
Abstract
Background
Viral gastroenteritis belongs to the major public health problems of infant and children worldwide. The largest proportion of morbidity and mortality occurs in Sub-Saharan Africa.
Purpose
Aimed to assess the burden and genetic diversity of enteric viruses among children with diarrhea.
Patients and Methods
A cross-sectional study was undertaken from December 2015 to April 2016 in Debre Tabor. A total of thirty-eight children, who presented with diarrhea at Debre Tabor health centers, were included. Fecal samples were collected and screened for enteric viruses by RT-PCR. Data were analyzed using SPSS software. Descriptive summary techniques were used to display the findings.
Results
Out of thirty-eight children screened, 52.6% were positive for at least one enteric virus. Six (30.0%) of the children had mixed enteric virus infections. Human adenovirus (HAdV) 7 (18.4%) was predominant followed by noroviruses (NoVs) 5 (13.2%), enterovirus (EV) 5 (13.2%), rotavirus A (RVA) 4 (10.5%), human astrovirus (HAstV) 2 (5.3%), and human parechovirus (HPeV) 1 (2.6%). Overall, nineteen different types of enteric virus genotypes were identified. Diverse adenovirus within species A (HAdV-12,-31), B (HAdV-3), C (HAdV-2), and F (HAdV-4) were detected. Norovirus II (GII.4 and GII.6) and norovirus I (GI.2, GI.3, and GI.5) genotypes were found. Sapovirus genotypes within genogroup II (GII.1, GII.5, and GII.6) were identified. Wild-type rotavirus G9 and P[8] genotypes were detected in one of the rotavirus positive samples. Non-polio enteroviruses within species A (coxsackie A virus (CAV) 5, CAV6, and CAV14) and C (enterovirus (EV-C) 99) were also identified. In two of the fecal samples classic HAstV-2 was detected.
Conclusion
Diverse enteric viruses were detected in fecal samples from under-five children with diarrhea. The detection of heterogeneous enteric viruses in this small data set highlights the need for extended multicenter studies to describe the burden and genetic diversity of enteric virus.
Introduction
Diarrheal disease is one of the leading causes of morbidity and mortality worldwide. It is accounted for approximately 499,000 deaths in under-five children worldwide in 2017, with the largest proportion of deaths in sub-Saharan Africa and Asia.Citation1,Citation2 Enteric viruses are the most common pathogens causing diarrhea in high-income as well as low-income countries.Citation3,Citation4 The most common agents are rotavirus (RV), human adenovirus (HAdV), noroviruses (NoV), sapovirus (SaV), and human astrovirus (HAstV). Human parechovirus (HPeV) and enterovirus (EV) have also been associated with gastroenteritis.Citation5,Citation6
Despite the availability of effective vaccines, rotavirus A (RVA) is still the leading cause of morbidity and mortality worldwide.Citation7 Rotaviruses are double stranded (ds) RNA viruses belonging to the family Reoviridae. To date, more than nine species of RV have been recognized.Citation8 Species RVA is accountable for more than 90% of the infections. The RVA is further classified into P and G-types based on the VP4 and VP7 segments, respectively.Citation9 As per the WHO recommendation, Ethiopia has introduced the Rotarix vaccine since November 2013.Citation10,Citation11 NoVs and SaVs are also recognized as the major viral pathogens among patients with diarrhea. They belong to the family of Caliciviridae, naked viruses with positive-sense single-stranded (SS) RNA genome.Citation12,Citation13 Both of them affect humans and animals and show extensive genetic diversity. Based on the VP1 gene, SaVs and NoVs are classified into 15 (GI–GXV) and ten (GI–GX) genogroups, respectively.Citation14,Citation15 NoV genogroup GI, GII, GIV, GVIII, and GIX infect humans, with GII and GI predominance.Citation14,Citation16,Citation17 Human pathogenic SaV strains are assigned to GI, GII, GIV, and GV genogroups.Citation14 Adenoviruses are non-enveloped, dsDNA viruses with more than 90 recognized serotypes. They are associated with a wide spectrum of symptoms, including gastroenteritis, respiratory infections, conjunctivitis, hemorrhagic cystitis, and meningoencephalitis.Citation18,Citation19 Types 40 and 41 are the most frequently reported cause of HAdV-associated gastroenteritis.Citation19–22 HAstV are non-enveloped and contain a positive sense of ssRNA.Citation24 The HAstVs are categorized into classical and novel or divergent groups. The classic group consists of eight genotypes, which are responsible for 2 to 9% of acute gastroenteritis cases in children worldwide.Citation23,Citation25 Enterovirus and HPeV are belonging to the family Picornaviridae. The family contains extensively diverse genotypes. They are associated with a wide array of clinical manifestations ranging from respiratory or gastrointestinal symptoms to neonatal sepsis and infections of the central nervous system.Citation26
In Ethiopia, investigation of diarrheal samples is largely limited to light microscopic and limited bacterial culture. Investigation of enteric viruses as causes for diarrhea is uncommon. Moreover, epidemiological studies on viral infections are rare. Those few available studies focus on RVs and NoVs. Hence, the contributions of other enteric viruses in case of diarrhea remain unknown. Therefore, this study was aimed to provide preliminary information on the burden and genetic diversity of enteric viruses in Debre Tabor, Northwest Ethiopia.
Materials and Methods
Study Area and Period
This cross-sectional study was conducted at the outpatient departments of Debre Tabor and Hidar Asrand Health Centers, Debre Tabor town, from December 1, 2015, to April 30, 2016. The town is an administrative center of South Gondar Zone, Amhara National Regional State, Northwest Ethiopia. Debre Tabor has situated 50 km to the East of Lake Tana. There are a referral hospital and two health centers in the town. The study was approved by the Ethical Review Board of the University of Gondar (O/V/RC/05/1180/2016). Parents or guardians of children provided their informed consent to participate in the study. The study was conducted in accordance with the Declaration of Helsinki.
Specimen Collection
During the study period thirty-eight fecal samples were collected from infants and under-five children with diarrhea attending the outpatient departments. Clinical information and demographic data such as age, sex, and residential area were recorded. Parents or guardians of children were instructed on how to collect fecal samples from their children. About 1–3 g of the fecal specimen was saved. The samples were stored at −20 °C at the site of collection, before being transported to the Institute of Virology, Leipzig University, Germany, for further viral screening and molecular characterization.
Nucleic Acid Extraction
Nucleic acid were extracted from 10% fecal suspensions in phosphate-buffered saline. Viral nucleic acids were extracted from 200 μL of the concentrated sample using MagNA Pure 96 Instrument (Roche, Germany) as per the instruction of the manufacturer. The extracted nucleic was stored at −80 °C until further use.
Detection of Enteric Viruses
The samples were screened for the presence of RVA, NoV, SaV, HAdV, HAstV, EV, and HPeV. Screening and quantification assays were run on a LightCycler® 2.0 Instrument (Roche Diagnostics, Mannheim, Germany) using previously published primers and probes.Citation27–Citation34 The HPeV screening was carried out by using new primer pairs and probe (PeV-5NTR-F 5’-CTGGTAACAGRTGCCYCTGG-3’, PeV-5NTR-R 5’-GCCCCARATCAGATCCA YAGTG-3’ and PeV-5NTR-LNA-FL 5’-ACACTAGTTGTAHGGCCC-3’). The reaction and amplification conditions were performed as per the in-house protocols of the institute of Virology, Leipzig University, Germany.
Genotyping of Enteric Viruses
Rotavirus strain was G and P genotyped by amplification of the VP7 and VP4 segments, respectively.Citation28 In brief, reverse transcription reactions were performed in 10 µL reaction for both VP4 and VP7 regions. The reaction mixture consisted of PCR grade water, 1X AMV buffer, 0.2 mM dNTPs mix, 2U/µL RNase inhibitor and AMV Reverse Transcriptase (Promega Corporation, WI, USA). Five µL of RNA was added to the reaction mixture and incubated at 50 °C for 1 hour. The generated cDNA were used for the subsequent amplification steps with primer pairs specific for each region. Components of the PCR reaction mixture were PCR grade water, 1X PCR buffer, 1.5 mM MgCl2, 200 µM dNTP mix, 0.2 µM reverse primer, 0.2 µM forward primer, and 2 U/µL Platinum® Taq DNA polymerase. The final reaction mixture was amplified in a block cycler with an initial step of denaturation at 95°C for 3 min followed by 40 cycles of denaturation at 95°C for 30s, annealing at 50°C for 40s and elongation at 72°C for 20s. The final amplification cycle had an additional step of extension at 72°C for 5 min.
For NoV capsid genotyping, a portion of the VP1 gene was amplified by using previously published protocols.Citation35 In order to generate cDNA for the PCR steps, SuperScript III reverse transcriptase (Invitrogen) was used in a 10 µL reaction mixture containing 0.5 µM of the reverse primer, 10 mM Dithiothreitol (DTT), 0.5 mM dNTPs mix and 1X first strand buffer. After this, 5 µL of temple RNA was added and the reaction mixture was incubated at 50 °C for 60 min. The generated cDNA were used for amplification and sequencing of a portion of NoV VP1 region using primers as previously described.Citation35 Sapovirus positive samples were characterized by amplifying the major capsid region (VP1) by using RT-PCR kit.Citation14
HAdV genotyping was performed by amplification of a portion of the hexon loop one region as previously described.Citation36 In brief, the 50-µL amplification reaction mixture consisted of 1X PCR buffer, 3 mM MgCl2, 400 µM dNTPs, 0.4 µM AdvHex1, 0.4 µM AdvHex2, 2 U of Platinum® Taq DNA polymerase per µL and 5 µL of viral nucleic acid (DNA). The viral DNA amplified with an initial step of denaturation at 95 °C for 3 min, followed by 44 amplification cycles: denaturation at 95 °C for 30s, annealing at 50 °C for 40s, and elongation at 72 °C for 20s. The last amplification cycle included an additional step of final extension at 72 °C for 5 min:
For the classic HAstV genotyping, a partial region of the 5` end of the open reading frame (ORF) 2 gene coding for the capsid protein precursor was amplified using primers as described previously.Citation32 In brief, the reverse transcription reaction was carried out in a 20-µL reaction volume, and the reaction mixture was composed of 1X buffer, 1.5 µM astrovirus reverse primer, 1.25 mM each deoxyribonucleotide mix, 2 U of RNase inhibitor, and 0.5 U of AMV reverse transcriptase per µL (Invitrogen). Then, a 50-µL PCR mixture was prepared, consisting of 1X buffer, 2 mM MgCl2, 0.2 µM astrovirus reverse primer, 0.2 µM forward primer, 400 µM each dNTP mix, 2 U of platinum Taq DNA polymerase per µL, and 5 µL of cDNA. The amplification reaction was performed with an initial denaturation at 95 °C for 3 min followed by 40 amplification cycles: denaturation at 95 °C for 20s, annealing at 60 °C for 20s, extension at 72 °C for 30s, and a final extension step at 72 °C for 3 min.
Samples positive for EVs were further amplified on the portion of the VP1 region using nested RT-PCR according to previously published primers. Samples positive for EV were further amplified by using a portion of the VP1 region using nested RT-PCR according to previously published primers.33 The reverse transcription reaction was carried out by using a mixture of four different reverse primers (AN32, AN33, AN34, and AN35). The first round PCR was carried out with the use of forward (224) and reverse (222) primers. The second round PCR was carried out with primers (AN89 and AN88).
The HPeV genotyping was performed by amplification and sequencing of the VP3/ VP1 transition region. Newly designed primers, HPeV-F (5’-CATAYTCNTTYTCHACHTGGATGRGGAA-3’) and HPeV-R (5’-GGN CCATCATCYTGW GCT GAN GT-3’) were used. Briefly, five microliters RNA extract was used for reverse transcription reaction to generate cDNA. The RT reaction was carried out in 10 µL reaction containing1X strand buffer, 0.01 M DTT, 1µM reverse primer, 200 µM dNTPs, 2 U/µL RNase inhibitor and 10 U/µL of Superscript III reverse transcriptase. The PCR reaction was performed in a 50 µL reaction volume containing 1X buffer, 1.5 mM MgCl2, 0.6 µM forward and reverse primers each, 200 µM each dNTPs mix, 2 U/µL of Platinum-Taq-DNA polymerase, and 10 µL of cDNA template. The amplification reaction was carried out with an initial denaturation at 95◦C for 3 min followed by 40 cycles: denaturation at 95 ◦C for 30s, annealing at 52◦C for 20s, extension at 72 ◦C for 1 min. The last elongation cycle had an extra step of final extension at 72 ◦C for 5 min.
Gel Electrophoresis and Sequencing
The PCR products for each of the enteric viruses were subjected to gel electrophoresis in a 2% agarose gel and purified using a Wizard® SV Gel and PCR Clean-Up System (Promega, Mannheim, Germany). Sequencing reactions were performed using BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems Corporation, Foster City, CA, USA). Finally, sequencing was performed on ABI PRISM3500 Genetic Analyzer as per the manufacturer’s instructions (Applied Biosystems).
Phylogenetic Analysis
Phylogenetic trees were constructed using MEGA software (version 7). The Maximum-likelihood (ML) method with Kimura’s two-parameter correction was selected to construct the trees. Bootstrap analysis was performed with 1000 replicates. The online norovirus and rotavirus genotypic tools were also used to identify norovirus and rotavirus genotypes. The nucleotide sequences of each of the enteric viruses were submitted and deposited to GenBank under the accession numbers: HAstV (OK48335-OK483336), NoV (OK493612-OK493616), and the other enteric viruses (OK500274-OK500289).
Statistical Analyses
Data were statistically analyzed using the Statistical Package for the Social Sciences (SPSS) version 16 software. Discrete variables were expressed as numbers and percentages. Data were compared using chi-square and fisher’s exact test. The level of statistical significance was set at a P-value of 0.05.
Results
Study Population
During the study period, thirty-eight infants and children with diarrhea were enrolled. More than half (52.6%) of the children were male. The majority, 27 (71.1%) of the children were from rural villages. The mean (SD) age of the children was 31.5 (± 17.7) months ().
Table 1 The Positivity Rate of Enteric Virus by Demographic and Clinical Characteristics of the Study Participants
Enteric Viruses Stool Positivity Rate
The positivity rate of children for at least one of the enteric viruses was 52.6%. There was no significant difference in the positivity rate of children younger than 1 year (60%) and 1-<5 years old (50%) (P= 0.719) and the distribution of the enteric viruses detected was similar. The EV and HPeV were exclusively detected in children older than 2 years. The detection rates of HAdV, NoV, EV, RV, SaV, HAstV and HPeV were 7 (18.4%), 5 (13.2%), 5 (13.2%), 4 (10.5%), 2 (5.3%), and 1 (2.6%), respectively (). Mixed infection was detected in 6 of the children (SaV + HAdV, SaV + EV, HAdV + EV (n = 2) and NoV + HAdV + HAstV).
Table 2 Genotypic Distribution of Enteric Viruses in Under-Five Children with Diarrhea
Genotypic Distribution and Phylogenetic Analysis of Enteric Viruses
Rotavirus
Rotavirus was detected in four (10.5%) of the fecal samples. Of these, only one sample was successfully genotyped and three could not be genotyped. Phylogenetic and sequence analyses of the VP7 and VP4 regions revealed the detection of G9 and P[8] RV genotypes, respectively. Both the G9 and P[8] sequences showed close phylogenetic relatedness with other similar sequences reported from Ethiopia in 2012 and 2016 ( and ).

Figure 1 Continued.
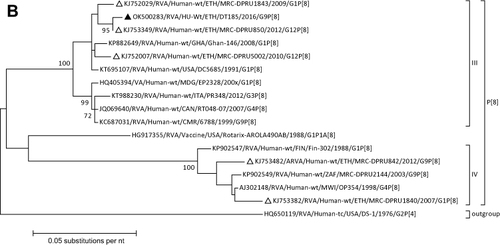
Figure 1 Continued.
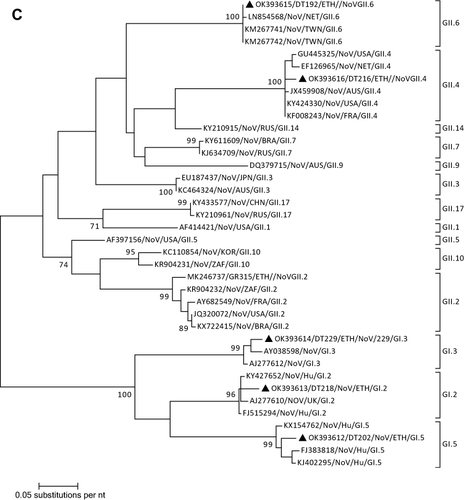

Figure 1 Continued.
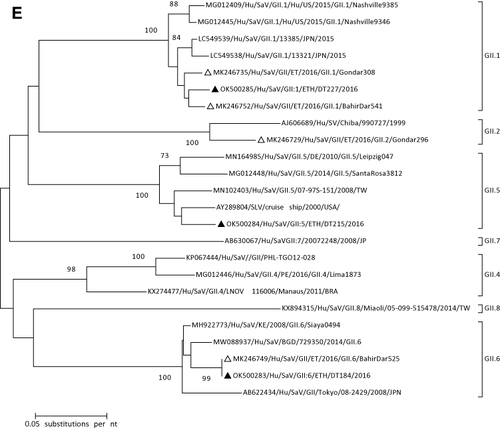
Figure 1 Continued.
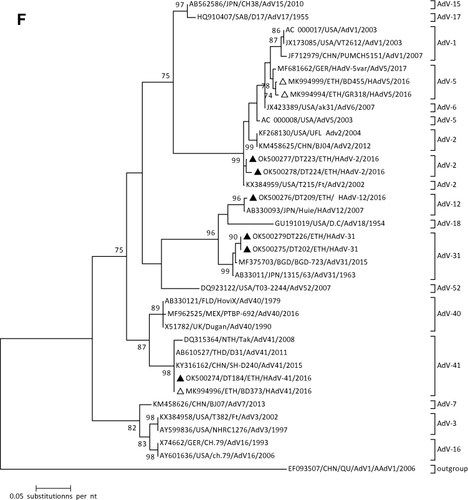
Figure 1 Continued.
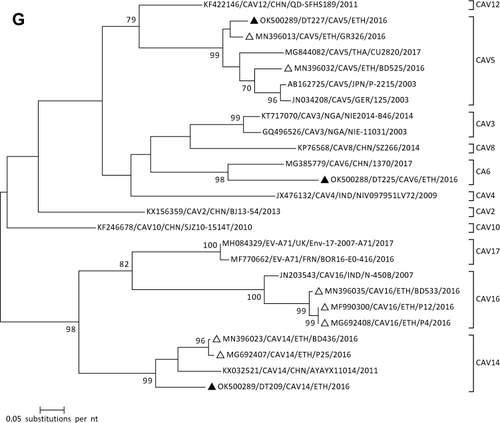
Figure 1 Phylogenetic analysis of RVA (A and B), NoV (C), HAstV (D), SaV (E), HAdV (F), EVA (G) and EVC (H). A portion the VP7 and VP4 regions were used for RVA. Partial sequences of hexon loop one and ORF2 were used for HAdV and HAstV, respectively. Portions of the VP1 coding regions were used for NoV, SaV and EV. The trees were constructed by using Maximum-likelihood (ML) method with a bootstrap analysis of 1000. Strains from the present study are labeled with black triangles and white triangles indicate previously reported strains from Ethiopia.

Norovirus
Norovirus was detected in five samples (13.2%). Sequencing and phylogenetic analysis of part of the VP1 region was successful in all positive samples. The phylogenic and BLAST search analysis showed the presence of NoV II (GII.6 and GII.4) and NoV I (GI.2, G.3, and GI.5) genotypes. The present GII.4 sequence was identified as GII.4 Sydney 2012 variants. The GII.4 and GII.6 genotypes showed clustering with previously published global sequences of GII.4 and GII.6, respectively. ().
Astrovirus
Phylogenetic analysis of the two positive cases from a partial region of the ORF2 gene revealed the detection of HAstV-2 genotypes. The current Ethiopian HAstV strains showed close phylogenetic relatedness with a similar HAsV-2 strain reported from Kenya in 1999 ().
Sapovirus
For characterization of SaV strains, the major capsid coding region (VP1) was phylogenetically analyzed (). All SaV positive samples were successfully genotyped and assigned into a single genogroup (Genogroup II). Three different GII genotypes (GII.1, GII.5, and GII.6) were found.
Adenoviruses
In this study, seven of the samples were positive for HAdV. The phylogenetic analysis revealed that the current Ethiopian strains belonged to the species HAdV-A (HAdV-12 and HAdV-31), HAdV- B (HAdV-3), HAdV-C (AdV-2), and HAdV-F (HAdV-41) species ().
Enterovirus and Parechovirus
Five and one of the samples were positive for EV and HPeV, respectively. Sequence analysis based on the VP1 gene revealed that the current Ethiopian EV isolates grouped into EV-A and EV-C. Of these, CAV5, CAV6, and CAV14 genotypes belongs to EV-A. Similarly, EV-C99 which belongs to EV-C was detected in one sample. One of the positive samples was not successfully amplified. Phylogenetic analysis of the species A EVs indicated that CAV5 sequences clustered most closely to previous Ethiopian sequences isolated in Gondar in 2016. Likewise, the current CAV6 sequence showed close relatedness with a similar strain reported from China in 2017. The CAV14 sequence of this study should distinct clustering from the global and previous Ethiopia isolates (). The EV-C99 sequence showed close phylogenetic relatedness to sequences isolated in Cameroon in 2009 (). One of the samples was positive for HPeV and its sequence analysis revealed that the identified genotype was HPeV-6.
Discussion
Although the burden of enteric viruses has been well characterized for industrialized countries, there is little information in resource-limited countries. In this preliminary assessment, based on direct molecular detection and characterization, we detected seven different and diverse types of enteric viruses. To the best of our knowledge, this is one of the very few assessments which considered the screening of multiple enteric viruses in Ethiopian under-five children.
The burden and genotypic distribution of RV in Ethiopia has been reviewed by considering studies published before 2020.Citation37 According to this review, the most common genotypes were G12P[8], G3P[6], G1P[8], and G3P[8]. The G3 and P[8] were the predominant G and P-types in Ethiopia.Citation37 In the present study, four of the samples were positive for RVA, with a positivity rate of 10.5%. The present low positivity rate of RV is consistent with a previous report from northwestern Ethiopia.Citation38 In our study, unfortunately, genotyping was not successful for three of the samples might be due to the low viral load observed during the RT-qPCR assay. Similar to other previous studies G9[P8] was detected.Citation39–41 Phylogenic analysis of both the VP7 and VP4 sequences demonstrated that the current rotavirus genotype is closely related to previously reported genotypes from Ethiopia in 2009 and 2012.
There is a paucity of data on the genetic diversity of caliciviruses in Ethiopia. In the present study, SaV was detected and sequenced. The phylogenic analysis based on sequences of a portion of the VP1 region confirmed the detection of GII.1, GII.5, and GII.6 genotypes. Except for GII.5, the present SaV genotypes were reported from previous studies in Northwest EthiopiaCitation42,Citation43 and Addis Ababa.Citation44 The reporting of the GII.5 genotype in this study is a new addition to the data on the genetic diversity of SaV in Ethiopia. In most of the previous studies in Africa, SaV-GI.1 followed by GI.2 was the predominant genotype detected in clinical samples.Citation45 A similar proportion of NoVs to that of SaVs positive cases was observed in this study. Extensively diverse NoV genotypes within genogroup I and genogroup II were reported from previous studies in Ethiopia.Citation40,Citation42–44 The present study also confirms the circulation of three genotypes in genogroup I (GI.2, GI.3, and G1.5) and two genotypes in genogroup II (GII.4, and GII.6) genotypes in Debre Tabor, Northwest Ethiopia. Except for GI.5, the other SaV genotypes were reported previously from Gondar.Citation41
In this study, HAdV was the most frequently detected enteric virus, with a detection rate of 18.4%. In this small data set, sequence analysis revealed that a wide variety of HAdV species (four) and types (five) were circulating in Debre Tabor, Northwest Ethiopia. The HAdV types were belonging to HAdV-A (HAdV-12 and −31), HAdV-B (HAdV-3), HAdV-C (HAdV-2), and HAdV-F (HAdV-41). This observation was consistent with a previous report in Ethiopia in 2016 (46). It has been documented that types 40 and 41 are the most frequently reported cause of adenovirus-associated gastroenteritis.Citation21,Citation22 A single HAdV-41 was detected in the present study and the phylogenetic analysis showed that this genotype has close relatedness with the previous genotype reported from Bahir Dar in 2016 and other global reports. Adenovirus types are known to infect respiratory tracts were also detected. These HAdV types might be shed from the respiratory tract and detected in the feces of the children. However, their contribution to the causation of diarrhea cannot be excluded as they have also been reported to cause diarrhea.Citation19,Citation20
Enteric viruses including EV, HPeV, and HAstV are responsible for a wide range of clinical diseases in humans. Unfortunately, the burden and genetic diversity of these enteric viruses in Sub-Saharan Africa remain under-documented. In the current study, a considerable number of EV (CAV5, CAV6, CAV14, and EV-C99), HPeV-6) and HAstV-2 genotypes were detected from the stool samples of children with diarrhea. Similar to our observation, the present EV genotypes have been reported from previous studies in Ethiopia and other sub-Saharan African countries.Citation42,Citation47–51 The phylogenetic and individual sequence analysis showed the genotypes are closely related to previous isolates from Ethiopia, Cameroon, and China. Similar to the present study, HPeV-2 and HAstV-2 genotypes were reported from Gondar.Citation46,Citation51
The present study has the following limitations. Case number is very low but it is from a single center. The positivity rates and genetic diversities of the different enteric viruses observed in this smaller study might be enlightening but not conclusive. Longitudinal studies with large sample sizes are required to evaluate the precise prevalence, genetic diversity, and their contributions to diarrheal disease in children in Ethiopia.
In conclusion, enteric viruses were detected in 52.6% of the samples obtained from under-five children with diarrhea. Phylogenic analysis revealed the circulation of extensively diverse genotypes in the study area. The detection of heterogeneous enteric viruses in this small data set highlights the need for extended multicenter studies to describe the burden and genetic diversity.
Data Sharing Statement
The dataset used during this study is available and can be shared upon request from the corresponding author. The nucleotide sequences for genotypes reported in this study are deposited in GenBank.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare they have no competing interests.
Acknowledgments
Our special thanks go to the study participants and the data collectors. We are also grateful to Sandra Bergs for the excellent technical assistance.
References
- Institute for Health Metrics and Evaluation (IHME). Findings from the global burden of disease study 2017. Seattle, WA, USA: IHME; 2018.
- Troeger C, Khalil IA, Rao PC, et al. Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5 years. JAMA Pediatr. 2018;172(10):958–965. doi:10.1001/jamapediatrics.2018.1960
- Liu L, Oza S, Hogan D, et al. (2016) Global, regional, and national causes of under-5 mortality in 2000-15: an updated systematic analysis with implications for the sustainable development goals. Lancet. 2016;388(10063):3027–3035. doi:10.1016/S0140-6736(16)31593-8
- Green RJ. Viral Infections in Children Volume II. Publishing SI, editor. Springer; Vol. 2, 2017:1–224
- Moore NE, Wang J, Hewitt J, et al. Metagenomic analysis of viruses in feces from unsolved outbreaks of gastroenteritis in humans. J Clin Microbiol. 2015;53(1):15–21. doi:10.1128/JCM.02029-14
- Holtz LR, Cao S, Zhao G, et al. Geographic variation in the eukaryotic virome of human diarrhea. Virology. 2014;468–470:556–564. doi:10.1016/j.virol.2014.09.012
- Tate JE, Burton AH, Boschi-Pinto C, Parashar UD. Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000–2013. Clin Infect Dis. 2016;62(2):S96–S105. doi:10.1093/cid/civ1013
- Bányai K, Kemenesi G, Budinski I, et al. Candidate new rotavirus species in Schreiber’s bats, Serbia. Infect Genet Evol. 2017;48:19–26. doi:10.1016/j.meegid.2016.12.002
- Kirkwood CD. Genetic and antigenic diversity of human rotaviruses: potential impact on vaccination programs. J Infect Dis. 2010;202(S1):S43–8. doi:10.1086/653548
- World Health Organization. Rotavirus vaccines: an update. WklyEpidemiol Rec. 2009;84:533–540.
- Mwenda JM, Burke RM, Shaba K, et al. Implementation of rotavirus surveillance and vaccine introduction-world health organization African region, 2007–2016. MMWR. 2017;66(43):1192–1196. doi:10.15585/mmwr.mm6643a7
- Oka T, Wang Q, Katayama K, Saif LJ. Comprehensive review of human sapoviruses. Clin Microbiol Rev. 2015;28(1):32–53. doi:10.1128/CMR.00011-14
- Robilotti E, Deresinski S, Pinsky BA. Norovirus. Clin Microbiol Rev. 2015;28(1):134–164. doi:10.1128/CMR.00075-14
- Oka T, Lu Z, Phan T, Delwart EL, Saif LJ, Wang Q. Genetic characterization and classification of human and animal sapoviruses. PLoS One. 2016;11(5):e0156373. doi:10.1371/journal.pone.0156373
- Chhabra P, de Graaf M, Parra GI, et al. Updated classification of norovirus genogroups and genotypes. J Gen Virol. 2019;100(10):1393–1406. doi:10.1099/jgv.0.001318
- Vinje J. Advances in laboratory methods for detection and typing of norovirus. J Clin Microbiol. 2015;53(2):373–381. doi:10.1128/JCM.01535-14
- de Graaf M, van Beek J, Koopmans MP. Human norovirus transmission and evolution in a changing world. Nat Rev Microbiol. 2016;14(7):421–433. doi:10.1038/nrmicro.2016.48
- Lynch J, Kajon A. Adenovirus, epidemiology, global spread of novel serotypes, and advances in treatment and prevention. Semin Respir Crit Care Med. 2016;37(04):586–602. doi:10.1055/s-0036-1584923
- Khanal S, Ghimire P, Dhamoon AS. The repertoire of adenovirus in human disease: the innocuous to the deadly. Biomedicines. 2018;6(1):30. doi:10.3390/biomedicines6010030
- Ghebremedhin B. Human adenovirus: viral pathogen with increasing importance. Eur J Microbiol Immunol. 2014;4(1):26–33. doi:10.1556/EuJMI.4.2014.1.2
- Reis TAV, Assis ASF, Do Valle DA, et al. The role of human adenoviruses type 41 in acute diarrheal disease in Minas Gerais after rotavirus vaccination. Braz J Microbiol. 2016;47(1):243–250. doi:10.1016/j.bjm.2015.11.011
- Ouedraogo N, Kaplon J, Bonkoungou IJO, et al. Prevalence and genetic diversity of enteric viruses in children with diarrhea in Ouagadougou, Burkina Faso. PLoS One. 2016;11(4):e0153652. doi:10.1371/journal.pone.0153652
- Perot P, Lecuit M, Eloit M. Astrovirus diagnostics. Viruses. 2017;9(1):10. doi:10.3390/v9010010
- Vu D-L, Bosch A, Pinto RM, Guix S. Epidemiology of classic and novel human astrovirus: gastroenteritis and beyond. Viruses. 2017;9(2):33. doi:10.3390/v9020033
- Bosch A, Pintó RM, Guix S. Human astroviruses. Clin Microbiol Rev. 2014;27(4):1048–1074. doi:10.1128/CMR.00013-14
- Tapparel C, Siegrist F, Petty TJ, Kaiser L. Picornavirus and enterovirus diversity with associated human diseases. Infect Genet Evol. 2013;14:282–293. doi:10.1016/j.meegid.2012.10.016
- Pietsch C, Schuster V, Liebert UG. A hospital based study on inter- and intragenotypic diversity of human rotavirus A VP4 and VP7 gene segments, Germany. J Clin Virol. 2011;50(2):136–141. doi:10.1016/j.jcv.2010.10.013
- Pietsch C, Ennuschat N, Härtel Liebert UG. Within-host evolution of virus variants during chronic infection with novel GII.P26-GII.26 norovirus. J Clin Virol. 2018;108:96–102. doi:10.1016/j.jcv.2018.09.013
- Hoehne M, Schreier E. Detection of Norovirus genogroup I and II by multiplex real-time RT- PCR using a 3′-minor groove binder-DNA probe. BMC Infect Dis. 2006;6(1):69. doi:10.1186/1471-2334-6-69
- Pietsch C, Liebert UG. Intrahost viral evolution during chronic sapovirus infections. J Clin Virol. 2019;113:1–7. doi:10.1016/j.jcv.2019.02.001
- Heim A, Ebnet C, Harste G, Pring-Akerblom P. Rapid and quantitative detection of human adenovirus DNA by real-time PCR. J Med. 2003;70:228–239.
- Logan C, O’Leary JJ, O’Sullivan N. Real-time reverse transcription PCR detection of norovirus, sapovirus and astrovirus as causative agents of acute viral gastroenteritis. J Virol Methods. 2007;146(1–2):36–44. doi:10.1016/j.jviromet.2007.05.031
- Nix WA, Oberste MS, Pallansch MA. Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J Clin Microbiol. 2006;44(8):2698–2704. doi:10.1128/JCM.00542-06
- Savolainen C, Mulders MN, Hovi T. Phylogenetic analysis of rhinovirus isolates collected during successive epidemic seasons. Virus Res. 2002;85(1):41–46. doi:10.1016/S0168-1702(02)00016-3
- Kroneman A, Vennema H, Deforche K, et al. An automated genotyping tool for enteroviruses and noroviruses. J Clin Virol. 2011;51(2):121–125. doi:10.1016/j.jcv.2011.03.006
- Allard A, Albinsson B, Wadell G. Rapid typing of human adenoviruses by a general PCR combined with restriction endonuclease analysis. J Clin Microbiol. 2001;39(2):498–505. doi:10.1128/JCM.39.2.498-505.2001
- Damtie D, Melku M, Tessema B, Vlasova AN. Prevalence and genetic diversity of rotaviruses among under-five children in Ethiopia: a systematic review and meta-analysis. Viruses. 2020;12(1):62. doi:10.3390/v12010062
- Azage M, Kumie A, Worku A, Bagtzoglou AC. Childhood diarrhea in high and low hotspot districts of Amhara Region, northwest Ethiopia: a multilevel modeling. J Health Popul Nutr. 2016;35(1):13. doi:10.1186/s41043-016-0052-2
- Abebe A, Teka T, Kassa T, et al. Hospital based surveillance for rotavirus gastroenteritis in children younger than 5 years of age in Ethiopia: 2007–2012. Pediatr Infect Dis J. 2014;33(1):S28–S33. doi:10.1097/INF.0000000000000048
- Yassin MA, Kirby A, Mengistu AA, et al. Unusual norovirus and rotavirus genotypes in Ethiopia. Paediatr Int Child Health. 2012;32(1):51–55. doi:10.1179/1465328111Y.0000000047
- Gelaw A, Pietsch C, Liebert UG. Molecular epidemiology of rotaviruses in Northwest Ethiopia after national vaccine introduction. Infect Genet Evol. 2018;65:300–307. doi:10.1016/j.meegid.2018.08.016
- Altan E, Aiemjoy K, Phan TG, et al. Enteric virome of Ethiopian children participating in a clean water intervention trial. PLoS One. 2018;13(8):e0202054. doi:10.1371/journal.pone.0202054
- Gelaw A, Pietsch C, Mann P, Liebert UG. Molecular detection and characterisation of sapoviruses and noroviruses in outpatient children with diarrhoea in Northwest Ethiopia. Epidemimol Infect. 2019;147(e218):1–7.
- Sisay Z, Djikeng A, Berhe N, et al. Prevalence and molecular characterization of human noroviruses and sapoviruses in Ethiopia. Arch Virol. 2016;161(8):2169–2182. doi:10.1007/s00705-016-2887-7
- Makhaola K, Moyo S, Kebaabetswe LP. Distribution and genetic variability of sapoviruses in Africa. Viruses. 2020;12(5):490. doi:10.3390/v12050490
- Gelaw A, Pietsch C, Liebert UG. Genetic diversity of human adenovirus and human astrovirus in children with acute gastroenteritis in Northwest Ethiopia. Arch Virol. 2019;164(12):2985–2993. doi:10.1007/s00705-019-04421-8
- Ayukekbong JA, Fobbing C, Lindh M, Nkuo-Akenji T, Bergström T, Norder H. Molecular analysis of enterovirus in Cameroon by partial 5ʹUTR-VP4 gene sequencing reveals a high genetic diversity and frequency of infections. J Med Virol. 2014;86(12):2092–2101. doi:10.1002/jmv.23926
- Faleye TOC, Adewumi MO, Coker BA, Nudamajo FY, Adeniji JA. Direct detection and identification of enteroviruses from faeces of healthy Nigerian children using a cell-culture independent RT-seminested PCR assay. Adv Virol. 2016;2016:1412838. doi:10.1155/2016/1412838
- Fernandez-Garcia MD, Kebe O, Fall AD, Ndiaye K. Identification and molecular characterization of non-polio enteroviruses from children with acute flaccid paralysis in West Africa, 2013–2014. Sci Rep. 2017;7(1):3808. doi:10.1038/s41598-017-03835-1
- Opanda SM, Wamunyokoli F, Khamadi S, Coldren R, Bulimo WD. Genotyping of enteroviruses isolated in Kenya from pediatric patients using partial VP1 region. Springer Plus. 2016;5(1):158. doi:10.1186/s40064-016-1834-0
- Gelaw A, Pietsch C, Tigabu Z, Liebert UG. Genotyping of enteroviruses and human parechoviruses highlights their diversity in Northwest Ethiopia. J Med Virol. 2020;92(12):2891–3895. doi:10.1002/jmv.25765