
Abstract
Inorganic polyphosphate (polyP) and its metabolic enzymes are important in several cellular processes related with virulence and antibiotic susceptibility. Accordingly, bacterial polyP synthesis has been proposed as a good target for designing novel antivirulence molecules as alternative to conventional antibiotics. In most pathogenic bacteria, polyphosphate kinase 1 (PPK1), in charge of polyP synthesis from ATP, is widely conserved. Current colorimetric and radioactive polyP synthesis enzymatic assays are not suitable for high-throughput screening of PPK1 inhibitors. Given the ability of polyP to modify the excitation-emission spectra of DAPI (4ʹ-6-diamidino-2-phenylindole), a fluorescence assay was previously developed by using a purified recombinant PPK1 enzyme from Escherichia coli. In this work we have developed a suitable methodology for high-throughput measurement of E. coli PPK1 activity. This platform can be used for the screening putative antimicrobial molecules for related enteropathogenic bacteria.
Inorganic polyphosphate (polyP) is a linear polymer ubiquitous among living organisms that presents a variety of biochemical functions. Studies of polyP-metabolizing enzymes are important for medicine and particularly infectious diseases,Citation1,Citation2 because polyP functions are directly related with virulence factor production in bacteria.Citation3–Citation5 In addition, mutants lacking polyP kinase 1 (PPK1), the main bacterial enzyme in charge of polyP synthesis, are more sensitive toward antibiotics.Citation6,Citation7
Given the fact that polyP synthesis is not essential for bacterial growth but for the regulation of cellular processes related to virulence, it has been proposed that polyP metabolism, and particularly polyP synthesis by the PPK1, is an attractive target for the design of novel antivirulence compounds.Citation8 However, a feasible polyphosphate kinase assay is currently not available for this purpose. We have therefore focused on developing a high-throughput fluorescence-based PPK1 enzymatic assay to facilitate the screening of putative inhibitor that could be used as antivirulence molecules.
For this, we took advantage that, when excited at 415 nm, fluorescence emitted by DAPI-polyP complex is detected at 550 nm and fluorescence emission from free DAPI and DAPI-DNA complex are minimal at this wavelength. In addition, the highly specific DAPI-polyP complex can be detected at polyP concentration as low as 25 ng/ml.Citation9
First, ppk1 gene of E. coli K12 was synthesized into pUC57, using Genescript (Piscataway, NJ, USA) service. In-frame insertion of an amino-terminal 6xHis-tag were included by cloning the NdeI/HindII fragment of the synthetic ppk1 gene into pET-TEV for affinity purification purpose.Citation10 The resulting plasmid pET-TEV:Ecppk1 was transformed into E. coli strain BL21-AI strain for protein overexpression. Transformant clones were randomly selected for growing and induction of the recombinant protein ().
Figure 1 Purification of E. coli PPK1-His6- tag by immobilized metal affinity chromatography (IMAC). Recombinant E. coli PPK1-His6-tag was purified by immobilized Ni-Sepharose affinity chromatography as previously reported.Citation8 Bacterial Protein Extraction Reagent B-PER® (Thermo Fisher Scientific, Waltham, MA, USA) was used for obtaining the recombinant proteins from the bacterial pellet. (A) Insoluble and soluble fractions of cell proteins respectively from 4 different colonies (lanes 2–9) were obtained by centrifugation and visualized in Tris/Glycine 12% SDS–PAGE stained with Coomassie Blue. (B) Insoluble fraction of selected colony (line 4) was discarded and the respective soluble fraction (line 5) was equilibrated and loaded onto the affinity column (His-Trap ®; GE Healthcare, Chicago, IL, USA) to obtain the purified PPK1-His6-tag protein (line 6). Insoluble and soluble protein extracts from negative expression control were loaded into lines 2 and 3. (C, D) Fractions were eluted with 200 mM de imidazole and were analyzed by Tris/Glycine 12% SDS–PAGE. Non-retained fraction (line 3) and eluted fraction (lines 4–9) were stained with Coomassie Blue (C) and also by Western-Blot (D) using Nickel-HRP, HisDetectorTM kit (KPL Inc., Milford, MA, USA).
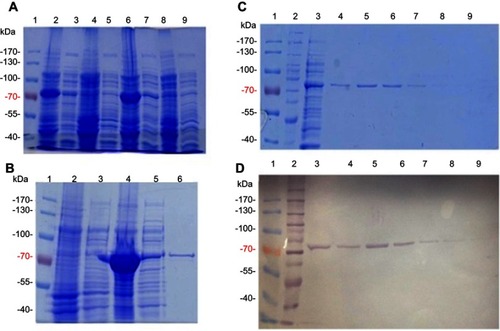
For PPK1 purification, E. coli BL21-AI cells transformed with the derived plasmid (pET-TEV:Ecppk1) were aerobically grown in Luria-Bertani (LB) medium at 37 °C and stopped after 2 hrs of induction with L-arabinose (0.4%). As shown in , an induced 80 kDa polypeptide was detected in both the soluble and insoluble fractions of two clones (). To obtain the purified PPK1, soluble fraction of bacterial cells obtained from large-scale cultures (500 mL) were used for further protein purification (). For this, cells were harvested by centrifugation and resuspended in B-PER™ Bacterial Protein Extraction Reagent (Life Technologies, Carlsbad, CA, USA). The soluble cellular fraction was loaded into a HisTrap column (GE Healthcare, Chicago, IL, USA) previously charged with NiSO4 (Merck, Kenilworth, NJ, USA), according to the instructions of the manufacturer. The bound proteins were eluted using a buffer Tris 50 mM MgCl2 10 mM pH 8.2 supplemented with stepwise increasing concentrations of imidazole from 40 mM to 600 mM. The eluted fractions were pooled and the purity and identity of the recombinant PPK1 were confirmed by SDS-PAGE and Western-blot, respectively (). Purified recombinant PPK1-His6-Tag was dialyzed with buffer D (20 mM Tris-HCl pH 8.2, 150 mM NaCl, 10 mM β-mercaptoethanol, 20% glycerol, 0.05% Tween 20, 50 mM ammonium sulfate and 10 mM MgCl2) and was kept at −20 °C until use. The purified PPK1 were then tested in vitro for polyP synthesis assays using DAPI ().
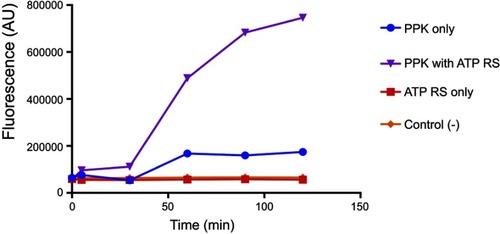
Figure 2 Polyphosphate kinase fluorescence enzymatic assay using DAPI. (A) PolyP kinase activity was measured using DAPI to quantify polyP production. Purified E. coli PPK1-His6-tag (16,5 µg/ml) was added to the reaction mix: 50 mM Hepes-KOH pH 7.2, 5 mM MgCl2, 50 mM ammonium sulfate, 1 mM ATP, 1 µg/ml poliP45, 2 mM phosphate creatine and 20 µg/ml creatine kinase. At each time reaction was stopped by adding 50 μM DAPI to 20 μL aliquots and fluorescence was read using SynergyTM Multi-Mode Microplate Reader (Biotek, Winooski, VT, USA) using excitation and emission filters of 400/10 nm and 540/25 nm respectively. (B) PPK activity of the purified PPK1-His6-Tag was assayed at different substrate (ATP) concentrations.
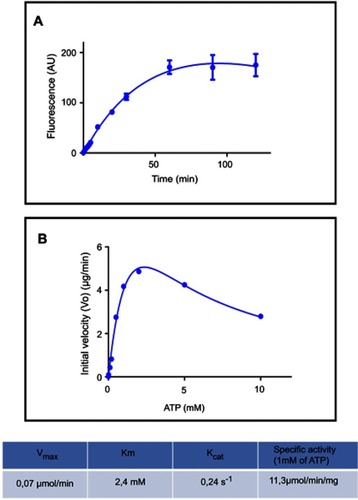
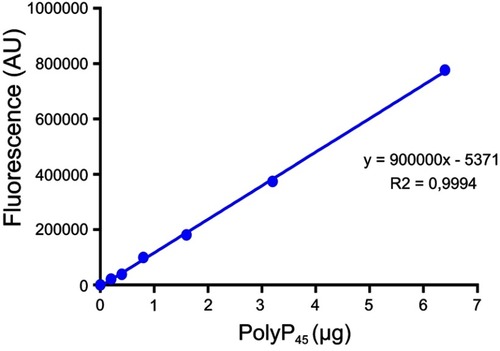
PolyP kinase activity was assessed by incubating the purified enzyme (5 µg protein/reaction) in assay buffer (50 mM HEPES-KOH, pH 7.5 40 mM ammonium sulfate, 4 mM magnesium sulfate, 60 mM creatine phosphate and 50 µg of creatine kinase) containing 5 mM ATP as substrate (total reaction volume = 200 µl) during 1 h at 37 °C.Citation11 This was followed by addition of DAPI at a final concentration of 50 µM. Fluorescence intensity measurements of DAPI-polyp complex were obtained using Synergy 2 Multi-Mode microplate reader (Biotek, Winooski, VT, USA) at an excitation wavelength of 400 nm (bandwidth of 10 nm). Fluorescence emission was measured at 540 nm (bandwidth of 25 nm). All experiments were performed using black fluorescence 96 well microplates and the values obtained were corrected by subtracting the fluorescence emitted by control wells containing all the ingredients, except the enzyme. Standard curves were prepared with known concentrations (1–10 µg) of poly-P45 (Merck) and the PPK activity was expressed as µg of polyP produced per min. (). Similar to the PPK radioactive assay, which requires an ATP regeneration system (RS), our DAPI fluorescence assay also requires the use of creatine kinase based regeneration system (). As shown in , kinetic parameters from E. coli PPK1 by using our assay were in agreement with those previously reported with radioactive assays.Citation11,Citation12
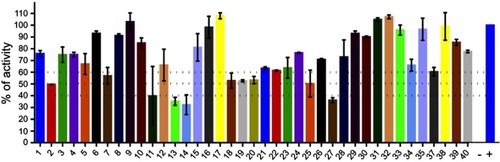
Finally, we evaluated 40 compounds that had been previously selected by virtual screening as inhibitors of PPK1 from P. aeruginosa PAO1 (PaPPK1).Citation8 The PPK1 activity was assayed in 96-well by measuring the polyP-DAPI fluorescence at 550 nm. As shown in , we have identified 9 compounds that inhibit around or more than 50% of E. coli PPK1 activity at 1 μM. Similar results for these compounds were obtained against PPK1 from P. aeruginosa PAO1.Citation8
Figure 3 Screening for PPK1-His6-tag inhibitors. Inhibitors of E. coli PPK1 activity were selected by measuring polyP production using DAPI fluorescence. Evaluated molecules (1 μM) were previously assayed toward PPK1 from P. aeruginosa PAO1 (PaPPK1).Citation8 These compounds were selected by virtual screening for inhibitors of PPK1 and for antivirulence activity toward P. aeruginosa PAO1.
In conclusion, we have developed a suitable PPK assay based on polyP-DAPI fluorescence emission. The assay was successfully validated using PPK1 from E. coli, which was obtained by affinity purification. The purified PPK1 was used for the screening of inhibitor molecules that can be tested in future antivirulence assays. This study will serve as a basis for the screening putative antimicrobial molecules for related enteropathogenic bacteria.
Highlights
We have synthetized and overexpressed PPK1 from Escherichia coli PPK1 fused to polyhistidine tag.
A novel fluorescence PPK1 enzymatic assay was developed using DAPI.
The fluorescence assay was used for screening of polyP synthesis inhibitors.
The assay will be useful in high-throughput screening of putative antivirulence molecules targeting enteric pathogens.
Acknowledgments
We are grateful to Nicole Molina for technical assistance at SysmicroLab.
Supplementary materials
Disclosure
FPC received grants from FONDECYT (1120209). MAV and JOS were supported by CONICYT fellowships. The authors report no other conflicts of interest in this work.
References
- Chávez FP, Lagos CF, Reyes-parada M, Guiliani N, Jerez CA. Polyphosphate synthesis as a target for novel antibiotics. Curr Enzym Inhib. 2011;7(3):163–168. doi:10.2174/157340811798807605
- Rashid MH, Rao NN, Kornberg A. Inorganic polyphosphate is required for motility of bacterial pathogens. J Bacteriol. 2000;182(1):225–227. Available from: http://jb.asm.org/cgi/content/abstract/182/1/22510613886
- Fraley CD, Rashid MH, Lee SSK, et al. A polyphosphate kinase 1 (ppk1) mutant of Pseudomonas aeruginosa exhibits multiple ultrastructural and functional defects. Proc Natl Acad Sci U S A. 2007;104(9):3526–3531. Available from: http://opus.bath.ac.uk/7641/17360677
- Varela C, Mauriaca C, Paradela A, Albar J, Jerez C, Chavez F. New structural and functional defects in polyphosphate deficient bacteria: A cellular and proteomic study. BMC Microbiol. 2010;10(1):7 Available from: http://www.biomedcentral.com/1471-2180/10/720067623
- Kim K-S, Rao NN, Fraley CD, Kornberg A. Inorganic polyphosphate is essential for long-term survival and virulence factors in Shigella and Salmonella spp. Proc Natl Acad Sci U S A. 2002;99(11):7675–7680. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=124319&tool=pmcentrez&rendertype=abstract12032342
- Ortiz-Severín J, Varas M, Bravo-Toncio C, Guiliani N, Chávez FP. Multiple antibiotic susceptibility of polyphosphate kinase mutants (ppk1 and ppk2) from Pseudomonas aeruginosa PAO1 as revealed by global phenotypic analysis. Biol Res. 2015;48:22. doi:10.1186/s40659-015-0012-025907584
- Nikel PI, Chavarría M, Martínez-García E, Taylor AC, de Lorenzo V. Accumulation of inorganic polyphosphate enables stress endurance and catalytic vigour in Pseudomonas putida KT2440. Microb Cell Fact. 2013;12(1):50 Available from: http://www.microbialcellfactories.com/content/12/1/50.23687963
- Bravo-Toncio C, Álvarez JA, Campos F, et al. Dictyostelium discoideum as a surrogate host–microbe model for antivirulence screening in Pseudomonas aeruginosa PAO1. Int J Antimicrob Agents. 2016:47(5):403–409. doi:10.1016/j.ijantimicag.2016.02.005.
- Aschar-Sobbi R, Abramov AY, Diao C, et al. High sensitivity, quantitative measurements of polyphosphate using a new DAPI-based approach. J Fluoresc. 2008;18(5):859–866. doi:10.1007/s10895-008-0315-418210191
- Rocco CJ, Dennison KL, Klenchin VA, Rayment I, Escalante-Semerena JC. Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid. 2008;59(3):231–237. doi:10.1016/j.plasmid.2008.01.00118295882
- Tzeng CM, Kornberg A. The multiple activities of polyphosphate kinase of Escherichia coli and their subunit structure determined by radiation target analysis. J Biol Chem. 2000;275(6):3977–3983. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10660553.10660553
- Ault-Riché D, Fraley CD, Tzeng C-M, Kornberg A. Novel assay reveals multiple pathways regulating stress-induced accumulations of inorganic polyphosphate in Escherichia coli. J Bacteriol. 1998;180(7):1841–1847. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=107098&tool=pmcentrez&rendertype=abstract9537383