCitation29 made a further outcome analysis of pre- and postoperative histological findings showing that improvements in clinical condition and biochemical indices of hepatobiliary injury after PEBD are followed by improvements in histological cholestasis and fibrosis.
PEBD may postpone or preclude liver transplantation in patients with PFIC but may fail in advanced stages of the disease and patients may be directed to transplantation.
Stoma prolapse is the main surgical complication in PEBD. The need for stoma revision, postoperative cholangitis and dehydration are among the disadvantages of external drainage techniques.
Partial internal biliary drainages (PIBD)
In partial internal biliary drainages, there is no external fistula/stoma but instead a jejunal, ileal or appendix conduit between gallbladder and colon (called cholecystojejunocolic, cholecystoileocolic, cholecystoappendicocolic anastomosis, respectively) or anastomosis between gall bladder and anti-reflux loop of colon (cholecystocolocolic anastomosis) is made either via open surgery or by laparoscopy.Citation31–Citation35
This technique is rather new and has limited follow-up long-term feedbacks. But reports all agree that regardless of the preferred internal drainage technique and postoperative liver functions, pruritus regressed significantly in all patients. Rectal bleeding may be due to irritation of bile acids in the first postoperative days. The risk of colon cancer due to bile acids is not yet known.
Ileal exclusion/bypass (IE)
In ileal bypass technique, an ileocolonic anastomosis is made bypassing the distal 15% of small intestine and interrupting the enterohepatic circulation of bile salts via decreasing the reuptake of bile components. It can be used in patients with previous cholecystectomy and aims to avoid an external stoma and related complications. The disadvantage is that ileal adaptation occurs in time and symptoms recur in the majority of patients by the end of first year.Citation36
Liver transplantation (LT)
Progressive familial intrahepatic cholestasis is among the five most common indications for orthotopic liver transplantation in children. When medical treatment or biliary diversions fail to enable relief of symptoms or end-stage liver disease (ESLD) develops, liver transplantation is considered. LT improves cholestasis and related symptoms in most patients, irrespective of the PFIC subtype and patients generally have good prognosis. Transplants from heterozygous parents are successful.Citation37–Citation40
Since PFIC type 1 is a multiorgan disease, extrahepatic manifestations such as diarrhea may persist despite LT and recurrent graft steatosis may occur in PFIC-1 patients. PEBD may be indicated to relieve these symptoms. Cholestiramin may also help in the treatment of posttransplantation diarrhea. Total internal biliary diversion during liver transplantation for these patients seems a reasonable alternative that may prevent recurrent graft steatosis following LT for PFIC type 1.
Post transplantation recurrence has been observed in up to 8% of patients with PFIC-2, due to formation of autoantibodies against BSEP. These patients can be treated with a diverse immunosuppressive therapy, plasmapheresis or immunoadsorption or with rituximab.
LT is advocated as the first therapeutic option for children with PFIC in some centers even in children without severe fibrosis or cirrhosis. But although it has an increasing success rate, LT still carries significant morbidity and mortality risks besides the complications as graft rejection, graft steatosis and disease recurrence and there is still need for lifelong immunosuppression.
Novel therapies
The apical sodium-dependent bile acid transporters take up almost all of the bile acids from the intestine, prevent their fecal loss and thus sustain the enterohepatic circulation of bile acids. Thus, the pharmacological inhibition of bile acid uptake transporters does have benefits. In a human phase I trial, ASBT inhibitors reduced the total serum bile acids with increased fecal bile acid excretion.Citation41–Citation47
Since FXR activation and increased production of the FGF19 hormone reduces endogenous bile acid synthesis, protects hepatocytes from bile acid toxicity and promotes secretion of bile acids, together with apical sodium-dependent bile acid transporter (ASBT) inhibitors, FXR agonists and FGF19 mimetics represent the most promising anticholestatic strategies and are being tested in several clinical trials.Citation44
FXR agonists have been successfully tested in animal models of cholestasis but dyslipidemia frequency rose among cholestatic patients.
Overexpression of FGF19 in mice has also been associated with HCC development. So a non-tumorigenic FGF19-like peptide has recently been designed to eliminate this negation.Citation47 But further studies are still required for effective treatment in the management of PFIC.
Being less invasive and repeatable, hepatocyte transplantation might be an alternative to liver transplantation. Since the donor hepatocytes are limited here, alternative cell sources are being investigated as hepatocytes generated by transdifferentiation or induced pluripotent stem cells (iPSCs) derived hepatocyte-like cells.Citation48,Citation49
PFICs subtypes
The disease has been classified into three subtypes. All the three types of PFIC are caused by defects in bile secretion from hepatocyte to canaliculi. PFIC types 1 and 2 have been reported in all races. PFIC3 has been seen in Western European, White, and North African Arabic populations.
PFIC-1 patients may present with short height, growth retardation, deafness, diarrhea, pancreatitis, increased sweat electrolyte concentration, hepatic steatosis and epistaxis despite bleeding diathesis while PFIC-2 may directly refer with a malignancy. PFIC-3 patients are likely to present with cirrhosis in late childhood and young adulthood.
Clinical presentation, liver biochemistry, radiological and histological evaluations all help to diagnose PFIC and help to distinguish the subtype.
PFIC type 1 (Byler Disease)
PFIC-1 occurs due to a mutation of the assumed aminophospholipid transporter FIC1/ATP8B1 gene on chromosome 18 resulting in impaired hepatocellular bile salt secretion.Citation50,Citation51 It starts during infantile period with episodes of cholestasis (average onset of 3 months of age) and leads to liver cirrhosis and rapidly progresses to ESLD requiring liver transplantation. It was defined as Byler’s disease by Clayton et alCitation7 in 1969.
Patients usually present with elevated alkaline phosphatase and serum primary bile acids (in particular chenodeoxycholic acid) while serum gamma-glutamyltransferase (GGT) activity is not elevated. Serum cholesterol levels are usually normal.
The mutated gene encoding for the protein FIC1, not only affects the protein expression in the liver but also in the pancreas, small intestine, urinary bladder, stomach, and prostate. That is why patients have extrahepatic manifestations, such as diarrhea, malabsorption, pancreatitis, short height, an increased sweat electrolyte concentration, and hepatic steatosis.
Benign recurrent intrahepatic cholestasis (BRIC-1, also known as Summerskill syndrome) is a milder form of PFIC-1 which is also related with FIC1 gene mutation, where recurrent episodes of cholestasis occur but not necessarily leading to liver cirrhosis. However there are rare reported BRIC-1 cases progressing into PFIC-1. Since FIC1 gene is widely expressed in extrahepatic tissues, both diseases have manifestations such as diarrhea, pancreatitis, bile acid malabsorption and nephrolithiasis.
PFIC type 2 (Byler Syndrome)
PFIC-2 occurs due to a mutation of the major canalicular BSEP gene on chromosome 2 (BSEP/ABCB11).Citation52 Expression of this gene is limited to liver. Therefore although the clinical course of PFIC-2 is similar to that for PFIC-1, extrahepatic manifestations are absent.
PFIC2 frequently presents as nonspecific giant cell hepatitis at a few months of age with recurrent or chronic jaundice which is undistinguishable from idiopathic neonatal giant cell hepatitis. But it usually progresses even more quickly to ESLD than PFIC-1. Patients require liver transplantation during the first decade of life.
Hepatocellular carcinoma or cholangiocarcinoma may develop in the first year of life. Therefore right after diagnosis, prompt screening for malignancies is very important. This patients must follow with alpha-fetoprotein levels and hepatic ultrasound.
As in PFIC-1, serum bile acids, bilirubin and transaminases with a marked increase in ALT level to more than five times normal but GGT levels are low. PFIC-2 also has a lighter variant called Benign intrahepatic recurrent cholestasis-2 (BRIC-2) where the course of the disease is more moderate. Also, other missense mutations of the ABCB11 gene, as can be proved by genetic testing, may follow a milder course not necessitating typical treatment for PFIC2.
PFIC type 3
PFIC-3 occurs due to a mutation of adenosine triphosphate-binding cassette, subfamily B, member 4 (ABCB4) gene encoding for the multidrug resistance class III (MDR3) protein related with the bile phospholipid export pump.Citation53
Bile phospholipids are required for the formation of mixed micelles with bile acids and cholesterol. They protect the bile duct epithelium from the detergent properties of bile acids. The bile flow is not impaired itself but impaired bile phospholipid excretion results in bile duct damage. The unmiscelle form bile acid monomers are toxic to cholangiocytes and hepatocytes. These patients may present with progressive cholestasis and cirrhosis later in childhood or young adulthood. Pruritus and short stature may be less apparent but variceal bleeding and portal hypertension are common in PFIC-3 patients.
New subtypes
Tight junction protein two gene (chromosome 9) has recently been associated with PFIC as a new subtype, PFIC-4. TJP2 codes for a protein involved in the organization of epithelial and endothelial intercellular junctions that separate bile from plasma in the liver.
Furthermore, a fifth subtype of PFIC with farnesoid X receptor (FXR) mutations is distinguished.
During fasting, bile acids are stored in the gallbladder and they are secreted into the small intestine postprandially to participate in digestion. Then they are reabsorbed through the portal circulation into the liver to be recycled and just 5% of them are newly synthesized in the liver daily to sustain a proper bile acid pool in the organism. The farnesoid X receptor (FXR), which is a nuclear receptor and a product of NR1H4 gene, balances the production and circulation of bile acids. It is presented as the master regulator of bile acid homeostasis. FXR also provides protection against hepatocarcinogenesis.
Genetic studies have revealed that defects in FXR pathway account for different clinical types of cholestatic liver diseases, such as intrahepatic cholestasis of pregnancy (ICP), drug-induced cholestasis (DIC) or PFIC.
Gomez-Ospina et alCitation54 presented evidence for NR1H4 mutations resulting in complete absence of BSEP expression in the bile canaliculi and leading to cholestasis. It is also an autosomal recessive disorder characterized with an onset of intralobular cholestasis and severe vitamin K-independent coagulopathy in the neonatal period. It is rapidly progressive, leading to liver failure and death unless liver transplantation is performed. Here, serum GGT levels is low-to-normal and serum alpha-fetoprotein is elevated.
Discussion
PFIC is one of the most distressing cholestatic liver diseases of childhood resulting in cirrhosis and end stage liver disease. Pruritus associated with the accumulation of excessive bile acids, is the most disturbing symptom. There is a variety of medical therapeutics having limited benefits. And there are some surgical options defined having positive effects on symptoms and liver histology. The surgical technique preference depends on the gastrointestinal anatomy of the patient and the surgeon’s preference and experience.
Since relief from symptoms is not sustained in an acute manner, the disposable percutaneous transhepatic biliary drainage catheters are not suitable for this disease group.
External diversion techniques include a stoma, thus have stoma-related complications.
Internal drainage techniques avoid a stoma. They have lower complication rates and provide a better quality of life. But since they are rather new, they have limited long-term outcome reports.
In a study comparing the inferior long-term outcome of ileal bypass to PEBD reported recurrence of symptoms in 50% of the patients which was probably related to the re-absorption of bile acids over time. The large amounts of bile salts entering the colon induce choleretic diarrhea and rectal bleeding – a potential complication of PIBD – which can be treated with oral cholestyramine.
Diversions are shown to be more successful if performed at the early stages of the disease. Uncontrolled pruritus despite diversions or advanced liver disease may finally indicate liver transplantation. So if a prior alternative surgery will be performed, the least challenging procedure should be chosen.
Living-donor transplantation is reported to be safe and effective.Citation55 Nevertheless, since liver transplantation still has significant morbidity and mortality rates, protocols to identify those patients early and to follow up properly are mandatory.
The knowledge on the biological role of bile acids in metabolic pathways is still evolving. Thus, novel pharmaceuticals affecting bile acid circulation/metabolism are also promising for PFIC patients.
Finally, the diagnosis and treatment of patients with PFIC are difficult. They may result in ESLD if not diagnosed before the development of cirrhosis and hepatic fibrosis. Therefore, cholestasis should be considered as a whole and the differential diagnosis should promptly be made. The treatment strategy must be planned before hepatic fibrosis and cirrhosis develop. External or internal biliary diversion and liver transplantation approaches should be performed in selected cases. Early diagnosis and biliary diversions may prevent significant morbidity and mortality from ESLD.
Disclosure
The authors report no conflicts of interest in this work.
References
- FawazRBaumannUEkongUGuideline for the evaluation of cholestatic jaundice in infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and NutritionJ Pediatr Gastroentero Nutr2017641154168
- GhentCNBloomerJRKlatskinGElevations in skin tissue levels of bile acids in human cholestasis: relation to serum levels and to pruritusGastroenterology197773511251130908491
- Davit-SpraulAGonzalesEBaussanCJacqueminEProgressive familial intrahepatic cholestasisOrphanet J Rare Dis2009411219133130
- WagnerMZollnerGTraunerMNew molecular insights into the mechanisms of cholestasisJ Hepatol200951356558019595470
- JaquimeneEProgressive familial intrahepatic cholestasisClin Res Hepatol Gastroentrol201236Suppl 1S26S35
- BullLNCarltonVEStrickerNLGenetic and morphological findings in progressive familial intrahepatic cholestasis (Byler disease [PFIC-1] and Byler syndrome): evidence for heterogeneityHepatology19972611551649214465
- ClaytonRJIberFLRuebnerBHMckusickVAByler disease. Fatal familial intrahepatic cholestasis in an Amish kindredAm J Dis Child196911711121245762004
- van der WoerdWLHouwenRHvan de GraafSFCurrent and future therapies for inherited cholestatic liver diseasesWorld J Gastroenterol201723576377528223721
- SrivastavaAProgressive familial intrahepatic cholestasisJ Clin Exp Hepatol201441253625755532
- Oude ElferinkRPKremerAEMartensJJBeuersUHThe molecular mechanism of cholestatic pruritusDig Dis2011291667121691108
- ZollnerGTraunerMMechanisms of cholestasisClin Liver Dis200812112618242495
- GongZXuWJTianGLZhangTLvZNeonatal intrahepatic cholestasis caused by citrin deficiency differentiated from biliary atresiaEur J Pediatr Surg201626325525925988746
- PouponRIntrahepatic cholestasis of pregnancy: from bedside to bench to bedsideLiver Int200525346746815910481
- PerHArslanDGümüşHCoskunAKumandaşSIntracranial hemorrhages and late hemorrhagic disease associated cholestatic liver diseaseNeurol Sci2013341515622327309
- ArnellHFischlerBBergdahlSHepatobiliary scintigraphy during cholestatic and noncholestatic periods in patients with progressive familial intrahepatic cholestasis after partial external biliary diversionJ Pediatr Surg201146346747221376194
- EvasonKBoveKEFinegoldMJMorphologic findings in progressive familial intrahepatic cholestasis 2 (PFIC2): correlation with genetic and immunohistochemical studiesAm J Surg Pathol201135568769621490445
- TogawaTSugiuraTItoKMolecular genetic dissection and neonatal/infantile intrahepatic cholestasis using targeted next-generation sequencingJ Pediatr201617117117726858187
- FeranchakAPRamirezROSokalRJMedical and nutritional management of cholestasiSuchyFJSokolRJBalistreriWFLiver Disease in Children2nd edPhiladephia, PALippincott Williams and Wilkins2001195238
- van DijkRKremerAESmitWCharacterization and treatment of persistent hepatocellular secretory failureLiver Int20153541478148824905729
- JacqueminEde VreeJMCresteilDThe wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthoodGastroenterology200112061448145811313315
- HasegawaYHayashiHNaoiSIntractable itch relieved by 4-phenylbutyrate therapy in patients with progressive familial intrahepatic cholestasis type 1Orphanet J Rare Dis201498925022842
- NaoiSHayashiHInoueTImproved liver function and relieved pruritus after 4-phenylbutyrate therapy in a patient with progressive familial intrahepatic cholestasis type 2J Pediatr201416451219122724530123
- WangKSTiaoGBassLMChildhood Liver Disease Research Network (ChiLDReN). Analysis of surgical interruption of the enterohepatic circulation as a treatment for pediatric cholestasisHepatology20176551645165428027587
- WhitingtonPFWhitingtonGLPartial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasisGastroenterology19889511301363371608
- GunaydinMTanderBDemirelDDifferent techniques for biliary diversion in progressive familial intrahepatic cholestasisJ Pediatr Surg201651338638926382286
- CiesJJGiamalisJNTreatment of cholestatic pruritus in childrenAm J Health Syst Pharm200764111157116217519457
- SchukfehNMetzelderMLPetersenCNormalization of serum bile acids after partial external biliary diversion indicates an excellent long-term outcome in children with progressive familial intrahepatic cholestasisJ Pediatr Surg201247350150522424345
- YangHPorteRJVerkadeHJde LangenZJHulscherJBPartial external biliary diversion in children with progressive familial intrahepatic cholestasis and Alagille diseaseJ Pediatr Gastroenterol Nutr200949221622119561545
- ArnellHPapadogiannakisNZemackHFollow-up in children with progressive familial intrahepatic cholestasis after partial external biliary diversionJ Pediatr Gastroenterol Nutr201051449449920683202
- HalaweishIChwalsWJLong-term outcome after partial external biliary diversion for progressive familial intrahepatic cholestasisJ Pediatr Surg201045593493720438930
- RamachandranPShanmugamNPSinaniSAOutcome of partial internal biliary diversion for intractable pruritus in children with cholestatic liver diseasePediatr Surg Int201430101045104925064227
- DiaoMLiLZhangJSYeMChengWLaparoscopic cholecystocolostomy: a novel surgical approach for the treatment of progressive familial intrahepatic cholestasisAnn Surg201325861028103323187749
- MousaviSAKaramiHPartial internal biliary diversion in progressive familial intrahepatic cholestasis: introduction of a new approachHepat Mon2014143e1354924693315
- MochizukiKObatakeMTakatsukiMPartial internal biliary diversion for patients with progressive familial intrahepatic cholestasis type 1Pediatr Surg Int2012281515422033772
- KalicińskiPJIsmailHJankowskaISurgical treatment of progressive familial intrahepatic cholestasis: comparison of partial external biliary diversion and ileal bypassEur J Pediatr Surg200313530731114618520
- MaliVPFukudaAShigetaTTotal internal biliary diversion during liver transplantation for type 1 progressive familial intrahepatic cholestasis: a novel approachPediatr Transplant201620798198627534385
- KeitelVBurdelskiMVojnisekZDe novo bile salt transporter antibodies as a possible cause of recurrent graft failure after liver transplantation: a novel mechanism of cholestasisHepatology200950251051719642168
- EnglertCGrabhornERichterALiver transplantation in children with progressive familial intrahepatic cholestasisTransplantation200784101361136318049123
- SieboldLDickAAThompsonRRecurrent low gamma-glutamyl transpeptidase cholestasis following liver transplantation for bile salt export pump (BSEP) disease (posttransplant recurrent BSEP disease)Liver Transpl201016785686320583290
- KubitzRDrögeCKlugeSAutoimmune BSEP disease: disease recurrence after liver transplantation for progressive familial intrahepatic cholestasisClin Rev Allergy Immunol2015482-327328425342496
- BaghdasaryanAFuchsCDÖsterreicherCHInhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitisJ Hepatol201664367468126529078
- StapelbroekJMvan ErpecumKJKlompLWHouwenRHLiver disease associated with canalicular transport defects: current and future therapiesJ Hepatol201052225827120034695
- MiethkeAGZhangWSimmonsJPharmacological inhibition of apical sodium-dependent bile acid transporter changes bile composition and blocks progression of sclerosing cholangitis in multidrug resistance 2 knockout miceHepatology201663251252326172874
- CarielloMPiccininEGarcia-IrıgoyenONuclear Receptor FXR, bile acids and liver damage: introducing the progressive familial intrahepatic cholestasis with FXR mutationsBiochim Biophys Acta2017S0925S4439173033430344
- PellicciariRFiorucciSCamaioniE6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activityJ Med Chem200245173569357212166927
- HegadeVSSpeightRAEtheringtonREJonesDENovel bile acid therapeutics for the treatment of chronic liver diseasesTherap Adv Gastroenterol201693376391
- ZhouMWangXPhungVSeparating tumorigenicity from bile acid regulatory activity for endocrine hormone FGF19Cancer Res201474123306331624728076
- MitryRRHughesRDDhawanAHepatocyte transplantationJ Clin Exp Hepatol20111210911425755322
- TakebeTSekineKEnomuraMVascularized and functional human liver from an iPSC-derived organ bud transplantNature2013499745948148423823721
- JansenPLMüllerMSturmEGenes and cholestasisHepatology20013461067107411731993
- BullLNvan EijkMJPawlikowskaLA gene encoding a P-type ATPase mutated in two forms of hereditary cholestasisNat Genet19981832192249500542
- StrautnieksSSBullLNKniselyASA gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasisNat Genet19982032332389806540
- DeleuzeJFJacqueminEDubuissonCDefect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasisHepatology19962349049088666348
- Gomez-OspinaNPotterCJXiaoRMutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasisNat Commun201671071326888176
- CutilloLNajimiMSmetsFSafety of living-related liver transplantation for progressive familial intrahepatic cholestasisPediatr Transplant200610557057416856993