Abstract
Hypertension (HTN) is an important factor in progressive loss of renal function. The kidney can be both a contributor to and a target of HTN. The functional integrity of the kidney is vital for the maintenance of cardiovascular homeostasis. Chronic activation of the renin system causes HTN and, ultimately, end-organ damage. Direct renin inhibitors (DRIs) inhibit plasma renin activity (PRA), thereby preventing the conversion of angiotensinogen to angiotensin I; consequently, the levels of both Ang I and Ang II are reduced. There is no compensatory increase in PRA activity with DRIs as seen with angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs). There are reasons to speculate that renin inhibition might prove to be a superior strategy for blocking the renin–angiotensin–aldosterone system compared with ACEIs or ARBs. Evidence for the efficacy of aliskiren (a DRI) is considered to be relatively strong, based on published, short-term, double-blind, randomized, controlled trials showing that aliskiren is as effective as other antihypertensive agents in reducing blood pressure (BP), with no rebound effects on BP after treatment withdrawal. When combined with diuretics, fully additive BP reduction is seen. When given with an ACEI or ARB, aliskiren produces significant additional BP reduction indicative of complimentary pharmacology and more complete renin–angiotensin system blockade.
Epidemiology of kidney disease in HTN
In the 18th century, Dr. Richard Bright, the father of nephrology who was physician to the Queen of England, and numerous other medical scientists, suspected that the kidney plays an important part in hypertension (HTN). A long-held view is that the kidney is somehow involved in the mechanisms of HTN. The precise stimulus that elicits HTN is still uncertain. But, there are vast amounts of valuable information about the mechanisms of HTN once this process has started. The kidney is an important target organ for HTN-related injury.Citation1
HTN is an important factor in progressive loss of renal function. Epidemiology has provided sound scientific evidence for the core concepts of progressive renal damage secondary to HTN. Multiple risk factors, such as age, atherosclerosis, diabetes, and race, are associated with progressive damage to the kidney. Because of its major contribution to HTN-related renal damage, diabetes has been a major focus of epidemiological studies. Improved medical care for patients with chronic renal disease has contributed to annual increases in the number of patients who survive with end-stage renal disease (ESRD). Most patients with ESRD have HTN, and most forms of renal disease are associated with HTN.Citation2,Citation3 HTN is most evident with glomerular diseases, in which 70%–80% of patients are affected, including diabetic nephropathy (DN), membranous glomerulonephritis, membranoproliferative glomerulonephritis, and focal segmental glomerulonephritis. Minimal change nephropathy is a notable exception. Tubulointerstitial disorders, such as analgesic nephropathy, chronic interstitial nephritis, medullary cystic diseases, and chronic reflux nephropathies, are less commonly associated with HTN.Citation4
Risk factors for progression of chronic kidney disease
Among the 332,544 men screened for the Multiple Risk Factor Intervention Trial from the year 1973–1975, older age, lower income, higher serum cholesterol concentrations, cigarette smoking, diabetes mellitus (DM), a history of HTN, and black race were also found to be associated with an increased risk of ESRD.
The third National Health and Nutrition Examination Survey has added new insights to serum creatinine and hemoglobin concentrations as risk factors for ESRD. Intervention trials for each risk factor that can be modified have not been completed. Accordingly, improvements in certain risk factors (eg, anemia) have not been shown to impact on the progression of renal disease.Citation2
HTN and chronic kidney disease: hemodynamic abnormalities
In parenchymal renal disease, multiple factors can influence cardiac output, systemic vascular resistance, or both. Blood pressure (BP) is the product of cardiac output multiplied by total peripheral vascular resistance. In most patients with chronic renal failure, studies suggest that cardiac output is normal or elevated, whereas overall extracellular fluid volume is expanded. Systemic vascular resistance is inappropriately elevated relative to cardiac output, reflecting a net shift in vascular control toward vasoconstriction.
Several factors affecting vascular tone are disturbed in patients with chronic renal failure, including increased adrenergic tone and activation of the renin–angiotensin system (RAS), endothelin, and vasoactive prostaglandins. An additional feature in some disorders appears to depend on reduced vasodilation, such as in impaired production of nitric oxide (NO).Citation3
HTN and chronic kidney disease: mechanisms
The functional integrity of the kidney is vital for the maintenance of cardiovascular homeostasis. The nephron, comprised of the glomerulus and the tubules, is the primary functional unit of the kidney. The kidney plays a critical role in the long-term regulation of BP. Thus, pathological abnormalities primary to the kidney may lead to an elevation of BP. As a corollary, HTN due to nonrenal causes can damage the kidney. The resulting loss of renal mass, in turn, can secondarily lead to further elevations in BP. The kidney can be both a contributor to and a target of HTN. Systemic BP elevation is associated with increased vascular tone due to decreased production or action of vasodilator molecules, such as endothelium-derived NO and prostacyclin (PgI2). At the same time, there is either maintenance or increased production of vasoconstrictors such as angiotensin II (Ang II) and endothelin-1. This imbalance is often termed endothelial dysfunction.
Increased renal sympathetic nerve activity may contribute to vasoconstriction and augment tubular reabsorption of sodium. Many of the molecules promoting vasoconstriction also cause long-term effects on the nephron by promoting growth of vascular smooth muscle cells (VSMCs) and mesangial cells, decreasing apoptosis, stimulating chemotaxis of vascular inflammatory cells into the vessel wall and the tubular interstitium, and stimulating the production of molecules that expand and alter the composition of the extracellular matrix in the mesangium and interstitium. The resulting glomerular HTN can lead to glomerular basement membrane damage and proteinuria.
Other conditions, such as DM, may also contribute to the glomerular basement membrane damage caused by HTN or may induce it independently through alteration of basement membrane proteins. Both glomerular HTN and proteinuria are associated with oxidative stress that can cause activation of circulating leukocytes and their diapedesis into the vessel wall and interstitium and can stimulate the release of cytokines and growth factors that lead to extracellular matrix formation, progressive sclerosis of both the glomerulus and tubules, and ultimately, loss of nephron units. When the compensatory capacity of the remaining nephrons is exceeded, renal function progressively deteriorates and renal failure develops.Citation5–Citation9
Consequences of renal damage in HTN
Functional changes that result from HTN include a decline in glomerular filtration rate (GFR) and abnormalities in tubular function, including new onset or worsening of proteinuria. These functional changes lead to structural changes in the glomerular basement, expansion of the mesangial and interstitial matrix, ultimately resulting in sclerosis of both glomerular and tubular elements.Citation5–Citation9 Vascular endothelium, under normal conditions, is an important source of vasodilators such as NO, PgI2, and one or more endothelium-derived hyperpolarizing factors (EDHFs) that are not yet chemically characterized. Endothelium-derived NO has many functions. Among them are VSMC relaxation, modulation of renal medullary blood flow, inhibition of VSMC growth, inhibition of platelet aggregation, inhibition of polymorphonuclear (PMN) leukocyte and monocyte (MF) adhesion molecule expression and immigration into the vascular wall and interstitium, and augmentation of apoptosis or programmed cell death. The formation or the actions of endothelium-derived NO are blunted in patients with HTN. This impairment may be more exaggerated in patients with concomitant DM. NO shares many of its actions in the vasculature and in the kidney with PgI2 or other EDHFs.Citation5–Citation9
Imbalance in factors affecting vascular tone and structure
HTN is associated with an altered balance in the elaboration or biological action of vasodilator and vasoconstrictor molecules. The production of vasodilators like PgI2 and NO is diminished in HTN, whereas the production of catecholamines, reactive oxygen species (ROS), Ang II, endothelin-1, and other endothelium-derived constricting factors is either maintained or increased.
Many of the molecules that augment vascular tone also have long-term mitogenic effects on VSMCs and glomerular mesangial cells, activate adhesion molecules on leukocytes and platelets with resulting influx of these cells into the vessel wall, decrease apoptosis, and stimulate extracellular (interstitial and mesangial) matrix formation, whereas molecules that promote vasodilation tend to inhibit these processes.Citation10–Citation14
ROS reduces the biological effects of NO
The potent vasodilator molecule, NO, is produced by constitutively expressed endothelial NO synthase from l-arginine via a 5-electron redox reaction. NO also inhibits VSMC proliferation and migration, mononuclear (MF) and PMN leukocyte adhesion molecule expression, and platelet aggregation. Injured VSMCs or endothelial cells; activated vascular wall mast cells, fibroblasts, macrophages, and leukocytes; and oxidation of norepinephrine (NE) from renal sympathetic nerves produce increased amounts of ROS that then interact with NO to form the potent cytotoxic peroxynitrite radical (OONO-). This radical interacts with proteins in the kidney that are important for normal glomerular and tubular functions to reduce their activities.Citation15–Citation17
Renin–angiotensin cascade
Activation of the renin system occurs following the release of renin from juxtaglomerular apparatus within the kidney. Renin cleaves the circulating angiotensinogen to angiotensin I (Ang I); Ang I itself is inactive, but is converted to the biologically active peptide Ang II by the ACE, which is produced in the lungs. Ang II is a vasoconstrictor; it binds with type I Ang II (AT1) receptors in the SMCs of the peripheral blood vessels causing vasoconstriction and, consequently, increased peripheral vascular resistance and increased BP. Activation of AT1 receptors by Ang II also stimulates the release of aldosterone from the adrenal gland, which promotes retention of sodium and water along the nephron, further increasing BP (). In pathologic conditions, the renin system can become chronically activated as increases in renin system activity lead to increased BP, and then chronic activation of the renin system causes HTN and ultimately end-organ damage.
Figure 1 The renin–angiotensin cascade and the three available approaches to pharmacologic inhibition of production or action of angiotensin II.
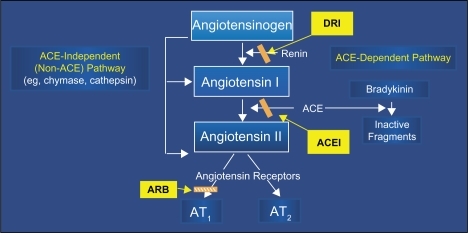
There are at least two alternative pathways for Ang II formation that do not rely on either renin or ACE. In the non-renin pathway, tissue plasminogen activator (tPA) forms Ang II directly from angiotensinogen, bypassing the renin-mediated production of Ang I as an intermediate.
A second alternative pathway involves enzymes like chymase that can form Ang II from Ang I via an ACE-independent mechanism. These alternative pathways are implicated in the gradual return toward pretreatment Ang II concentrations during treatment of patients with angiotensin-converting enzyme inhibitors (ACEIs) and provide a rationale for considering angiotensin receptor blockers (ARBs) that directly inhibit the binding of Ang II to the AT1 receptor either in conjunction with or as an alternative to ACEI therapy.Citation18,Citation19
Ang II and kidney diseases
Ang I formed in the afferent arteriole via the action of renin synthesized and secreted by the modified VSMCs in the arteriolar tunica media (juxtaglomerular cells) then interacts with ACE, a membrane bound enzyme on the luminal plasma membrane of the endothelial cells to form Ang II. Ang II can bind to and activate angiotensin AT1 receptors located on VSMCs, endothelial cells, tubular cells, and mesangial cells. Binding of Ang II to afferent arteriolar juxtaglomerular cell AT1 receptor further diminishes renin production, thus, providing an intrarenal feedback loop for the control of renin secretion and Ang II generation.Citation20 Ang II plays a pivotal role in pathological processes in HTN that ultimately leads to renal glomerular and tubular destruction and renal failure. Ang II, acting either through signal transducing mechanisms or directly on cells to stimulate the production or activation of mediators, causes infiltration of inflammatory cells or increased production of mesangial and interstitial matrix with resultant glomerular and tubular injuries and destruction (nephron loss). The remaining normal nephrons are forced to compensate by increasing filtration rate, which (in the presence of increased Ang II) increases glomerular capillary pressure and perpetuates this progressive spiral of deteriorating renal function.Citation21 Ang II stimulates several key enzymes or cytokines in vascular and tubular cells within the kidney to produce increased amounts of ROS. In addition, Ang II activates NF-kB, a nuclear factor pivotal to the production of inflammatory cytokines. These cytokines exert their effects, in part, through oxidative mechanisms.Citation12
Aldosterone promotes renal fibrosis by multiple mechanisms
The potent mineralocorticoid, aldosterone, is produced by the adrenal zona glomerulosa in response to Ang II stimulation. There is also evidence that the myocardium, vascular endothelial cells and SMCs are capable of synthesizing aldosterone. Aldosterone has been implicated in vascular, myocardial, and renal fibrosis that occurs in patients with arteriosclerosis, congestive heart failure, and renal failure, particularly if HTN is also present and is accompanied by activation of the renin–angiotensin–aldosterone system (RAAS).
The mechanisms by which aldosterone contributes to vascular and renal fibrosis are not well understood, but there are several experimental findings that support the mechanisms. Aldosterone has been reported to augment affinity of Ang II for the AT1 receptor, thereby enhancing the pressor effects of Ang II; augment the influx of sodium into VSMCs, contributing to VSMC hypertrophy;Citation22 potentiate the action of NE by inhibiting its uptake into VSMCs; contribute to endothelial dysfunction by inhibiting NO synthesis; stimulate the production of plasminogen activator inhibitor-1, which promotes fibrosis; and promote synthesis of Type I collagen by interstitial fibroblasts.Citation23–Citation29
Pathways leading to progressive renal failure
Both hypertensive and nonhypertensive injuries that cause loss of single nephron units result in HTN in the remaining glomeruli (glomerular HTN).
Glomerular HTN can lead to injury to the glomerular basement membrane, causing it to leak plasma proteins in the urine. The proximal tubules attempt to reabsorb this filtered protein. This causes injury to the tubular cells, activates an inflammatory response, and is associated with the development of lipid metabolic abnormalities that create further oxidative stress on the already compromised glomerulus. The resultant tubular inflammatory response and renal microvascular injury activate pathways that lead to fibrosis and scarring of both glomerular and tubular elements of the nephron.
An additional consequence of glomerular HTN and resultant reduction in GFR activates growth factors and cytokines. This promotes an influx of monocytes and macrophages into the vessel wall and into the renal interstitium as well as differentiation of renal cells into the fibroblasts. Monocytes, macrophages, and fibroblasts are capable of producing those growth factors and cytokines that activate pathways leading to expansion of extracellular matrix, fibrosis, and loss of both tubular and glomerular structures.
HTN and renal parenchymal diseases are closely interrelated. Primary renal diseases eventually disrupt sodium and volume control to produce clinical HTN. Activation of the RAS, adrenergic nervous system, and endothelin can enhance vasoconstriction and mediate tissue injury by the promotion of inflammatory cytokines and interstitial fibrosis. Arterial HTN accelerates renal disease and hastens the progression to end-stage renal failure.
The dual significance of proteinuria
In patients with proteinuric renal diseases, the rate of progression of renal insufficiency is determined by the level of BP and proteinuria. The reduction of BP and proteinuria has a beneficial impact on renal risk. Observational studiesCitation30,Citation31 have also demonstrated that proteinuria is a marker for systemic vascular injury. It is hoped that efforts to reduce proteinuria and control BP will improve cardiovascular risk and reduce mortality. In this belief, the National Kidney Foundation and the American Diabetes Association have revised their recommendations for management of high-risk patients for tighter BP control to <130/80 mmHg.Citation32–Citation39,Citation33
Renal disease in HTN: core concepts of treatment
BP levels are reliable predictors of renal outcome. Absolute systolic and diastolic BP levels correlate with renal risk. In patients with renal disease, BP reduction clearly affects renal outcome. Proteinuria is an independent predictor of progression of renal disease, independent of absolute BP levels. In patients with and without diabetes, BP reduction provides protection against a progressive decline in renal function. The absolute level of BP reduction is important. African Americans may be at greater risk compared with other racial or ethnic groups. Relative renal hypoperfusion during initial stages of HTN correlates with a transient limited increase in serum creatinine; however, this phenomenon has become a deterrent to effective therapy rather than a hallmark of appropriate antihypertensive management. The reduction of BP and proteinuria has an impact on both cardiovascular and renal risk for, thus far, unexplained reasons.
Management of risk factors in HTN and chronic kidney disease
Current National Kidney Foundation recommendations for the management of patients with HTN and chronic kidney disease (CKD) include antihypertensive therapy to reduce BP to goal values, use of lipid-lowering drugs to reduce low-density lipoprotein cholesterol and triglycerides to goal values, and smoking cessation. Recent evidence indicates that anemia may also have a deleterious influence on the incidence of cardiovascular events and on death in patients with chronic renal insufficiency. Similar observations have been made for abnormalities of calcium and phosphorus. The beneficial consequences of treating the last two risk factors for cardiovascular events in patients with chronic renal disease are less well defined than are the beneficial effects of aggressively managing the first three risk factors.Citation40–Citation42
Proteinuria reflects injury to glomerular capillary endothelial cell basement membranes, allowing the extravasation of protein from the vascular compartment into the urine. In patients with HTN and diabetes, this process probably also occurs in other vascular beds in addition to the renal vascular bed. This pathological process may explain why patients with proteinuria are at increased cardiovascular risk. Although important, aggressive management of HTN in patients with chronic renal disease and proteinuria is not sufficient. In addition to the landmark trial by Lewis et al demonstrating that the ACEI captopril reduced both the combined primary end point (time to doubling of serum creatinine, death or the need for dialysis) and proteinuria in patients with type 1 diabetes, other clinical trials involving fewer numbers of patients have also demonstrated a beneficial effect of ACEIs and ARBs on proteinuria in patients with diabetes and in hypertensive and normotensive patients without diabetes with immunoglobulin nephropathy.Citation49 Although the dose-dependency of this effect of ACEIs and ARBs on protein has not been extensively examined, dose-titration of agents from either class seems warranted in patients with proteinuria, to monitor for possible adverse effects as the dosage is increased.
There are limited but encouraging data to suggest that combination of an ACEI and ARB may be more effective than either agent alone in reducing proteinuria. Drugs from other antihypertensive classes appear to have less effect on proteinuria than ACEIs and ARBs. In fact, certain dihydropyridine calcium channel blockers (CCBs) have been associated with worsening of proteinuria. Management of identified cardiovascular risks is just as important in the hypertensive patient with chronic renal disease as in the patient with diabetes. This includes drugs and diet to maintain a normal lipid profile; adequate control of BP, dietary protein, and sodium restriction; smoking cessation; reduction of overweight to normal body mass index; and a regular aerobic exercise program. Patients with anemia are at increased cardiovascular risk, and early initiation of erythropoietin and iron therapies for those that are erythropoietin and iron deficient, respectively, is warranted. Patients with hyperhomocysteinemia may be candidates for folic acid therapy.Citation43–Citation47,Citation33
The management of HTN and chronic renal disease in patients with diabetes should be even more aggressive than in the patients without diabetes. The goal of antihypertensive therapy should be BP values <130/80 mmHg, except in those patients with diabetes who have >1 g/d of proteinuria. In the latter patients, the goal BP should be <125/75 mmHg. Multiple antihypertensive drugs will usually be required to achieve these BP goals. Drugs or drug combinations that are most effective in reducing or abolishing proteinuria should be used and the doses should be modified based on frequent determination of urine protein, until the desired effect is achieved.
Effective risk factor management of patients with diabetes also includes control of hyperlipidemia and hyperglycemia. Modest dietary protein restriction is recommended for the hypertensive patients with diabetes whose creatinine clearance is already diminished. Sodium restriction is indicated in patients with CRD and impaired ability to excrete sodium. Smoking cessation is a key to successful cardiovascular risk reduction.Citation43–Citation47,Citation33
CKD: initial treatment recommendations
In randomized clinical trials of patients with both diabetic and nondiabetic chronic renal disease, ACEIs and ARBs have frequently been more effective than other antihypertensive agents in ameliorating a progressive decline in renal function and reducing proteinuria even when comparable reduction in BP is achieved. In Microalbuminuria, Cardiovascular, and Renal Outcomes – Heart Outcomes Prevention Evaluation (MICRO-HOPE), Citation48 which was a substudy of the HOPE study, among patients with diabetes at high risk of cardiovascular events, treatment with ramipril lowered the incidence of the development of overt nephropathy. In Irbesartan Diabetic Nephropathy TrialCitation49 among patients with type 2 diabetes, the ARB irbesartan demonstrated a significantly decreased incidence of end points assessing the progression of renal disease compared with amlodipine and placebo. These effects appeared to be independent of irbesartan’s effects in lowering BP. In Renal and Cardiovascular Outcomes in Patients with Type 2 Diabetes and Nephropathy Trial,Citation50 losartan conferred significant renal benefits in patients with type 2 diabetes and nephropathy. The observed decrease in progression of renal failure corresponded to an average of 2-year delay in need for dialysis or transplantation in losartan group vs placebo group.Citation50
For these reasons, it is currently recommended that initial therapy of patients with microalbuminuria or macroalbuminuria, laboratory evidence by either creatinine clearance or serum creatinine measurements of renal insufficiency or with diabetes regardless of the level of renal function include an ACEI or ARB. Numerous studies have suggested benefits of dual RAS blockade in patients with albuminuria when compared with either an ARB or an ACEI monotherapy.Citation51 In a thorough meta-analysis of 49 studies involving over 6,000 patients, Kunz et alCitation52 found “encouraging” evidence that dual RAS blockade reduced proteinuria by 20%–25% more than either drug alone.Citation52 The most recent landmark study, the Renal Outcomes with Telmisartan, Ramipril, or Both, in People at High Vascular Risk study,Citation53 is no exception in this regard. The increase in albuminuria was reduced with a combination of telmisartan and ramipril when compared with monotherapy, but worsened major renal outcomes overall. This raises many questions about the relationship between proteinuria and renal failure that must be answered in future trials.
Escape of Ang II despite ACE inhibition
A growing body of evidence suggests that despite effective inhibition of Ang II activity, the non-ACE synthetic pathways still permit Ang II generation via serine proteases such as chymase, cathepsin G, and tPA. ARBs block the AT1 receptor that transduces many of the known physiologic actions of Ang II. Other Ang II receptors, whose functions are less well defined, exist and are not blocked by ARBs. In this study, 19 hypertensive patients were treated with increasing doses of enalapril. The investigators concluded that the use of enalapril 20 twice daily effectively reduced BP but did not constantly suppress plasma Ang II and aldosterone. This phenomenon of the return of plasma Ang II to pretreatment values despite continued inhibition of ACE has been called angiotensin escape.Citation54
Uniqueness of direct renin inhibition
The emergence of the direct renin inhibitors (DRIs) has afforded clinicians a new strategy for RAS blockade. In outcomes studies that have used ACEIs or ARBs, there has been a relatively high residual event rate in the treatment arm, and this has been ascribed to the fact that neither ACEIs nor ARBs completely repress RAS. For this reason, combined RAS blockade with an ACEI and ARB has emerged as a therapeutic option. In HTN, combined RAS blockade elicits only a marginal incremental drop in BP and it does not further lower the risk for cardiovascular events.Citation55
DRIs represent a new class of antihypertensive agents that act on the renin system at its point of activation. DRIs have a unique effect on the components of the system. DRIs inhibit the activity of renin, thereby preventing the conversion of angiotensinogen to Ang I and reducing plasma renin activity (PRA) consequently; DRIs reduce the level of both Ang I and Ang II. Similar to ACEIs and ARBs, the activity of negative feedback loop is reduced with DRIs and renin release is increased; however, unlike with ACEIs and ARBs, the activity of the released renin is inhibited by DRIs. Consequently, there is no compensatory increase in renin system activity with DRIs, and this is reflected by the level of PRA, which decreases in the presence of DRIs. The clinical implications of the reduction in PRA with DRIs vs an increase with ACEIs and ARBs are not known at present. Aliskiren (DRI) is a newer agent that inhibits renin, the rate-limiting step in the RAAS. There are several theoretical reasons to suggest that aliskiren may have a protective action superior to those of ACEIs and ARBs.
Brunner et alCitation56 reported an association between high levels of PRA (as measured by the renin–sodium profile) and the incidence of myocardial infarction in patients with HTN. Parikh et alCitation57 based on the Framingham Heart Study provided some insights that all-cause mortality increased with higher plasma renin at baseline in the whole sample (standardized hazard ratio [HR] 1.14; P = 0.046) and in hypertensive patients (HR 1.16; P = 0.046) but that the strength of these associations decreased with the length of follow-up. On the other hand, plasma renin did not predict hard cardiovascular or coronary events.
RAS inhibition, limiting the hemodynamic effects of Ang II, reduces proteinuria and is renoprotective in the long term. The hemodynamic changes caused by RAS inhibitors partially contribute to the antiproteinuric effect. Loriga et alCitation58 in a trial of benazepril or valsartan showed direct correlation between PRA and decrease in proteinuria, indicating that renin may contribute to renal vasoconstriction and could be a further determinant of kidney damage besides a promising target for renoprotection.Citation58,Citation59 Renal vascular damage involving the afferent arteriole may then cause the level of PRA to increase,Citation60 thus, establishing a relation between increasing levels of PRA and the risk of a major coronary event. Baldoncini et alCitation61 investigated the relationship between PRA and the urinary albumin excretion rate (UAER), an early marker of HTN-related renal changes, in human essential HTN. Their study demonstrated that the UAER is elevated in high-renin essential hypertensive patients, suggesting that high PRA accelerates the onset of early renal changes in human essential HTN.
The study by Yeyati and AdroguéCitation62 support the existence of inappropriate activation of the RAS in humans with chronic renal disease. Such stimulation might play a critical role in the pathophysiology of advanced renal injury. Their investigation examined the PRA and the ratio of this parameter to the simultaneously measured GFR (PRA/GFR) in normal volunteers and in patients with chronic renal disease. A mean 10-fold increase in the PRA/GFR ratio was observed in patients with chronic renal disease as compared with normal volunteers. The observed augmentation in PRA was not caused by the physiologic mechanisms aimed at conserving urinary sodium since a positive correlation was found between PRA/GFR and the fractional excretion of sodium as opposed to that of normal controls.
PRA and serum aldosterone levels are elevated in hypertensive left ventricular (LV) hypertrophy and relate to LV mass index. In addition to BP, activation of the RAAS may be an important nonhemodynamic mechanism in the control of LV hypertrophy.Citation63
Increased PRA plays an important role in the development of HTN in subjects with central obesity. Licata et alCitation64 demonstrated that PRA was significantly higher in obese hypertensive than in obese normotensive subjects.
Aliskiren neutralized the increase in PRA associated with agents that stimulate renin release. This was studied in 1,093 patients with mild to moderate HTN and diastolic BP <95 mmHg and >110 mmHg at randomization. Pooled analysisCitation65 was done from four randomized, double-blind, multicenter trials, lasting 8 weeks, in patients who had PRA measured at the baseline and at the end of the study. Aliskiren, 150–600 mg, therapy caused significant reductions in plasma inactivity of greater than 70%, vs placebo. In contrast, ramipril, hydrochlorothiazide (HCTZ), and valsartan stimulated increases in PRA. When combined with ramipril, HCTZ, or valsartan, aliskiren continued to provide significant reductions in PRA, thus, neutralizing the affect of antihypertensive agents that lead to increased activity of this biomarker.
Aliskiren was also studied alone or in combination with valsartan, in 1,797 patients with mild to moderate HTN, and showed reduced PRA and plasma aldosterone levels, both as monotherapy and as combination therapy, in an 8-week, randomized, double-blind, placebo-controlled study.Citation66,Citation67
Aliskiren provides dose-dependent BP-lowering efficacy
Evidence for the efficacy of aliskiren is considered to be relatively strong, based on published, short-term, double-blind, randomized, and controlled trials showing that aliskiren is as effective as other antihypertensive agents in reducing BP. When added to other antihypertensive agents, aliskiren provided additional BP lowering effects. Several trials have shown 150 mg or 300 mg daily dose of aliskiren (given for up to 8 weeks) to be superior to placebo in lowering BP.Citation68
The efficacy of aliskiren has been tested in various clinical trials, including placebo-controlled studies and comparator studies with other antihypertensive agents, and in combination with other antihypertensive agents. Several studies have compared aliskiren with other antihypertensive agents, including HCTZ diuretics,Citation69 ARBs (valsartan,Citation70 irbesartan,Citation71 and losartan),Citation72 ACEIs (ramipril and enalapril),Citation73,Citation74 and dihydropyridine CCBs (amlodipine),Citation75 in patients with uncomplicated HTN. There are no comparative or combination trials with loop diuretics, α-blockers, α-agonists, or direct vasodilators. As mentioned earlier, HCTZ diuretics, ACEIs, and ARBs each cause reactive increases in PRA. Therefore, it is hypothesized that these drugs used in combination with aliskiren will inhibit the increase in renin activity.
Patients with mean sitting diastolic BP (MSDBP) of 95–109 mmHg were randomized to aliskiren 150, 300, or 600 mg or placebo once daily for 8 weeks.Citation71 Patients completing this treatment phase entered into a 2-week, treatment-free withdrawal period. Office BP was recorded at baseline, weeks 2, 4, 6, and 8 of treatment, and 4 days and 2 weeks after cessation of treatment. A subgroup of patients underwent ambulatory BP monitoring. In total, 672 patients were randomized to treatment. After 8 weeks, aliskiren 150, 300, and 600 mg significantly reduced mean sitting BP (systolic/diastolic) by 13.0/10.3, 14.7/11.1, and 15.8/12.5 mmHg, respectively, vs 3.8/4.9 mmHg with placebo (all P < 0.0001 for systolic and diastolic BP). The BP-lowering effects of aliskiren persisted for up to 2 weeks after treatment withdrawal. Aliskiren significantly reduced the mean 24-hour ambulatory BP (P < 0.0001 vs placebo with all doses) exhibiting smooth and sustained effects and high trough-to-peak ratios. Aliskiren was well tolerated; overall adverse event rates were 40.1%, 46.7%, and 52.4% with aliskiren 150, 300, and 600 mg, respectively, and 43.0% with placebo. Few patients discontinued the treatment due to adverse events.Citation71
Aliskiren monotherapy or in combination with HCTZ
Aliskiren monotherapy provided significant BP-lowering effects, which was greater when combined with HCTZ. The compensatory increase in PRA induced by HCTZ can be neutralized by renin inhibition with aliskiren. This was studied with varying doses of aliskiren (75, 150, or 300 mg) and HCTZ (6.25, 12.5, or 25 mg) alone and in combination in an 8-week, randomized, placebo-controlled trial of 2,776 patients with mild to moderate HTN.Citation76 The primary objectives were to assess the changes in MSDBP and mean sitting systolic BP (MSSBP) at baseline. The effects on PRA were also assessed. BP reductions were greater with the combinations than with monotherapy. Responders were defined as achieving a MSDBP of 90 mmHg and/or a > 10 mmHg reduction from baseline. BP control was defined as a MSDBP of 90 mmHg and a MSSBP of 140 mmHg. Superior responder rates occurred with aliskiren 300 mg (63.9%; P = 0.0005), HCTZ 12.5 and 25 mg (60.6% and 59.0%, respectively; for both P = 0.02), and all combination doses compared with placebo. Better control and responder rates for all combinations of aliskiren (75–300 mg) with HCTZ 25 mg and aliskiren 300 mg/HCTZ 12.5 mg were superior to both monotherapies (P = 0.05). Aliskiren reduced the PRA by up to 65% from baseline. PRA decreased in all of the combination groups; however, HCTZ alone increased PRA by up to 72%.Citation76
Aliskiren monotherapy or in combination with ARBs
Aliskiren used in combination with valsartan may provide additive and tolerable BP-lowering effects. Aliskiren and valsartan were investigated as monotherapy and in combination in an 8-week, randomized, placebo-controlled, dose-escalation trial of 1,797 patients with uncomplicated HTN.Citation65,Citation77 Patients received either aliskiren 150 mg, valsartan 160 mg, the combination of aliskiren 150 mg and valsartan 160 mg, or placebo. After 4 weeks, all medication doses were doubled, and therapy was continued for an additional 4 weeks. Seated trough cuff BP was measured at baseline, 4th week, and 8th week. BP reductions with the combinations were greater than the reductions with both monotherapies. The combination of aliskiren 300 mg and valsartan 320 mg had statistically superior reductions in MSSBP and MSDBP compared with aliskiren 300 mg alone and valsartan 320 mg alone at week 8.Citation77
Multiple studies have evaluated the efficacy and safety of aliskiren in comparison with various ARBs (irbesartan,Citation71 losartan).Citation72 In a 4-week study that included ambulatory BP monitoring, there was no statistical difference between the change in daytime systolic BP with 100 mg/d losartan and 300 mg aliskiren. In another study, the antihypertensive efficacy of aliskiren 150 mg was found to be similar to that of irbesartan 150 mg/d, which at this dose increased both PRA (+116.3%) and plasma renin concentration (PRC, +107.0%).Citation78
Aliskiren alone vs in combination with ramipril
Two trials have compared the BP-lowering effects of aliskiren with that of the ACEI ramipril. In hypertensive patients with diabetes, aliskiren (300 mg) was equally effective in lowering MSDBP (the primary efficacy variable) compared with ramipril (10 mg). A significantly greater reduction in sitting systolic BP was seen in aliskiren-treated patients. Similar results were obtained in a longer term (6 months) comparison of aliskiren and ramipril in hypertensive patients without diabetes. Ramipril also produced RAS activation. In one study, ramipril 10 mg increased both PRA (+110.6%) and PRC (+67.9%).Citation79
Aliskiren monotherapy vs combination therapy with amlodipine
One study has assessed the efficacy and tolerability of aliskiren combined with a CCB. Patients showing an inadequate response to amlodipine 5 mg/d were randomized to continued therapy with amlodipine 5 mg, up-titration to amlodipine 10 mg, or the addition of aliskiren 150 mg to amlodipine 5 mg. Up-titration of amlodipine or addition of aliskiren resulted in significantly greater BP reduction than continuation of amlodipine 5 mg/d, with no difference between the low-dose combination and high-dose amlodipine. The 10-mg amlodipine dose was associated with a higher incidence of treatment-related peripheral edema (a dose-dependent side effect of CCBs) compared with the low-dose combination (11.2% vs 2.1%).Citation80
Dual RAS blockade and kidney diseases
Dual blockade of the RAS with losartan and aliskiren was beneficial in reducing albuminuria among patients with type 2 diabetes having HTN and nephropathy, as seen in aliskiren in the Evaluation of Proteinuria in Diabetes (AVOID) Trial,Citation81 in which 599 subjects were initially administered losartan for 3 months, following which they were randomized to receive either aliskiren or placebo for 6 months along with the ARB. The dose of aliskiren was 150 mg for the first 3 months and was doubled during the subsequent 3 months. The early morning urine sample was evaluated for albumin–creatinine ratio. It was observed that the main albumin–creatinine ratio reduced by 20% in subjects on aliskiren as compared with placebo. Also, a 50% reduction in the ratio was noted in almost 1 of 4 patients on aliskiren. Aliskiren also appeared to have a modest BP-lowering effect; however, the reduction in albuminuria was preserved after controlling for this change in BP. This is an important finding since many patients who receive angiotensin-receptor blockers or ACEIs still have progression of renal disease. These results are promising, and it will be of interest to see whether an ongoing clinical outcomes study of patients with DN confirms the superiority of dual RAS blockade with an ARB and aliskiren vs ARB-based therapy alone.Citation82
Conclusion
There are reasons to speculate that renin inhibition might prove to be a superior strategy for blocking the RAS compared with existing drugs. Evidence for the efficacy of aliskiren (a DRI) is considered to be relatively strong, based on published, short–term, double-blind, randomized, controlled trials showing that aliskiren is a potent inhibitor of renin and its bioavailability is sufficient to produce sustained suppression of PRA and BP reduction after chronic oral dosing. This suppression is present whether the drug is given alone or in combination and persists after drug discontinuation. It is logical to assume that PRA suppression also leads to downstream suppression of Ang II formation. Although there are supportive data from animal studies, it is also unknown whether aliskiren will prove to be superior, equal, or inferior to ACEIs or ARBs in terms of end-organ protection. These are the key outstanding questions, the answers to which will ultimately determine the place of renin inhibition and aliskiren in the treatment of HTN and related cardiovascular disorders. A landmark clinical trials program, Aspire Higher, investigates the potential of aliskiren to move cardio-renal protection beyond that offered by ACEIs and ARBs. It includes 4 morbidity and mortality trials for aliskiren, in heart failure, DN, and prevention of cardiovascular outcomes in elderly. Given the well-demonstrated effectiveness of many antihypertensive agents in reducing cardiovascular end points, most clinicians need proof of either a significant superiority in preventing these end points or a compelling drug class benefit to identify the role of renin inhibition in the treatment of HTN.
Acknowledgements
The author would like to sincerely thank Ms. Ruthie York for her diligent work and secretarial assistance.
Disclosure
The author reports no conflict of interest in this work.
References
- TraubeLUeber den zusammenhang von herz und nierenkrankeiten. Page IH. Effect on renal efficiency of lowering blood pressure in cases of essential hypertension and nephritisJ Clin Invest19341390916694258
- KlagMJWheltonPKRandallBLBlood pressure and end-stage renal disease in menN Engl J Med1996334113187494564
- TextorSCRenal Parenchymal disease and hypertension atlas of diseases of the kidneySchrierRWISN Commission on Nephrology Informatics and NKF cyberNephrology20013 Chapter 2.4, (Figure 2.7).
- SmithMCDunnMJHypertension in renal parenchymal diseaseLaraghJHBrennerBMHypertension: Pathophysiology, Diagnosis and ManagementNew YorkRaven Press199520812102
- VanhouttePMBoulangerCMEndothelium-dependent responses in hypertensionHypertens Res19951887987584924
- LüscherTFBockHAThe endothelial L-arginine/nitric oxide pathway and the renal circulationKlin Wochenschr1991696036091753683
- LlinasMTGonzalezJDRodriguezFNavaETaddeiSSalazarFJRenal changes induced by nitric oxide and prostaglandin synthesis reduction: effects of trandolapril and verapamilHypertension19983126576649461237
- ZouAPWuFCowleyAWJrProtective effect of angiotensin II-induced increase in nitric oxide in the renal medullary circulationHypertension199831Pt 22712769453315
- StecDEMattsonDLRomanRJInhibition of renal outer medullary 20-HETE production produces hypertension in Lewis ratsHypertension199729Pt 23153199039121
- CampeseVMNeurogenic factors and hypertension in renal diseaseKidney Int200057Suppl 75S2S6
- AsahiKIchimoriKNakazawaHNitric oxide inhibits the formation of advanced glycation end productsKidney Int2000581780178711012913
- KlahrSMorrisseyJJThe role of vasoactive compounds, growth factors and cytokines in the progression of renal diseaseKidney Int200057Suppl 75S7S14
- LeeheyDJSinghAKAlaviNSinghRRole of angiotensin II in diabetic nephropathyKidney Int200058Suppl 77S93S98
- TharauxPLChatziantoniouCCasellasDFouassierLArdaillouRDussauleJCVascular endothelin-1 gene expression and synthesis and effect on renal type I collagen synthesis and nephroangiosclerosis during nitric oxide synthase inhibition in ratsCirculation1999992185219110217661
- SzilvassyZCsontTPaliTDroy-LefaixMTFerdinandyPNitric oxide, peroxynitrite and cGMP in atherosclerosis-induced hypertension in rabbits: beneficial effects of cicletanineJ Vasc Res200138394611173993
- ThuraisinghamRCNottCADoddSMYaqoobMMIncreased nitrotyrosine staining in kidneys from patients with diabetic nephropathyKidney Int2000571968197210792615
- WalkerLMWalkerPDImamSZAliSFMayeuxPREvidence for peroxynitrite formation in renal ischemia-reperfusion injury: studies with the inducible nitric oxide synthase inhibitor L-N(6)-(1-Iminoethyl) lysineJ Pharmacol Exp Ther200029541742210992009
- BalcellsEMengQCJohnsonWHJrOparilSDell’ItaliaLJAngiotensin II formation from ACE and chymase in human and animal hearts: methods and species considerationsAm J Physiol19972734 Pt 2H1769H17749362242
- PetrieMCPadmanabhanNMcDonaldJEHillierCConnellJMMcMurrayJJAngiotensin converting enzyme (ACE) and non-ACE dependent angiotensin II generation in resistance arteries from patients with heart failure and coronary heart diseaseJ Am Coll Cardiol2001371056106111263608
- GriendlingKKSorescuDUshio-FukaiMNAD(P)H oxidase: role in cardiovascular biology and diseaseCirc Res20008649450110720409
- HaugenENCroattAJNathKAAngiotensin II induces renal oxidant stress in vivo and heme oxygenase-1 in vivo and in vitroKidney Int20005814415210886559
- HatakeyamaHMiyamoriLFujitaTTakedaYTakedaRYamamotoHVascular aldosterone: biosynthesis and a link to angiotensin II-induced hypertrophy of vascular smooth muscle cellsJ Biol Chem199428924316243207929089
- SilvestreJSRobertVHeymesCMyocardial production of aldosterone and corticosterone in the rat. Physiological regulationJ Biol Chem1998273488348919478930
- UllianMESchellingJRLinasSAldosterone enhances angiotensin II receptor binding and inositol phosphate responsesHypertension19922067731618554
- KornelLSmoszna-KonaszewskaBAldosterone (ALDO) increases trans-membrane influx of Na+ in vascular smooth muscle (VSM) cells through increased synthesis of Na+ channelsSteroids1995601141197792795
- WeberMAPurdyRECatecholamine mediated constrictor effects of aldosterone on vascular smooth muscleLife Sci198230200920177109834
- IkedaUKanbeTNakayamaIKawaharaYYokoyamaMShimadaKAldosterone inhibits nitric oxide synthesis in vascular smooth muscle cells induced by interleukin-1bEur J Pharmacol199529069738575535
- BrownNJNakamuraSMaLAldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivoKidney Int2000581219122710972684
- EpsteinMAldosterone as a mediator of progressive renal disease: pathogenetic and clinical implicationsAm J Kidney Dis200137467768811273866
- American Diabetes AssociationStandards of medical care for patients with diabetes mellitusDiabetes Care200124Suppl 1S33S66
- BakrisGLWilliamsMDworkinLPreserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working GroupAm J Kidney Dis200036364666110977801
- RamsayLWilliamsBJohnstonGGuidelines for management of hypertension: report of the third working party of the British Hypertension SocietyJ Hum Hypertens199913956959210482967
- FeldmanRDCampbellNLarochelleP1999Canadian recommendations for the management of hypertension. Task force for the development of the 1999 Canadian recommendations for the management of hypertensionCMAJ1999161Suppl 12S1S1710624417
- ChalmersJMacMahonSManciaG1999 World Health Organization-International Society of Hypertension Guidelines for the management of hypertension. Guidelines sub-committee of the World Health OrganizationClin Exp Hypertens1999215–61009106010423121
- JNC-VIThe sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressureArch Intern Med199715721241324469385294
- LazarusJMBourgoignieJJBuckalewVMAchievement and safety of a low blood pressure goal in chronic renal disease. The modification of diet in Renal Disease Study GroupHypertension19972926416509040451
- EstacioROJefersBWHiattWRBiggerstaffSLGiffordNSchrierRWThe effect of nisoldipine as compared with enalapril on cardiovascular outcomes in patients with non-insulin-dependent diabetes and hypertensionN Engl J Med1998338106456529486993
- UK Prospective Diabetes Study GroupTight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38 UK Prospective Diabetes Study GroupBMJ199831771607037139732337
- HanssonLZanchettiACarruthersSGEffects of intensive blood pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomized trial. HOT Study GroupLancet19983519118175517629635947
- KauszATObradorGTPereiraBJAnemia management in patients with chronic renal insufficiencyAm J Kidney Dis2000366 Suppl 3S39S5111118157
- LevinAThompsonCREthierJLeft ventricular mass index increase in early renal disease: impact of decline in hemoglobinAm J Kidney Dis199934112513410401026
- BlockGAHulbert-ShearonTELevinNWPortFKAssociation of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national studyAm J Kidney Dis19983146076179531176
- RossingPHommelESmidtUMParvingHHReduction in albuminuria predicts a beneficial effect on diminishing the progression of human diabetic nephropathy during antihypertensive treatmentDiabetologia19943755115168056190
- GansevoortRTde ZeeuwDde JongPELong-term benefits of the antiproteinuric effect of angiotensin-converting enzyme inhibition in nondiabetic renal diseaseAm J Kidney Dis19932212022068322784
- RussoDPisaniABallettaMMAdditive antiproteinuric effect of converting enzyme inhibitor and losartan in normotensive patients with IgA nephropathyAm J Kidney Dis19993385185610213639
- CrepaldiGCartaQDeferrariGEffects of lisinopril and nifedipine on the progression to overt albuminuria in IDDM patients with incipient nephropathy and normal blood pressure. The Italian Microalbuminuria Study Group in IDDMDiabetes Care19982111041109538979
- RuggenentiPPernaAGherardiGRenoprotective properties of ACE-inhibition in non-diabetic nephropathies with non-nephrotic proteinuriaLancet1999354917635936410437863
- Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICROHOPE substudy. Heart Outcomes Prevention Evaluation Study InvestigatorsLancet200035525325910675071
- LewisEJHunsickerLGClarkeWRfor the Collaborative Study GroupRenoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetesN Engl J Med200134585186011565517
- BrennerBMCooperMEde ZeeuwDfor the RENAAL Study InvestigatorsEffects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathyN Engl J Med200134586186911565518
- MacKinnonMShurrawSAkbariAKnollGAJaffeyJClarkHDCombination therapy with an angiotensin receptor blocker and an ACE inhibitor in proteinuric renal disease: a systematic review of the efficacy and safety dataAm J Kidney Dis20064882016797382
- KunzRFriedrichCWolbersMMannJFMeta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal diseaseAnn Intern Med2008148304817984482
- MannJFSchmiederREMcQueenMRenal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trialLancet200837254755318707986
- BakrisGLSlataperRVicknairNSadlerRACE inhibitor mediated reductions in renal size and microalbuminuria in normotensive, diabetic subjectsJ Diabetes Complications199481268167383
- EpsteinBJSmithSMChoksiRRecent changes in the landscape of combination RAS blockadeExpert Rev Cardiovasc Ther200971373138419900020
- BrunnerHRLaraghJHBaerLEssential hypertension: renin and aldosterone, heart attack and strokeN Engl J Med19722864414484257928
- ParikhNIGonaPLarsonMGWangTJPlasma renin activity and risk of cardiovascular disease and mortality: the Framingham Heart StudyEur Heart J2007282644265217895253
- LorigaGVidiliGRuggenentiPRenal hemodynamics and renoprotectionNephron Clin Pract2008110c213c21918974652
- NussbergerJGradmanAHSchmiederREPlasma renin and the antihypertensive effect of the orally active renin inhibitor aliskiren in clinical hypertensionInt J Clin Pract200761146117590217
- BrownMJAliskirenCirculation200811877378418695203
- BaldonciniRDesideriGBelliniCHigh plasma renin activity is combined with elevated urinary albumin excretion in essential hypertensive patientsKidney Int1999561499150410504501
- YeyatiNLAdroguéHJInappropriately high plasma renin activity accompanies chronic loss of renal functionAm J Nephrol1996164714778955757
- MalmqvistKOhmanKPLindLRelationships between left ventricular mass and the renin-angiotensin system, catecholamines, insulin and leptinJ Intern Med200225243043912528761
- LicataGScaglioneRCorraoSHeredity and obesity-associated hypertension: impact of hormonal characteristics and left ventricular massJ Hypertens1995136116187594417
- WeirMRBushCAndersonDRAntihypertensive efficacy, safety, and tolerability of the oral direct renin inhibitor aliskiren in patients with hypertension: a pooled analysisJASH20071426427720409858
- BirkenhagerWHStaessenJADual inhibition of the renin system by aliskiren and valsartanLancet200737019519617658374
- TaylorAAAndersonDRAroraVRenin system suppression with the oral direct renin inhibitor, aliskiren administered alone or in combinationJ Am Soc Nephrol200415312631333415579516
- OhB-HMitchellJHerronJRChungJKhanMKeefeDLAliskiren, an oral renin inhibitor, provides dose-dependent efficacy and sustained 24-hour blood pressure control in patients with hypertensionJ Am Coll Cardiol200749111157116317367658
- SchmiederREPhilippTGuerediagaJLong-term antihypertensive efficacy and safety of the oral direct renin inhibitor aliskiren: a 12-month randomized, double-blind comparator trial with hydrochlorothiazideCirculation2009119341742519139391
- OparilSYarowsSPatelSFangHZhangJSatlinAEfficacy and safety of combined use of aliskiren and valsartan in patients with hypertension: a randomised, double-blind trialLancet2007370958322122917658393
- GradmanAHSchmiederRELinsRLNussbergerJChiangYBedigianMPAliskiren, a novel orally effective renin inhibitor, provides dose-dependent antihypertensive efficacy and placebo-like tolerability in hypertensive patientsCirculation20051111012101815723979
- FramptonJECurranMPAliskiren: a review of its use in the management of hypertensionDrugs200767121767179217683174
- UresinYTaylorAAKiloCEfficacy and safety of the direct renin inhibitor aliskiren and ramipril alone or in combination in patients with diabetes and hypertensionJ Renin Angiotensin Aldosterone Syst20078419020018205098
- StrasserRHPuigJGFarsangCCroketMLiJvan IngenHA comparison of the tolerability of the direct renin inhibitor aliskiren and lisinopril in patients with severe hypertensionJ Hum Hypertens20072178078717541390
- DrummondWMungerMARafiqueMAntihypertensive efficacy of the oral direct renin inhibitor Aliskiren as add-on therapy in patients not responding to amlodipine monotherapyJ Clin Hypertens2007910742750
- VillamilAChrysantSGCalhounDRenin inhibition with aliskiren provides additive antihypertensive efficacy when used in combination with hydrochlorothiazideJ Hypertens20072521722617143194
- PoolJLSchmiederREAziziMAliskiren, an orally effective renin inhibitor, provides antihypertensive efficacy alone and in combination with valsartanAm J Hypertens200720112017198906
- O’BrienEBartonJNussbergerJAliskiren reduces blood pressure and suppresses plasma renin activity in combination with a thiazide diuretic, an angiotensin-converting enzyme inhibitor, or an angiotensin receptor blockerHypertension20074927628417159081
- AndersenKWeinbergerMHConstanceCMComparative effects of aliskiren-based and ramipril-based therapy on the renin system during long-term (6 months) treatment and withdrawal in patients with hypertensionJ Renin Angiotensin Aldosterone Syst20091015716719617271
- MungerMADrummondWEssopMRAliskiren as add-on to amlodipine provides significant additional blood pressure lowering without increased edema associated with doubling the amlodipine dose [abstract]Eur Heart J200627Suppl11716267068
- ParvingHHPerssonFLewisJBLewisEJHollenbergNKAVOID Study InvestigatorsAliskiren combined with losartan in type 2 diabetes and nephropathyN Engl J Med20083582433244618525041
- ParvingHHBrennerBMMcMurrayJJAliskiren trial in type 2 diabetes using cardio-renal endpoints (ALTITUDE): rationale and study designNephrol Dial Transplant2009241663167119145003