
Abstract
Chronic hepatitis B is a numerically important cause of cirrhosis and hepatocellular carcinoma, despite an effective prophylactic vaccine and well-tolerated and effective oral antivirals. Both the incapacity of the immune system to clear hepatitis B virus (HBV) infection and the unique replication strategies adopted by HBV are considered key determinants of HBV chronicity. In this regard, the formation of the HBV DNA minichromosome, the covalently closed circular DNA (cccDNA), in the nucleus of infected hepatocytes, is essential not only for the production of all viral proteins but also for HBV persistence even after long-term antiviral therapy. Licensed polymerase inhibitors target the HBV reverse transcriptase activity, control the disease with long-term therapy but fail to eliminate the cccDNA. Consequently, the production of viral RNAs and proteins, including the hepatitis B surface antigen (HBsAg), is not abolished. Novel therapeutic efforts that are in the pipeline for early clinical trials explore novel targets and molecules. Such therapeutic efforts focus on achieving a functional cure, which is defined by the loss of HBsAg and undetectable HBV DNA levels in serum. Since a true cure of HBV infection requires the elimination of the cccDNA from infected cells, comprehension of the mechanisms implicated in cccDNA biogenesis, regulation and stability appears necessary to achieve HBV eradication. In this review, we will summarize the state of knowledge on cccDNA metabolism, focusing on insights suggesting potential weak points of the cccDNA that may be key for the development of therapeutic approaches and design of clinical trials aiming at lowering cccDNA loads and activity.
Introduction
Hepatitis B virus infection can be prevented due to the availability of a safe and effective vaccine, while approved oral antivirals improve survival by efficiently reducing HBV replication and hepatic inflammation in HBV chronic carriers. However, resolution of chronic HBV infection (CHB) is rarely achieved. Consequently, CHB still represents a major global health burden with more than 250 million people persistently infected worldwide and every year around 780,000 people die due to the consequences of an HBV-associated liver disease.Citation1 Approved antiviral treatments mostly rely on the use of nucleos(t)ide analogs (NAs) and to a much lesser extent on pegylated interferon alpha (peg-IFNα). By suppressing HBV replication, NAs successfully keep the infection under control and improve the patients’ quality of life. Moreover, NAs have been shown to prevent the progression of liver disease in most patients and to reduce the risk of developing hepatocellular carcinoma (HCC) in non-cirrhotic and cirrhotic patients.Citation2 However, discontinuation of NAs therapy often leads to the relapse of HBV. This is mainly due to the fact that NAs efficiently hinder the production of new virions, but they do not target the cccDNA directly or specifically lower HBV RNAs. In contrast, peg-IFNα can achieve stable HBV suppression and is the only approved finite treatment for CHB, although it is effective in only a minority of patients and its use is limited due to side effects. The overall goal of therapy is to achieve a sustained off-treatment loss of hepatitis B surface antigen (HBsAg) and undetectable HBV DNA levels and normalization of transaminases in serum; parameters that are associated with a long-term improvement of clinical outcome and hence defining an ideal end-point for antiviral therapy. However, such a functional cure is rarely achieved with existing treatments.Citation3 Thus, HBV cure research has become a top priority within the scientific and medical community and has triggered the WHO to call for actions to significantly reduce disease burden by 2030. A curative therapy for CHB is thought to require both restoration of the HBV-specific antiviral immune responses and destruction or silencing of the cccDNA minichromosome. Drugs targeting the cccDNA may even achieve a complete cure of HBV. Therefore, understanding the molecular mechanisms regulating cccDNA formation, transcriptional activity, intracellular stability and turnover appear mandatory to find a true cure for CHB infection enabling eradication of the virus.
Establishment of HBV Infection and cccDNA Formation
HBV is transmitted by percutaneous exposure to infected blood or body fluids. The virus infects the hepatocytes, which are the only target cells susceptible for infection. The infectious particle, the virion, contains a 3.2-kb partially double-stranded relaxed circular DNA (rcDNA) genome that is covalently linked to the polymerase and packaged within the nucleocapsid, which is wrapped by the envelope membrane presenting the three envelope proteins (large, medium and small). Characteristic of HBV is not only its high tissue and species specificity but also the unique genomic organization and replication mechanism. The process of cell entry involves an initial reversible interaction of the virion with glycosaminoglycans, such as heparan sulfate proteoglycans,Citation4 which is followed by an irreversible binding of the virion to the Na+-taurocholate co-transporting polypeptide (NTCP),Citation5 a bile acid receptor exclusively expressed on the hepatocytes. After entering the cell, the viral genome is transferred into the nucleus to build the cccDNA molecule,Citation6 and hence to initiate productive infection ().
Figure 1 HBV life cycle with antiviral treatments and conditions affecting cccDNA.
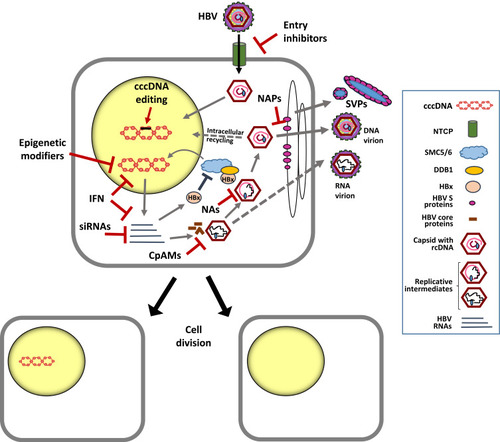
Even if the events following viral entry are not yet well characterized, they were shown to involve endocytosis of the nucleocapsids, which are then conveyed to the nuclear envelope.Citation7 By interacting with the nuclear pore complex, the release of the rcDNA and core capsid subunits takes place.Citation8 The molecular mechanisms determining the conversion of the rcDNA to the supercoiled episome that constitutes the cccDNA molecule also remain largely unknown. Clearly, establishment of the HBV minichromosome is a multi-step process requiring the cellular DNA repair machinery.Citation9,Citation10 First, the viral polymerase that is covalently attached to the HBV genome needs to be removed to form a protein-free rcDNA (PF-rcDNA) intermediate.Citation11 Formation of the cccDNA requires then the removal of both an RNA primer from the positive strand and of terminally redundant sequences from the negative strand, as well as the repair of the incomplete positive strand before both DNA strands can be ligated.Citation12 Distinct cellular components and nuclear enzymes appear to participate in cccDNA formation. For instance, the DNA polymerase K was shown to participate in completing the positive strand DNA of the rcDNA,Citation13 while the cellular DNA repair enzyme tyrosyl-DNA-phosphodiesterase 2 (TDP2) was shown to be involved in the release of the viral polymerase from rcDNA in vitro.Citation14
Establishment of the cccDNA as an episomal plasmid-like structure organized as a minichromosome involves its association with histone and non-histone proteins,Citation15–Citation17 including proteins of host and viral origin.Citation10 Resembling the host chromatin, the cellular transcriptional machinery enables the transcription of the viral RNAs needed for viral replication. On the HBV genome, all open reading frames (ORFs) largely overlap, are identically oriented and encoded by the negative DNA strand. Essential for the production of new HBV genomes is the transcription of an over-length pregenomic RNA (pgRNA), which encodes both the viral polymerase and the core proteins that serve for encapsidation and reverse transcription of the pgRNA. Of note, different studies showed that not only rcDNA-containing nucleocapsids but also particles containing HBV RNAs can be enveloped and secreted from the cell,Citation18–Citation20 although their impact on HBV life cycle and HBV-associated liver disease remains to be elucidated. The cccDNA also transcribes distinct subgenomic RNAs for the production of the three envelope proteins (large, medium and small) that all share the carboxyterminal ends and contain the HBsAg, which is commonly detected by tests for HBV infection (for a review seeCitation6). The high sensitivity of such tests relies on the fact that infected hepatocytes secrete subviral particles (SVPs) () in far higher numbers than infectious particles. SVPs are composed of the HBsAg and host-derived lipids, but do not contain the capsid and the HBV genome.Citation21 High amounts of SVPs are thought to contribute to generate a state of immune tolerance on both innate and adaptive immunity against HBV.Citation22 Apart from HBsAg, HBV produces two non-structural proteins, the secreted HBeAg and the small regulatory X protein (HBx). HBeAg is a non-particulate form of the nucleoprotein that appears to have tolerogenic functions in the neonates of viremic mothersCitation23 but that is not essential for viral replication. In clinical practice, the secreted HBeAg is used as a marker of viral replication. In contrast, the non-structural HBx plays a key role in HBV replication, since numerous in vitro and in vivo studies demonstrated a dramatic impairment of cccDNA-driven HBV RNA transcription in cells inoculated with an HBV-X-minus mutant.Citation24–Citation27
Identifying the diverse host factors determining or limiting HBV entry and cccDNA formation will be essential to progress in the development of new therapies targeting the early steps of infection and cccDNA establishment. However, therapies targeting the host may also have more adverse effects than strategies targeting viral components, which may also be essential to establish an active cccDNA minichromosome.
Regulation of cccDNA Activity
The function of the HBV minichromosome depends on the activity of numerous transcription factors and chromatin-modifying enzymes.Citation28–Citation30 Since HBV infects primary human hepatocytes, it is not surprising that the viral genome bears binding sites both for ubiquitous and liver-specific transcription factors.Citation31 Thus, both the recruitment and dynamic interplay of several factors, including coactivators and corepressors, appear essential to yield efficient HBV gene expressionCitation28,Citation32 (reviewed inCitation33). Apart from cellular factors, viral elements such as the HBV core (HBc) and HBx proteins are central in cccDNA biology and activity. The viral core protein appears to act as a structural component of the cccDNA and its physical association with the minichromosome was suggested to be accountable for the reduced nucleosomal spacing determined on the cccDNA in comparison to the spacing generally found in the cellular chromatin.Citation16 Although the role of HBc as positive regulator of cccDNA transcription is not yet clear, a reduced capacity to associate with the cccDNA and to recruit the histone acetyltransferases has been recently determined in the presence of HBc mutants.Citation34,Citation35
Numerous reports described the ability of the small regulatory HBx to interfere with several cellular pathways and transcription factors and HBx over-expression was shown to cause transactivation of a wide range of viral elements and cellular promoters.Citation36 Although the mechanism by which HBx promotes HBV replication is not fully elucidated, Belloni et al used a cccDNA-specific chromatin immunoprecipitation (ChIP) assay to show the recruitment of HBx onto the cccDNA minichromosome, thus providing first evidence that HBx can control cccDNA-driven HBV transcription by acting at epigenetic level.Citation37 However, HBx bears the capacity to affect a variety of cellular factors of hepatocytes, thus impairing our understanding of the hierarchy of interactions that are key in HBV infection of the hepatocytes.
A breakthrough regarding the impact of HBx on cellular proteins came from the study of Decorsiere et alCitation38 showing that HBx effectively degrades the ‘structural maintenance of chromosomes’ (Smc) complex SMC5/6 by hijacking the damaged DNA binding protein 1 (DDB1). Such binding, which was also confirmed in additional independent studies,Citation39 promotes the interaction of the SMC5/6 complex with a component of the ubiquitin–proteasome system, the so-called E3 ubiquitin ligase enzyme. Ubiquitination and degradation of this complex by the host proteasome machinery was demonstrated both in cultured human hepatocytes and in humanized mice and represents a new mechanism by which HBx can antagonize the ability of SMC5/6 to associate with the cccDNA (). Binding of SMC5/6 complex with the cccDNA can abrogate the transcription of the major HBV RNAs (pgRNA, precore mRNA and surface mRNAs). However, it has been recently hypothesized that such block may be ineffective for HBx mRNA. According to this scenario, the early production of the small HBV X RNA and HBx translation could promote SMC5/6 degradation to ensure then the transcription of the major HBV RNAs.Citation40 Such SMC5/6 antagonism mediated by HBx to ensure productive viral replication appears as an evolutionarily conserved strategy, since several viruses have evolved mechanisms to manipulate the ubiquitin–proteasome system.Citation41
HBx was also described to increase the expression of DNA methyltransferases (DNMTs),Citation42 suggesting that sustained HBx production may promote epigenetic changes on viral and host genes.Citation43,Citation44 Transcriptional silencing is often associated with the methylation of the DNA within CpG dinucleotides. Depending on the HBV genotype, the cccDNA contains two to three putative CpG islands that are strategically located within the regulatory elements of the genome. However, the methylation rate of these CpG islands was shown to correlate with the repression of cccDNA gene transcriptionCitation45,Citation46 and with lower levels of HBV viremia and HBsAg.Citation47 Of note, higher levels of HBV DNA methylation have been determined in HCC tissues compared to infected and cirrhotic tissues.Citation48 The methylation of the viral genome is regarded as a host defence mechanism attempting to silence viral gene expression. Thus, further studies are needed to investigate if and to which extent such mechanisms may be exploited for therapeutic purposes (see also section impact of antiviral treatments on the cccDNA), since these approaches bear the risk to affect also the host genome.Citation49
A variety of histone-modifying enzymes can also affect viral transcription by altering the histones associated to the cccDNA (see alsoCitation50). In this regard, analysis of liver biopsies from CHB patients indicated that histone hypoacetylation of the minichromosome correlates with low HBV viremia,Citation51 while acetylation of cccDNA-bound H4 was reported to correlate with higher levels of HBV replication.Citation51 Along this line, data revealing the association of HBx with the cccDNA minichromosome also indicated that HBx-mediated transcriptional activity relays on the recruitment of histone acetyltransferases (HATs)Citation37 and of coactivators involved in chromatin modulation,Citation52 such as the host CREB-binding protein (CBP), p300 and the p300/CBP-associated factor (PCAF). Moreover, in the absence of HBx, cccDNA silencing was associated with the decrease of histone 3 acetylation (H3), increase of repressive markers and recruitment of the heterochromatin protein 1 (HP1), a factor that correlates with condensed chromatin.Citation26
Of note, a recent report indicated that not only the expression levels of acetyltransferases, such as HAT-1, which is mostly involved in the acetylation of newly synthesized histone 3 and 4 but also the levels of the histone chaperone chromatin assembly factor 1 (CAF-I) appeared significantly elevated in distinct experimental settings of HBV infection.Citation53 Both HAT-1 and CAF-1 play a key role in nucleosome assembly. Notably, siRNA-mediated depletion of HAT-1, which was administered at the time of HBV infection, was reported to affect cccDNA establishment in vitro.Citation53 The study of Yang et al also revealed an increase of the long non-coding RNA (lncRNA) HULC (highly upregulated in liver cancer), which served to recruit HAT-1 to the cccDNA. This long non-coding RNA appeared to act as a scaffold promoting the assembly of different transcriptional regulators. Intriguingly, also the HBV core protein appeared to be involved in the HULC-HAT1 interaction,Citation53 thus highlighting the complexity of the events, which document the involvement of both virological and host factors in the formation and regulation of active cccDNA.
Concerning the organization of the cccDNA chromatin, Tropberger et al reported that the levels and distribution of active histone modifications appeared comparable to those determined on cellular chromatinCitation30 and that active histones where particularly enriched at sites of HBV promotors. However, this study also revealed an underrepresentation of repressive marks on the cccDNA.Citation30 Adding complexity to our understanding of cccDNA regulation, Flecken et al recently analyzed the profile of histone modifications on HBV DNA sequences obtained from liver biopsies of patients at different stages of CHB.Citation54 This study indicated the existence of strong differences in the deposition of posttranslational modifications (PTMs) among individuals. It remains to be investigated whether such heterogeneity of histone modifications reflects intrahepatic variations of HBV transcriptional levels or rather a non-canonical role of histone modifications on HBV DNA sequences. Keeping in mind the remarkably different organization of the cccDNA, such as its circular conformation, small size and compact organization of regulatory elements, differences in terms of epigenetic regulation among viral and host genome are not surprising. Understanding such differences appears mandatory to assess whether interventions based on epigenetic regulation of the cccDNA may become achievable and suitable therapeutic goals to accelerate HBV cure.
The cccDNA Pool and Its Stability
Studies in ducks and woodchucks led to the notion that newly synthesized DNA-containing nucleocapsids are efficiently transported into the nucleus of the hepatocytes to build a large pool of cccDNA molecules, where a high number of cccDNA molecules (up to 50 copies/cell) can be detected.Citation6,Citation55,Citation56 However, such a rapid intracellular accumulation of cccDNA molecules is generally not observed in human hepatocyte cultures.Citation57,Citation58 Despite the technical challenges in quantifying cccDNA amounts and reports indicating up to 15 copies/cell,Citation59,Citation60 most available evidence suggests that the amount of HBV minichromosomes per cell is generally low.Citation57,Citation61 Lower cccDNA copies per cell are often estimated in liver biopsies of CHB patientsCitation62–Citation64 and in the liver of human chimeric mice,Citation65,Citation66 where an average of 1 to 5 copies per cell is commonly determined although nearly all human hepatocytes stain HBV positive (for instance for HBcAg). Regarding the mechanisms by which a cccDNA pool can be amplified, it should be noted that in contrast to other larger DNA viruses, such as herpesviruses and papillomaviruses, the HBV cccDNA does not contain a so-called origin of replication (ORI), thus the HBV minichromosome cannot undergo semiconservative replication (.Citation6 Consequently, the cccDNA pool can be formed either by incoming rcDNA containing virions building new cccDNA molecules or by the generation of new rcDNA molecules that are redirected to the cell nucleus. Intriguingly, different mechanisms, both host and viral derived, appear to control the pool size of HBV cccDNA among hepadnaviruses.Citation67 In vitro studies showed that a slow infection process is required to establish the cccDNA pool and that its maintenance in NTCP-HepG2 cells depended both on intracellular recycling of HBV genomes and on infection of naïve cells by newly formed virions.Citation58 On the other hand, in vivo studies in humanized mice indicated that the amplification of the cccDNA pool mainly depends on new rounds of infection. In that study, administration of the entry inhibitor Myrcludex-B during the rump-up phase of infection blocked both further intrahepatic viral spreading among uninfected cells and new infection events in cells that already harbored the cccDNA.Citation61 Consequently, the expansion of cccDNA molecules within individually infected cells (i.e. by increasing cccDNA copy number per cell from two to five) could only derive from the intracellular amplification pathway and, if taking place, lead to a substantial increase of cccDNA loads in the whole liver of humanized mice, which should be appreciated even by qPCR methods. However, intrahepatic cccDNA increases and hence expansion of the cccDNA pool within already infected human hepatocytes could not be determined in the presence of Myrcludex-B.Citation61,Citation66 These in vivo studies indicate that in contrast to observations made with HBV-related viruses, the intracellular cccDNA amplification pathway may be less efficient in HBV-infected human cells.
The half-life of individual cccDNA molecules in vivo is not clearly defined. Some studies indicated that the cccDNA pool is very stable in the nucleus of non-dividing cells, apparently persisting for the life span of the hepatocyte.Citation68,Citation69 However, studies in hepatoma cell cultures estimated the half-life of the cccDNA to be around 40 days,Citation58 while studies in ducks and woodchucks indicated an average of 30 to 50 days for DHBV and WHV cccDNA molecules.Citation70,Citation71 Yet, the slow decay of cccDNA loads determined in CHB patients receiving nucleoside analogs (NAs)Citation64,Citation72–Citation76 contributed to extrapolate mathematical models suggesting that decades may be needed to achieve complete cccDNA clearance. Intriguingly, Huang et al recently employed a well-known lamivudine resistance mutation as a cccDNA molecular signature to monitor the kinetic of decay of these mutated cccDNA molecules within a complex pool of cccDNA quasi-species present in liver biopsies of patients from two distinct clinical trials.Citation77 Supported by the strong correlation determined between cccDNA loads, intrahepatic and serum HBV RNA levels, the authors concluded that cccDNA half-life may be significantly shorter (several months instead of years), at least in patients switching therapy (different NA or interferon) after having experienced virological breakthrough. Despite the small number of patients analyzed and the challenge to detect and quantify distinct cccDNA species with high accuracy, the study highlights the importance to investigate the kinetics of cccDNA decay in patients, as well as the need to explore further the potential of surrogate biomarkers able to reflect intrahepatic cccDNA amounts and activity. Despite such encouraging advances, elimination of the cccDNA from the whole liver remains challenging and it is likely to require substantial destruction of the infected cells or substantial cccDNA destabilization. Yet, keeping in mind that low cccDNA amounts are found even in the liver of patients who resolved acute infection,Citation78 complete elimination of the intrahepatic cccDNA reservoir may remain an unreachable goal not required to gain immunological control and resolve HBV infection.
The Contribution of Cell Division to HBV Resolution
Immune cells have the ability to destroy the cccDNA together with the infected cell, as well as to induce compensatory hepatocyte proliferation.Citation79,Citation80 Previous studies performed in animal models based on hepadnaviruses and their natural hosts (ducks and woodchucks),Citation71,Citation81 as well as using patient liver biopsies,Citation82 indicated an inverse relationship between cell turnover and intrahepatic cccDNA loads. Furthermore, cccDNA-negative cell clones containing HBV DNA integrations into the host genome demonstrated that cccDNA clearance without cell destruction can occur in chronically infected livers.Citation83
The cccDNA is an episomal, plasmid-like, structure lacking centromeres. Reports indicated that cell division may lead to an equalCitation84 or unequal distribution of the cccDNA molecules among daughter cells or even cause their loss during mitosisCitation85 (). In fact, cytosolic nucleases may promote efficient destruction of DNA molecules that are released into the cytoplasm.Citation86 Recent studies in human liver chimeric mice showed that the proliferation of HBV-infected human hepatocytes provoked a strong reduction of intrahepatic cccDNA loads.Citation66 Moreover, cell division caused not only dilution of the HBV cccDNA among daughter cells but also a substantial loss of intrahepatic cccDNA amounts. In spite of the fact that cccDNA could be efficiently purged from the great majority of the human hepatocytes, complete viral clearance was not achieved since HBV survived in sporadic, apparently non-proliferating human hepatocytes.Citation66 Consequently, virological markers rebounded as hepatocyte expansion relented and such rebound was largely due to reinfection of quiescent primary human hepatocytes, since treatment with the entry inhibitor Myrcludex-B blocked viral spread and intrahepatic cccDNA accumulation. The persistence of very few dispersed non-proliferating hepatocytes expressing high levels of HBV markers (HBcAg and HBV RNA) indicated that viral infection might hamper cell division. This observation is in line with previous studies pointing out the lower capacity of hepatocytes to proliferate in HBV-transgenic mice during liver regeneration.Citation87 The lower ability of HBV-positive cells to undergo mitosis might have significant clinical implications, since cells unable or that are less prone to divide may serve as viral reservoir. These experimental observations suggest that the development of therapeutic interventions targeting such persisting HBV-producing cells may be critical to achieve viral elimination. Moreover, the impact of cell division on cccDNA maintenance suggests that curative therapeutic approaches may need the involvement of controlled destruction of infected cells, for instance by boosting HBV-specific immune responses, whereas strategies preventing occurrence of new infection events would protect cured hepatocytes from re-infection. In this regard, an attractive strategy aiming at restoring viral immune responses may rely on the adaptive transfer of engineered HBV-specific T cells. Recent studies have shown how T cells expressing an HBV-specific T cell receptor (TCR) can specifically target HBV-infected hepatocytes both in vitro and in vivo, leading to significant elimination and perhaps also silencing of the cccDNA.Citation88–Citation90
In sum, increasing lines of evidence support the concept that both killing of infected cells and compensatory proliferation play a key synergistic role in resolving HBV infection. The fast recovery from acute HBV infection in adult patients suggests that multiple factors, including cytolytic cell killing and non-cytolytic mechanisms, such as compensatory hepatocyte proliferation and cytokine-mediated destabilization of the cccDNA (see section Impact of antiviral treatments on the cccDNA – interferons), contribute to HBV clinical resolution with loss of HBsAg while the liver remains functional. Such synergistic processes might also be active in CHB patients that stopped long-term NA therapy before reaching loss of HBsAg and that were reported to achieve a more favorable treatment outcome after experiencing a transitory hepatic flare.Citation75,Citation91,Citation92 Notably, reports indicated that low cccDNA amounts can be detected even in the liver of patients that resolved acute HBV infection,Citation78 thus augmenting the assumption that we may not need to eliminate the intrahepatic cccDNA reservoir completely in order to gain sustained off-treatment control resembling natural resolution of HBV infection.
Impact of Antiviral Treatments on the cccDNA
Targeting HBV Replication
Currently approved treatments based on nucleoside analogs (NAs) can block HBV reverse transcription with high efficiency, thus leading to the reduction of viremia even below detection limits. However, this class of compounds does not target directly the HBV minichromosome () and various studies indicated that long-term antiviral therapy is needed to achieve significant intrahepatic cccDNA reduction.Citation64,Citation72,Citation73,Citation93–Citation95 Persistence of the HBV minichromosome within the hepatocytes in spite of undetectable levels of HBV DNA viremia and HBsAg in the serum (functional cure) is the reason for the viral rebound determined after cessation of NA treatments. Moreover, recent studies indicated that even upon use of potent polymerase inhibitors, suppression of viral replication during NA therapy remains incomplete.Citation76 Such phenomenon could also lead to new infection events and so contribute substantially to the maintenance and renewal of the cccDNA pool. Thus, it will be important to determine whether stronger suppression of intracellular HBV replication can be achieved by developing more potent polymerase inhibitors or by combining NAs with strategies affecting different steps of HBV replication, such as inhibitors of the HBV RNAseHCitation96 or capsid assembly inhibitors (see below). A more effective block of HBV replication shall stop cccDNA replenishment and may reveal a faster decay of the cccDNA pool or of distinct cccDNA species, as recently proposed.Citation77
Prevention of HBV Entry
The use of an entry inhibitor in combination with drugs reducing HBV production, such as NAs, represents an increasingly attractive strategy to block efficiently the replenishment of the cccDNA pool via new infection events. Entry inhibition is efficiently achieved using Myrcludex B (Bulevirtide/Hepcludex), a small myristoylated lipopeptide containing the same aminoacid sequence of the preS1 domain of the HBV envelope protein and a first-in-class entry inhibitor blocking the NTCP cellular receptor.Citation97 Both preclinical studies in human liver chimeric mice and clinical trials performed in patients coinfected with Hepatitis Delta virusCitation61,Citation98,Citation99 demonstrated the safety profile and the potency of this peptide to prevent HBV and HDV intrahepatic spreading and cccDNA formation. Of note, the drug received Orphan Drug Designations for the treatment of HDV infection from the European Medicines Agency (EMA) and from the US Food & Drug Administration (FDA) and has been just approved by the EMA for the treatment of chronic HBV/HDV infection.
Viral entry can also be affected by small molecules, such as cyclosporine derivatives, or by using neutralizing antibodies against HBsAg epitopes.Citation100 Such antibodies may not only prevent HBV entry and cccDNA formation but they may also trigger additional antiviral and immunomodulatory activities, such as interference with viral secretion and phagocytosis of viral particles-immune complex. These studies indicate that the use of humanized HBsAg-specific neutralizing monoclonal antibodies, possibly in combination with NAs, may provide a valuable therapeutic approach to accelerate HBsAg reduction and to control cccDNA loads in chronic CHB patients.
Targeting Viral Proteins
Antiviral agents targeting viral replication at the level of protein production, assembly or release may also achieve block of de novo HBV infection. Among these are capsid inhibitors and inhibitors of HBV and HBsAg secretion.
The HBV core protein is essential for HBV genome packaging, reverse transcription, intracellular trafficking and re-shuttling of newly synthetized HBV genomes into the nucleus to form new cccDNA molecules. Compounds that interfere with capsid formation are core protein allosteric modulators (CpAMs), such as heteroaryldihydropyrimidines (i.e. GLS4) and capsid assembly modulators (i.e. NVR-3778, JNJ6379, ABI-H0731). While the first type promotes capsid mis-assembly, the second type form capsids devoid of nucleic acids. These compounds were shown to reduce HBV DNA and HBcAg levels in preclinical in vitro and in vivo models,Citation101,Citation102 as well as in clinical trials.Citation103,Citation104 Although these compounds are expected to interfere with the intracellular cccDNA replenishment, treatment of HBV-infected mice with humanized livers showed strong reduction of HBV DNA in serum, while levels of HBsAg were reduced significantly only when mice received the compound (NVR-3778) in combination with peg-IFN. Of note, levels of cccDNA were not affected by six weeks of treatment with this capsid inhibitor.Citation102
Several small molecules with the capacity to inhibit HBV secretion, such as a benzimidazole compound, have been reported to decrease HBsAg levels by inhibiting the intracellular re-localization of the HBV envelope proteins to the Golgi apparatus.Citation105 HBsAg release inhibitors such as nucleic acid polymers (NAPs) seem to inhibit HBV secretion too, although the exact mechanism is not fully elucidated and effects on the status of cccDNA need to be explored.Citation106
Among novel small molecules that selectively reduce viral antigen expression, RG7834 showed to reduce viremia and the levels of viral antigens (HBsAg).Citation107 However, these are also not expected to have a strong impact on the cccDNA. High levels of viral antigens are believed to contribute to the maintenance of HBV chronicity and therapies aiming at reducing viral RNAs and proteins are receiving increasing attention. It is however not yet clear how the reduction of certain viral antigens may impact the cccDNA. The mode of actions of distinct drugs needs to be carefully investigated.
Given the key role of HBx in cccDNA transcription, drugs targeting HBx may have the potential to inactivate cccDNA transcription. Nitazoxanide (NTZ), a thiazolide a broad-spectrum anti-infective agent that has been approved by the Food and Drug Administration for treating parasites, was shown to inhibit the binding of HBx to DDB1 and prevent the degradation of the SMC5/6 complex. Consequently, viral transcription could be reduced in cultured hepatocytes.Citation39 Whether efficient inhibition of cccDNA transcription could be achieved also in vivo remains to be investigated.
Targeting Viral RNAs
The potential of small interfering RNAs against HBV has been investigated in preclinical and clinical studies. These studies have shown the great potential of siRNAs to lower the levels of circulating HBsAg.Citation108 Recent studies in cultured HBV-infected hepatocytes showed that treatment with small interfering RNAs (siRNAs) targeting all HBV RNA levels, including the HBx mRNA, can promote the decrease of HBx protein levels and reappearance of the SMC5/6 complex.Citation109 Thus, targeting HBV replication with siRNAs not only enables the reduction of viremia and antigenemia but also bears the capacity to lower intracellular levels of core proteins and HBx production (). Whether the reappearance of the SMC5/6 complex induced by siRNA treatment can contribute to cccDNA silencing also in vivo is currently under investigation.
Targeting the cccDNA
As mentioned above, the cccDNA is the key viral molecule in HBV life cycle and curative therapy for CHB will require substantial reduction of cccDNA loads and likely silencing of persisting cccDNA molecules. Different gene therapy strategies are currently developed and under investigation either to edit or destroy the minichromosome or to epigenetically modify it to achieve transcriptional inactivation.
Strategies editing the cccDNA: Inhibiting cccDNA formation has the potential to limit intrahepatic accumulation of cccDNA loads. However, such strategies may not have substantial impact on the half-life of already formed molecules in infected hepatocytes. Nucleases have, however, the potential to target directly the cccDNA. The “clustered regularly interspaced short palindromic repeats” (CRISPR)/Cas9 system is an attractive approach because of the capacity of the single-guide RNAs (gRNA) design to target specific DNA sequences. Preclinical experiments have presented proof-of-concept for the use of the CRISPR/Cas9 system and showed cleavage and degradation of cccDNA molecules, but also occurrence of deletions leading to cccDNA inactivation.Citation110–Citation113 In these studies, different markers of viral replication were significantly reduced without evidence of toxicity, suggesting that the CRISPR/Cas9 system could be recruited to the HBV-expressing vectors. Although simultaneous targeting of different loci might improve efficiency of cccDNA cleavage and degradation, the CRISPR/Cas system can also target HBV DNA integrations and induce DNA double-stranded breaks on viral and host genome. Thus, it bears the risk of generating genomic rearrangements and an increased rate of integration and genomic instability. Moreover, hepatocellular delivery and possible off-target effects, including the occurrence of cleavage and unpredictable recombination of host genome sequences harboring HBV DNA integrations, need further studies. In this regard, a recent study employed dCas9-based approaches to inactivate HBV cccDNA by suppressing viral gene expression without DNA cleavage.Citation114 Despite all these safety concerns, the gene-editing field is constantly evolving by investigating novel nucleases with lower off-target effects and increased efficacy and safe delivery. Thus, understanding the biology of HBV after gene editing remains an essential topic.
Epigenetic modifiers promoting cccDNA silencing: As a DNA molecule associated with histones, transcription of the cccDNA minichromosome can be affected by inducing specific epigenetic modifications.Citation50 The development of such drugs has gained great attention and some epigenetic modifiers have been approved for the treatment of selected cancers. From a technical point of view, epigenetic therapies can focus on (i) targeting cellular enzymes that create epigenetic marks, like DNA methyltransferases (DNMTs) and histone acetyltransferases (HATs), or (ii) on targeting enzymes that recognize such marks and can act as mediators by attracting transcription factors; or (iii) by interfering with enzymes, like the histone deacetylases (HDACs) that remove certain epigenetic marks. For instance, AGK2 is an inhibitor of the HDAC SIRT2 and was shown to suppress cccDNA transcription in tissue culture.Citation115 Remarkably, the study also showed that transcription from integrated HBV DNA sequences appeared enhanced, thus indicating the existence of a different epigenetic regulation of transcription between episome and integrated HBV DNA. Despite first encouraging examples, it must be kept in mind that modulation of epigenetic modifiers by targeting cellular enzymes has not only the potential to suppress HBV transcription, but is likely to induce pleiotropic effects affecting cell homeostasis. Thus, the targeting of specific viral factors is expected, at least in principle, to cause less adverse effects. In this regard, the link reported between HBx expression and cccDNA epigenetic control deserves further evaluation. The mechanism of HBx-mediated cccDNA regulation through interaction with cellular partners is an interesting avenue for the use of small molecules to target either directly HBx or its interaction with the cccDNA. Anyhow, a better understanding of the mode of action of HBx in the setting of HBV infection and on epigenetic regulation of cccDNA is urgently needed to identify factors that could be specifically targeted to permanently silence the HBV reservoir.
Interferons: Cytokines and agonists of pattern recognition receptors (i.e. TLRs, RIG-I) are known to activate both the innate arm of immune responses (macrophages, dendritic cells, natural killer cells) and adaptive immune responses. Innate immune activation can trigger the production of inflammatory cytokines, activate myeloid cells and NK cell lysis, alter antigen presentation and affect HBV viral replication in hepatocytes (seeCitation116 for more details). The production of interferons will also boost activation of adaptive immune responses and eventually lead to destruction of infected hepatocytes, thus affecting also cccDNA amounts.
Cytokines like interferon alpha (IFN-α) deserve particular attention since an increasing number of reports pointed out the ability of some interferons to act not only as immune modulators but also to impact HBV replication and even cccDNA.Citation65,Citation117–Citation120 In particular IFN-α, which is used for the treatment of CHB, was reported to accelerate the degradation of pgRNA and core particles in HBV transgenic mice.Citation121–Citation123 Furthermore, studies in HBV-infected humanized mice revealed that administration of the therapeutic pegylated form (peg-IFNα) efficiently lowered the levels of both pregenomic and subgenomic HBV RNAsCitation29,Citation65 (). The underlying molecular mechanisms leading to the rapid decrease of genomic and subgenomic HBV RNAs are not yet elucidated. Nevertheless, IFNα was shown to induce epigenetic modifications of the histones bound to the cccDNA minichromosome,Citation29 while involvement of the chromatin remodelling polycomb repressive complex 2 (PRC2) indicated epigenetic suppression of the cccDNA.Citation29,Citation51 Further Chromatin Immunoprecipitation-Sequencing (ChIP-Seq) experiments also demonstrated the reduction of active histone marks upon IFNα administration and such decrease could be even achieved by using a small molecule inhibitor of the responsible histone acetyltransferase, thus proving a functional link between reduced HBV replication and reduced levels of active histone marks.Citation30 While these studies explain how IFN-α can directly contribute to the decline of viral antigen levels also without involvement of immune cells,Citation65 recent studies have also indicated that administration of peg-IFNα in HBV-infected human liver chimeric mice promotes the reappearance of the SMC5/6 complex in human hepatocytes. Thus, in line with the study of Decorsiere,Citation38 it appears that IFNα can promote cccDNA silencing by acting at different levels, including the induction of SMC5/6-mediated suppression of cccDNA transcription.Citation124
Besides promoting silencing, IFN-α was shown to induce partial cccDNA degradation by up-regulating nuclear cytidine deaminases and the nuclear factor κ-light-chain-enhancer of activated B cells (NFκB) pathways.Citation117 Of note, even other cytokines, like tumor necrosis factor α, were shown to contribute to a non-cytolytic cccDNA destabilization.Citation120 Studies in patients also indicated that one year of combination therapy with NAs and IFN-α promoted a stronger cccDNA reduction (2-log) than in the setting of NA monotherapy.Citation73 However, despite such encouraging evidence, interferon treatment is effective in only a minority of individuals and HBsAg loss rates remain low.Citation125 Keeping also in mind the systemic side effects and contraindications that are associated with IFN-based therapy, it seems important to concentrate research efforts in gaining knowledge of the molecular mechanisms by which some cytokines can achieve cccDNA silencing and destabilization.
Conclusions
Elimination of the HBV minichromosome remains challenging and more research is needed to advance our understanding in cccDNA biology and to develop therapeutic strategies efficiently targeting the cccDNA, as well as approaches boosting HBV-specific immune responses. Based on the current knowledge available, it appears clear that cell division represents a weak point for the virus in HBV persistence. At the same time, certain cytokines produced by immune cells, like IFNα, can also contribute to cccDNA silencing and destabilization. The growing awareness of the importance of the regulatory HBx protein in cccDNA activity encourages efforts toward the development of agents able to interfere with HBx, since these may silence the transcription of the HBV matrix and hinder the accumulation of various HBx-mediated host alterations that have been associated with CHB pathogenesis and liver cancer progression.
It is conceivable that targeting distinct viral components and steps of HBV life cycle, possibly by silencing the cccDNA, while protecting the hepatocytes from de novo infection, will likely be needed to achieve a functional cure for the majority of CHB patients. Moreover, the improvement of strategies enhancing HBV-specific adaptive immune responses may eliminate a substantial amount of HBV-infected cells and trigger compensatory cell proliferation; events that should further lower cccDNA loads, possibly achieving levels comparable to those determined in the setting of resolved HBV infection. To achieve such goals, we need to improve our comprehension of cccDNA biology and regulation in the setting of infection. Taking into advantage the availability of new technologies and experimental systems, the development of curative strategies may become a goal at reach in the near future.
Disclosure
MD was supported by the German Research Foundation (DFG: SFB 841), by the German Center for Infection Research (DZIF-BMBF; TTU-hepatitis 05.815) and by the State of Hamburg with the Research Program (LFF-FV44: EPILOG), during the conduct of the study. The authors report no other potential conflicts of interest in this work.
References
- who. Hepatitis B key facts. 2018.
- Dandri M, Petersen J. Mechanism of hepatitis B virus persistence in hepatocytes and its carcinogenic potential. Clin Infect Dis. 2016;Suppl 62(suppl 4):S281–S288. doi:10.1093/cid/ciw023
- Cornberg M, Suk-Fong Lok A, Terrault NA, et al. Guidance for design and endpoints of clinical trials in chronic hepatitis B - report from the 2019 EASL-AASLD HBV treatment endpoints conference. J Hepatol. 2020;71(3):1070–1092.
- Leistner CM, Gruen-Bernhard S, Glebe D. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell Microbiol. 2008;10:122–133.18086046
- Yan H, Zhong G, Xu G, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife. 2012;3:e05570.
- Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015;64(12):1972–1984. doi:10.1136/gutjnl-2015-30980926048673
- Rabe B, Glebe D, Kann M. Lipid-mediated introduction of hepatitis B virus capsids into nonsusceptible cells allows highly efficient replication and facilitates the study of early infection events. J Virol. 2006;80(11):5465–5473. doi:10.1128/JVI.02303-0516699026
- Gallucci L, Kann M. Nuclear import of hepatitis B virus capsids and genome. Viruses. 2017;9(1):21. doi:10.3390/v9010021
- Guo H, Xu C, Zhou T, et al. Characterization of the host factors required for hepadnavirus covalently closed circular (ccc) DNA formation. PLoS One. 2012;7(8):e43270. doi:10.1371/journal.pone.004327022912842
- Schreiner S, Nassal M. A role for the host DNA damage response in hepatitis B virus cccDNA formation-and beyond? Viruses. 2017;9. doi:10.3390/v9050125
- Guo H, Jiang D, Zhou T, et al. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol. 2007;81(22):12472–12484. doi:10.1128/JVI.01123-0717804499
- Baumert TF, Verrier ER, Nassal M, et al. Host-targeting agents for treatment of hepatitis B virus infection. Curr Opin Virol. 2015;14:41–46. doi:10.1016/j.coviro.2015.07.00926262886
- Qi Y, Gao Z, Xu G, et al. DNA polymerase κ is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog. 2016;12(10):e1005893. doi:10.1371/journal.ppat.100589327783675
- Koniger C, Wingert I, Marsmann M, et al. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc Natl Acad Sci U S A. 2014;111(40):E4244–E4253. doi:10.1073/pnas.140998611125201958
- Bock C-T, Schranz P, Schroder CH, et al. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes. 1994;8(2):215–229. doi:10.1007/BF17030797975268
- Bock CT, Schwinn S, Locarnini S, et al. Structural organization of the hepatitis B virus minichromosome. J Mol Biol. 2001;307(1):183–196. doi:10.1006/jmbi.2000.448111243813
- Newbold JE, Xin H, Tencza M, et al. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol. 1995;69(6):3350–3357. doi:10.1128/JVI.69.6.3350-3357.19957745682
- Giersch K, Allweiss L, Volz T, et al. Serum HBV pgRNA as a clinical marker for cccDNA activity. J Hepatol. 2017;66(2):460–462. doi:10.1016/j.jhep.2016.09.02827826059
- Wang J, Shen T, Huang X, et al. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J Hepatol. 2016;65(4):700–710. doi:10.1016/j.jhep.2016.05.02927245431
- Liu Y, Jiang M, Xue J, et al. Serum HBV RNA quantification: useful for monitoring natural history of chronic hepatitis B infection. BMC Gastroenterol. 2019;19(1):53. doi:10.1186/s12876-019-0966-430991954
- Glebe D, Urban S. Viral and cellular determinants involved in hepadnaviral entry. World J Gastroenterol. 2007;13:22–38. doi:10.3748/wjg.v13.i1.2217206752
- Dembek C, Protzer U, Roggendorf M. Overcoming immune tolerance in chronic hepatitis B by therapeutic vaccination. Curr Opin Virol. 2018;30:58–67. doi:10.1016/j.coviro.2018.04.00329751272
- Visvanathan K, Lewin SR. Immunopathogenesis: role of innate and adaptive immune responses. Semin Liver Dis. 2006;26(2):104–115. doi:10.1055/s-2006-93975516673289
- Zoulim F, Saputelli J, Seeger C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol. 1994;68(3):2026–2030. doi:10.1128/JVI.68.3.2026-2030.19948107266
- Lucifora J, Arzberger S, Durantel D, et al. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol. 2011;55(5):996–1003. doi:10.1016/j.jhep.2011.02.01521376091
- Riviere L, Gerossier L, Ducroux A, et al. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J Hepatol. 2015;63(5):1093–1102. doi:10.1016/j.jhep.2015.06.02326143443
- Tsuge M, Hiraga N, Akiyama R, et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J Gen Virol. 2010;91(7):1854–1864. doi:10.1099/vir.0.019224-020219897
- Levrero M, Pollicino T, Petersen J, et al. Control of cccDNA function in hepatitis B virus infection. J Hepatol. 2009;51(3):581–592. doi:10.1016/j.jhep.2009.05.02219616338
- Belloni L, Allweiss L, Guerrieri F, et al. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest. 2012;122(2):529–537. doi:10.1172/JCI5884722251702
- Tropberger P, Mercier A, Robinson M, et al. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc Natl Acad Sci U S A. 2015;112(42):E5715–E5724. doi:10.1073/pnas.151809011226438841
- Quasdorff M, Protzer U. Control of hepatitis B virus at the level of transcription. J Viral Hepat. 2019;156(8):1392–1403.e7. doi:10.1111/j.1365-2893.2010.01315.x
- Bar-Yishay I, Shaul Y, Shlomai A. Hepatocyte metabolic signalling pathways and regulation of hepatitis B virus expression. Liver Int. 2011;31(3):282–290. doi:10.1111/j.1478-3231.2010.02423.x21281428
- Mohd-Ismail NK, Lim Z, Gunaratne J, et al. Mapping the interactions of HBV cccDNA with host factors. Int J Mol Sci. 2019;20:4276.
- Guo Y-H, Li Y-N, Zhao J-R, et al. HBc binds to the CpG islands of HBV cccDNA and promotes an epigenetic permissive state. Epigenetics. 2016;62(6):S281–S288. doi:10.4161/epi.6.6.15815
- Chong CK, Cheng CYS, Tsoi SYJ, et al. Role of hepatitis B core protein in HBV transcription and recruitment of histone acetyltransferases to cccDNA minichromosome. Antiviral Res. 2017;144:1–7. doi:10.1016/j.antiviral.2017.05.00328499864
- Slagle BL, Bouchard MJ. Hepatitis B virus X and regulation of viral gene expression. Cold Spring Harb Perspect Med. 2010;91(3):a021402. doi:10.1101/cshperspect.a021402
- Belloni L, Pollicino T, De Nicola F, et al. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci U S A. 2009;106:19975–19979. doi:10.1073/pnas.090836510619906987
- Decorsiere A, Mueller H, van Breugel PC, et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. 2016;531(7594):386–389. doi:10.1038/nature1717026983541
- Sekiba K, Otsuka M, Ohno M, et al. Inhibition of HBV transcription from cccDNA with nitazoxanide by targeting the HBx-DDB1 interaction. Cell Mol Gastroenterol Hepatol. 2019;7:297–312. doi:10.1016/j.jcmgh.2018.10.01030704981
- Mitra B, Guo H. Hepatitis B virus X protein crosses out Smc5/6 complex to maintain covalently closed circular DNA transcription. Hepatology. 2016;64(6):2246–2249. doi:10.1002/hep.2883427639252
- Abdul F, Filleton F, Gerossier L, et al. Smc5/6 antagonism by HBx is an evolutionarily conserved function of hepatitis B virus infection in mammals. J Virol. 2018;92(16). doi:10.1128/JVI.00769-18
- Fu X, Song X, Li Y, et al. Hepatitis B virus X protein upregulates DNA methyltransferase 3A/3B and enhances SOCS-1CpG island methylation. Mol Med Rep. 2018;92(1):301–308. doi:10.3892/mmr.2015.4545
- Andrisani OM. Deregulation of epigenetic mechanisms by the hepatitis B virus X protein in hepatocarcinogenesis. Viruses. 2013;5(3):858–872. doi:10.3390/v503085823507839
- Vivekanandan P, Daniel HD-J, Kannangai R, et al. Hepatitis B virus replication induces methylation of both host and viral DNA. J Virol. 2010;84(9):4321–4329. doi:10.1128/JVI.02280-0920147412
- Kim J-W, Lee SH, Park YS, et al. Replicative activity of hepatitis B virus is negatively associated with methylation of covalently closed circular DNA in advanced hepatitis B virus infection. Intervirology. 2011;54(6):316–325. doi:10.1159/00032145021242658
- Zhang X, Hou J, Lu M. Regulation of hepatitis B virus replication by epigenetic mechanisms and microRNAs. Front Genet. 2013;4:202. doi:10.3389/fgene.2013.0020224133502
- Zhang Y, Mao R, Yan R, et al. Transcription of hepatitis B virus covalently closed circular DNA is regulated by CpG methylation during chronic infection. PLoS One. 2014;9(10):e110442. doi:10.1371/journal.pone.011044225337821
- Jain S, Chang -T-T, Chen S, et al. Comprehensive DNA methylation analysis of hepatitis B virus genome in infected liver tissues. Sci Rep. 2013;5(1):10478. doi:10.1038/srep10478
- Hong X, Kim ES, Guo H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: implications for epigenetic therapy against chronic hepatitis B. Hepatology. 2017;66(6):2066–2077. doi:10.1002/hep.2947928833361
- Dandri M. Epigenetic modulation in chronic hepatitis B virus infection. Semin Immunopathol. 2020;42(2):173–185. doi:10.1007/s00281-020-00780-632185454
- Pollicino T, Belloni L, Raffa G, et al. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology. 2006;130(3):823–837. doi:10.1053/j.gastro.2006.01.00116530522
- Cougot D, Allemand E, Riviere L, et al. Inhibition of PP1 phosphatase activity by HBx: a mechanism for the activation of hepatitis B virus transcription. Sci Signal. 2006;130(205):ra1. doi:10.1126/scisignal.2001906
- Yang G, Feng J, Liu Y, et al. HAT1 signaling confers to assembly and epigenetic regulation of HBV cccDNA minichromosome. Theranostics. 2019;9(24):7345–7358. doi:10.7150/thno.3717331695772
- Flecken T, Meier M-A, Skewes-Cox P, et al. Mapping the heterogeneity of histone modifications on hepatitis B virus DNA using liver needle biopsies obtained from chronically infected patients. J Virol. 2019;93(9). doi:10.1128/JVI.02036-18
- Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell. 1986;47(3):451–460. doi:10.1016/0092-8674(86)90602-13768961
- Wu -T-T, Coates L, Aldrics CE, et al. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology. 1990;175(1):255–261. doi:10.1016/0042-6822(90)90206-72155510
- Lucifora J, Protzer U. Attacking hepatitis B virus cccDNA – the holy grail to hepatitis B cure. J Hepatol. 2016;64(1):S41–S48. doi:10.1016/j.jhep.2016.02.00927084036
- Ko C, Chakraborty A, Chou WM, et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J Hepatol. 2018;69:1231–1241. doi:10.1016/j.jhep.2018.08.01230142426
- Balagopal A, Hwang HS, Grudda T, et al. Single hepatocyte hepatitis B virus transcriptional landscape in HIV co-infection. J Infect Dis. 2020;221(9):1462–1469.31740931
- Huang J-T, Yang Y, Hu Y-M, et al. A highly sensitive and robust method for hepatitis B virus covalently closed circular DNA detection in single cells and serum. J Mol Diagn. 2018;20(3):334–343. doi:10.1016/j.jmoldx.2018.01.01029656833
- Volz T, Allweiss L, ḾBarek MB, et al. The entry inhibitor myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J Hepatol. 2013;58(5):861–867. doi:10.1016/j.jhep.2012.12.00823246506
- Laras A, Koskinas J, Dimou E, et al. Intrahepatic levels and replicative activity of covalently closed circular hepatitis B virus DNA in chronically infected patients. Hepatology. 2006;44(3):694–702. doi:10.1002/hep.2129916941694
- Volz T, Lutgehetmann M, Wachtler P, et al. Impaired intrahepatic hepatitis B virus productivity contributes to low viremia in most HBeAg-negative patients. Gastroenterology. 2007;133(3):843–852. doi:10.1053/j.gastro.2007.06.05717854594
- Werle–Lapostolle B, Bowden S, Locarnini S, et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy1. Gastroenterology. 2004;126(7):1750–1758. doi:10.1053/j.gastro.2004.03.01815188170
- Allweiss L, Volz T, Lutgehetmann M, et al. Immune cell responses are not required to induce substantial hepatitis B virus antigen decline during pegylated interferon-alpha administration. J Hepatol. 2014;60(3):500–507. doi:10.1016/j.jhep.2013.10.02124398036
- Allweiss L, Volz T, Giersch K, et al. Proliferation of primary human hepatocytes and prevention of hepatitis B virus reinfection efficiently deplete nuclear cccDNA in vivo. Gut. 2018;67(3):542–552. doi:10.1136/gutjnl-2016-31216228428345
- Kock J, Rosler C, Zhang -J-J, et al. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 2015;64(9):e1001082. doi:10.1371/journal.ppat.1001082
- Dandri M, Burda MR, Will H, et al. Increased hepatocyte turnover and inhibition of woodchuck hepatitis B virus replication by adefovirIn vitro do not lead to reduction of the closed circular DNA. Hepatology. 2000;32(1):139–146. doi:10.1053/jhep.2000.870110869302
- Moraleda G, Saputelli J, Aldrich CE, et al. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J Virol. 1997;71:9392–9399.9371599
- Zhu Y, Yamamoto T, Cullen J, et al. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J Virol. 2001;75(1):311–322. doi:10.1128/JVI.75.1.311-322.200111119601
- Addison WR, Walters K-A, Wong WWS, et al. Half-life of the duck hepatitis B virus covalently closed circular DNA pool in vivo following inhibition of viral replication. J Virol. 2002;76(12):6356–6363. doi:10.1128/JVI.76.12.6356-6363.200212021368
- Lai C-L, Wong D, Ip P, et al. Reduction of covalently closed circular DNA with long-term nucleos(t)ide analogue treatment in chronic hepatitis B. J Hepatol. 2017;66(2):275–281. doi:10.1016/j.jhep.2016.08.02227639844
- Wursthorn K, Lutgehetmann M, Dandri M, et al. Peginterferon alpha-2b plus adefovir induce strong cccDNA decline and HBsAg reduction in patients with chronic hepatitis B. Hepatology. 2006;44(3):675–684. doi:10.1002/hep.2128216941693
- Lutgehetmann M, Volzt T, Quaas A, et al. Sequential combination therapy leads to biochemical and histological improvement despite low ongoing intrahepatic hepatitis B virus replication. Antivir Ther. 2008;13:57–66.18389899
- Bowden S, Locarnini S, Chang T-T, et al. Covalently closed-circular hepatitis B virus DNA reduction with entecavir or lamivudine. World J Gastroenterol. 2006;80(15):5465–5473. doi:10.3748/wjg.v21.i15.4644
- Boyd A, Lacombe K, Lavocat F, et al. Decay of ccc-DNA marks persistence of intrahepatic viral DNA synthesis under tenofovir in HIV-HBV co-infected patients. J Hepatol. 2015;21(4):4644–4651. doi:10.1016/j.jhep.2016.05.014
- Huang Q, Zhou B, Cai D, et al. Rapid turnover of HBV cccDNA indicated by monitoring emergence and reversion of signature-mutation in treated chronic hepatitis B patients. Hepatology. 2020. doi:10.1002/hep.31240
- Rehermann B, Ferrari C, Pasquinelli C, et al. The hepatitis B virus persists for decades after patients‘ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T–lymphocyte response. Nat Med. 1996;2(10):1104–1108. doi:10.1038/nm1096-11048837608
- Bertoletti A, Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut. 2012;61(12):1754–1764. doi:10.1136/gutjnl-2011-30107322157327
- Dandri M, Petersen J. Hepatitis B virus cccDNA clearance: killing for curing? Hepatology. 2005;42(6):1453–1455. doi:10.1002/hep.2097616317676
- Reaiche-Miller GY, Thorpe M, Low HC, et al. Duck hepatitis B virus covalently closed circular DNA appears to survive hepatocyte mitosis in the growing liver. Virology. 2013;446(1–2):357–364. doi:10.1016/j.virol.2013.08.01424074600
- Mason WS, Gill US, Litwin S, et al. HBV DNA Integration and clonal hepatocyte expansion in chronic hepatitis B patients considered immune tolerant. Gastroenterology. 2016;151(5):986–998.e4. doi:10.1053/j.gastro.2016.07.01227453547
- Mason WS, Low H-C, Xu C, et al. Detection of clonally expanded hepatocytes in chimpanzees with chronic hepatitis B virus infection. J Virol. 2009;83(17):8396–8408. doi:10.1128/JVI.00700-0919535448
- Li M, Sohn JA, Seeger C. Distribution of hepatitis B virus nuclear DNA. J Virol. 2018;92.
- Chen F, Zhang J, Wen B, et al. HBV/HCV dual infection impacts viral load, antibody response, and cytokine expression differently from HBV or HCV single infection. Sci Rep. 2016;6(1):39409. doi:10.1038/srep3940928009018
- Lechardeur D, Sohn K-J, Haardt M, et al. Metabolic instability of plasmid DNA in the cytosol: a potential barrier to gene transfer. Gene Therapy. 1999;6(4):482–497. doi:10.1038/sj.gt.330086710476208
- Quetier I, Brezillon N, Duriez M, et al. Hepatitis B virus HBx protein impairs liver regeneration through enhanced expression of IL-6 in transgenic mice. J Hepatol. 2013;59(2):285–291. doi:10.1016/j.jhep.2013.03.02123542345
- Kah J, Koh S, Volz T, et al. Lymphocytes transiently expressing virus-specific T cell receptors reduce hepatitis B virus infection. J Clin Invest. 2017;127(8):3177–3188. doi:10.1172/JCI9302428737510
- Bertoletti A, Le Bert N. Immunotherapy for chronic hepatitis B virus infection. Gut Liver. 2018;12(5):497–507. doi:10.5009/gnl1723329316747
- Wisskirchen K, Kah J, Malo A, et al. T cell receptor grafting allows virological control of hepatitis B virus infection. J Clin Invest. 2019;129(7):2932–2945. doi:10.1172/JCI12022831039136
- Hadziyannis E, Sialevris K, Georgiou A, et al. Analysis of serum alpha-fetoprotein-L3% and des-gamma carboxyprothrombin markers in cases with misleading hepatocellular carcinoma total alpha-fetoprotein levels. Oncol Rep. 2013;29:835–839. doi:10.3892/or.2012.214723174906
- Honer Zu Siederdissen C, Rinker F, Maasoumy B, et al. Viral and host responses after stopping long-term Nucleos(t)ide analogue therapy in HBeAg-negative chronic hepatitis B. J Infect Dis. 2016;214(10):1492–1497. doi:10.1093/infdis/jiw41227609808
- Sung JJ, Wong ML, Bowden S, et al. Intrahepatic hepatitis B virus covalently closed circular DNA can be a predictor of sustained response to therapy. Gastroenterology. 2005;128:1890–1897. doi:10.1053/j.gastro.2005.03.00915940624
- Wong DK, Yuen MF, Ngai VW, et al. One-year entecavir or lamivudine therapy results in reduction of hepatitis B virus intrahepatic covalently closed circular DNA levels. Antivir Ther. 2006;11:909–916.17302253
- Zheng Q, Zhu YY, Chen J, et al. Decline in intrahepatic cccDNA and increase in immune cell reactivity after 12 weeks of antiviral treatment were associated with HBeAg loss. J Viral Hepat. 2014;21:909–916. doi:10.1111/jvh.1226124888640
- Tavis JE, Zoidis G, Meyers MJ, et al. Chemical approaches to inhibiting the hepatitis B virus ribonuclease H. ACS Infect Dis. 2005;128(5):1890–1898. doi:10.1021/acsinfecdis.8b00045
- Petersen J, Dandri M, Mier W, et al. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat Biotechnol. 2019;5(3):655–658. doi:10.1038/nbt1389
- Lutgehetmann M, Mancke LV, Volz T, et al. Humanized chimeric uPA mouse model for the study of hepatitis B and D virus interactions and preclinical drug evaluation. Hepatology. 2012;55(3):685–694. doi:10.1002/hep.2475822031488
- Bogomolov P, Alexandrov A, Voronkova N, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: first results of a phase Ib/IIa study. J Hepatol. 2016;65(3):490–498. doi:10.1016/j.jhep.2016.04.01627132170
- Zhang TY, Yuan Q, Zhao JH, et al. Prolonged suppression of HBV in mice by a novel antibody that targets a unique epitope on hepatitis B surface antigen. Gut. 2016;65:658–671. doi:10.1136/gutjnl-2014-30896426423112
- Brezillon N, Brunelle MN, Massinet H, et al. Antiviral activity of bay 41-4109 on hepatitis B virus in humanized Alb-uPA/SCID mice. PLoS One. 2011;6:e25096. doi:10.1371/journal.pone.002509622162746
- Klumpp K, Shimada T, Allweiss L, et al. Efficacy of NVR 3–778, alone and in combination with pegylated interferon, vs entecavir in uPA/SCID mice with humanized livers and HBV infection. Gastroenterology. 2018;154:652–62 e8. doi:10.1053/j.gastro.2017.10.01729079518
- Wang XY, Wei ZM, Wu GY, et al. In vitro inhibition of HBV replication by a novel compound, GLS4, and its efficacy against adefovir-dipivoxil-resistant HBV mutations. Antivir Ther. 2012;17:793–803. doi:10.3851/IMP215222668794
- Yuen MF, Gane EJ, Kim DJ, et al. Antiviral activity, safety, and pharmacokinetics of capsid assembly modulator NVR 3–778 in patients with chronic HBV infection. Gastroenterology. 2019;156:1392–403 e7. doi:10.1053/j.gastro.2018.12.02330625297
- Xu YB, Yang L, Wang GF, et al. Benzimidazole derivative, BM601, a novel inhibitor of hepatitis B virus and HBsAg secretion. Antiviral Res. 2014;107:6–15. doi:10.1016/j.antiviral.2014.04.00224746457
- Bazinet M, Pantea V, Placinta G, et al. Safety and efficacy of 48 weeks REP 2139 or REP 2165, tenofovir disoproxil, and pegylated interferon Alfa-2a in patients with chronic HBV infection naive to Nucleos(t)ide therapy. Gastroenterology. 2020;158:2180–2194. doi:10.1053/j.gastro.2020.02.05832147484
- Mueller H, Wildum S, Luangsay S, et al. A novel orally available small molecule that inhibits hepatitis B virus expression. J Hepatol. 2018;68:412–420. doi:10.1016/j.jhep.2017.10.01429079285
- Thi EP, Dhillon AP, Ardzinski A, et al. ARB-1740, a RNA interference therapeutic for chronic hepatitis B infection. ACS Infect Dis. 2019;5:725–737. doi:10.1021/acsinfecdis.8b0019130403127
- Kornyeyev D, Ramakrishnan D, Voitenleitner C, et al. Spatiotemporal analysis of hepatitis B virus X protein in primary human hepatocytes. J Virol. 2019;93. doi:10.1128/JVI.00248-19
- Lin SR, Yang HC, Kuo YT, et al. The CRISPR/cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol Ther Nucleic Acids. 2014;3:e186. doi:10.1038/mtna.2014.3825137139
- Kostyushev D, Brezgin S, Kostyusheva A, et al. Orthologous CRISPR/Cas9 systems for specific and efficient degradation of covalently closed circular DNA of hepatitis B virus. Cell Mol Life Sci. 2019;76(9):1779–1794. doi:10.1007/s00018-019-03021-830673820
- Seeger C, Sohn JA. Targeting hepatitis B virus with CRISPR/cas9. Mol Ther Nucleic Acids. 2014;3:e216. doi:10.1038/mtna.2014.6825514649
- Kennedy EM, Kornepati AV, Cullen BR. Targeting hepatitis B virus cccDNA using CRISPR/cas9. Antiviral Res. 2015;123:188–192. doi:10.1016/j.antiviral.2015.10.00426476375
- Yang YC, Chen YH, Kao JH, et al. Permanent Inactivation of HBV genomes by CRISPR/cas9-mediated non-cleavage base editing. Mol Ther Nucleic Acids. 2020;20:480–490. doi:10.1016/j.omtn.2020.03.00532278307
- Yu H-B, Jiang H, Cheng S-T, et al. AGK2, A SIRT2 inhibitor, inhibits hepatitis B virus replication in vitro and in vivo. Int J Med Sci. 2018;15(12):1356–1364. doi:10.7150/ijms.2612530275764
- Fanning GC, Zoulim F, Hou J, et al. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat Rev Drug Discov. 2019;18:827–844.31455905
- Lucifora J, Xia Y, Reisinger F, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343(6176):1221–1228. doi:10.1126/science.124346224557838
- Wieland SF, Spangenberg HC, Thimme R, et al. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc Natl Acad Sci USA. 2004;101(7):2129–2134. doi:10.1073/pnas.030847810014764900
- Murray JM, Wieland SF, Purcell RH, et al. Dynamics of hepatitis B virus clearance in chimpanzees. Proc Natl Acad Sci U S A. 2005;102(49):17780–17785. doi:10.1073/pnas.050891310216306261
- Xia Y, Stadler D, Lucifora J, et al. Interferon-γ and tumor necrosis factor-α produced by T cells reduce the HBV persistence form, cccDNA, without cytolysis. Gastroenterology. 2016;150(1):194–205. doi:10.1053/j.gastro.2015.09.02626416327
- Asabe S, Wieland SF, Chattopadhyay PK, et al. The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J Virol. 2009;83(19):9652–9662. doi:10.1128/JVI.00867-0919625407
- Wieland SF, Eustaquio A, Whitten-Bauer C, et al. Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc Natl Acad Sci U S A. 2005;102(28):9913–9917. doi:10.1073/pnas.050427310215994231
- Uprichard SL, Wieland SF, Althage A, et al. Transcriptional and posttranscriptional control of hepatitis B virus gene expression. Proc Natl Acad Sci U S A. 2003;100(3):1310–1315. doi:10.1073/pnas.25277359912552098
- Allweiss L, Giersch K, Volz T, et al. PS-155-HBV entry inhibition after interferon alpha treatment hinders HBV rebound in hepatocytes that became negative for all HBV markers during interferon treatment. J Hepatol. 2019;70(1):e98. doi:10.1016/S0618-8278(19)30173-2
- Vigano M, Grossi G, Loglio A, et al. Treatment of hepatitis B: is there still a role for interferon? Liver Int. 2018;38(Suppl 1):79–83. doi:10.1111/liv.1363529427498