Abstract
Hyperammonemia is a true neonatal emergency with high toxicity for the central nervous system and developmental delay. The causes of neonatal hyperammonemia are genetic defects of urea cycle enzymes, organic acidemias, lysinuric protein intolerance, hyperammonemia–hyperornithinemia– homocitrullinemia syndrome, transient hyperammonemia of the newborn, and congenital hyperinsulinism with hyperammonemia. In some of these conditions the high blood ammonia levels are due to the reduction of N-acetylglutamate, an essential cofactor necessary for the function of the urea cycle, or to the reduction of carbamoyl-phosphate synthase-I activity. In these cases, N-carbamylglutamate (carglumic acid) can be administered together with the conventional therapy. Carglumic acid is an analog of N-acetylglutamate that has a direct action on carbamoyl-phosphate synthase-I. Its effects are reactivation of the urea cycle and reduction of plasma ammonia levels. As a consequence it improves the traditional treatment, avoiding the need of hemodialysis and peritoneal dialysis. In this review we evaluate the possible field of application of carglumic acid and its effectiveness and safety.
Introduction
In humans catabolism of proteins and other nitrogen-containing molecules results in the production of nitrogen. Waste nitrogen is excreted largely as urea with smaller amounts as creatinine, ammonium ion, amino acids, and other nitrogenous compounds. In the brain, ammonia is an important product of the metabolism of the neurotransmitters glutamate and aspartate and of the neurotransmitter monoamines. It is also the product of the synthesis of glutamate from glutamine in nerve endings. At least some of the glutamate released from the nerve endings is neutralized via uptake by the astrocytes where it is combined with ammonia to form the electrophysiologically inactive molecule glutamine. Despite its central importance in metabolism, high concentrations of ammonia are generally toxic, particularly to the central nervous system, and the compound must be maintained at relatively low levels in the tissues.
Many inborn errors of metabolism cause increases in blood ammonia levels. Hyperammonemia is a condition that can cause neurological complications, coma, and even death. Indeed, high concentrations of ammonia may partially saturate the “enzymatic detoxifier” of the astrocytes, impeding the brain’s capacity for self- protection, which contributes to the neurological dysfunction. Therefore, high levels of ammonia are a real emergency and should be treated promptly. First-line treatment consists of reducing catabolism and promoting anabolism, by a protein-restriction diet and substituting an oral or intravenous high energy source, such as glucose infusion, and parenteral lipids administration after exclusion of fatty acid oxidation disorder. Because the kidneys clear ammonia poorly, its removal from the body must be expedited by formation of compounds with high renal clearance. Ammoniascavenging drugs (sodium benzoate, sodium phenylbutyrate, and arginine hydrochloride) are at present considered the first-line drugs for the treatment of neonatal hyperammonemia. However, if the foregoing therapies fail to produce any appreciable change in blood ammonia level within a few hours, the neonate should be transferred without delay to a center with hemodialysis capacity, and continuous venovenous hemofiltration, in particular, should be performed.
The administration of N-carbamylglutamate (carglumic acid, NCG) has been reported to be of benefit in several different conditions presenting with hyperammonemia.Citation1 NCG is a synthetic analog of N-acetylglutamate (NAG) that activates carbamoyl-phosphate synthase I (CPS-I),Citation2 the enzyme of the first and rate-limiting step of the urea cycle stimulating ureagenesisCitation3 (). NCG is able to enter into the mitochondria and is not inactivated by cytosolic acylases.Citation4 For this reason it is an optimal pharmacological substitute; on the contrary, the natural NAG is hydrolyzed in vivo by acylamino acid acylase.Citation5
Figure 1 Schematic representation of the urea cycle and related pathways.
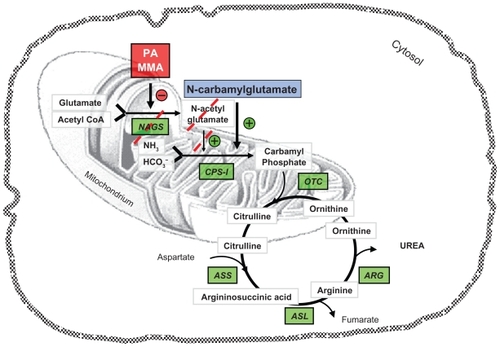
In this review we evaluate the field of application and the possibility for the treatment of hyperammonemia with NCG.
N-carbamylglutamate in urea cycle disorders
The urea cycle represents the principal mechanism of ammonia detoxification. Five enzymes are required for ureagenesis: CPS-I, ornithine transcarbamylase, argininosuccinate synthase, argininosuccinate lyase, and arginase. The urea cycle is also regulated by NAG, an essential cofactor necessary for the function of CPS-I. NAG is produced in the mitochondrial matrix from glutamate and acetyl coenzyme A by N-acetylglutamate synthase (NAGS). Deficiency in any one of these enzymes causes a urea cycle disorder which results in ammonia accumulation.Citation6
N-acetylglutamate synthase deficiency
A NAGS deficiency is a rare, autosomal recessive urea cycle disorder, and was described for the first time by Bachmann and colleagues in 1981.Citation7 In this congenital error there is a wide range of severity of symptoms, as well as in the age of onset. Most frequently, the onset occurs in the early neonatal period with the signs and symptoms of hyperammonemia: poorly fed, vomiting, and neurological alteration such as hypotonia, abnormal movements of limbs, irritability, generalized seizures, afterward lethargy, and coma. Late forms may present with acute attacks of hyperammonemia in a previously healthy individual or with chronic neurological, gastrointestinal, and psychiatric clinical signs. The symptoms could be chronic nausea, vomiting, ataxia, confusion, and irritability, and in acute cases could lead to coma. The laboratory findings show an increase in blood levels of ammonia and markedly elevated concentrations of glutamine and alanine. In contrast to most of the other urea cycle disorders, plasma citrulline and urinary orotic acid are normal as well as urine organic acids and blood acylcarnitines. These clinical and biochemical features are present both in NAGS and CPS-I deficiencies. The diagnosis could be confirmed either with determination of NAGS and CPS-I activity in liver tissue in presence of arginine or by molecular diagnostic analysis.8 The majority of early forms have <5% NAGS residual activity and often have frameshift or nonsense mutations. Indeed, the late form presentation is associated with partial NAGS deficiency and hypomorphic alleles with single amino acid substitutions.Citation8–Citation11 The treatment of acute hyperammonemia consists of an intravenous administration of adequate calories, fluids, and electrolytes and in a restriction of protein intake but providing a mixture of essential amino acids. NCG is a specific treatment of hyperammonemia due to NAGS deficiency and should be administered as early possible when it is suspected.Citation12 Also sodium benzoate (200–500 mg/kg/24 h), sodium phenylacetate (250–500 mg/kg/24 h), and arginine (200–600 mg/kg/24 h) should be considered. In case of failure of the above treatment, peritoneal dialysis must be initiated. In 1995, Guffon et al described a case of a newborn with NAGS deficiency treated by NCG; this treatment was initiated on the 25th day of life with a total dose of 80–100 mg/kg/day, with optimal results. A 1-year follow-up showed an infant with normal somatic and neurological development and a good metabolic balance.Citation13 In the following years, 3 other cases, 1 neonatal form and 2 cases of partial NAGS deficiency, were described.Citation14–Citation16 NCG was administered (60–100 mg/kg/day in 4 single doses) and a decrease of blood ammonia was observed: this enabled an increase in protein intake. In 2004, Caldovic et alCitation17 studied how the incorporation of an isotopic label from ammonium chloride into urea increased after the treatment with NAG. This study provided strong evidence that NCG therapy restores ureagenesis in NAGS deficiency. Later, in 2008, Tuchman et al presented a stable isotope 13C tracer method to measure ureagenesis and evaluate the effect of NCG in humans. The results of these investigations showed the reduction of plasma ammonia and glutamine levels and confirmed that NCG could be an effective treatment for inherited and secondary NAGS deficiency.Citation18 Nordenström et alCitation19 and Gessler et alCitation12 reported other clinical cases of NAGS deficiency and concluded that the immediate treatment of hyperammonemia with NCG entails a favorable long-term outcome.
Currently NCG is approved in the European Union and the United States for the treatment of hyperammonemia due to NAGS deficiency. The initial daily dose should be 100 mg/kg, up to 250 mg/kg if necessary. It should then be adjusted individually in order to maintain normal ammonia plasma levels.
Carbamoyl-phosphate synthase I deficiency
The therapy with NAG in the kinetic variant or partial CPS-I deficiency has been discussed by O’Conner et al in 1985.Citation2 In 1996 Kuchler et alCitation20 administered NCG in an infant on suspicion of a NAGS deficiency. After the treatment, the levels of ammonia and glutamine decreased to the normal range. The results of liver biopsy showed a normal activity of NAGS and a decrement of activity of CPS-I. The authors concluded that NCG stimulates the remaining CPS-I and increases ureagenesis, for this reason NCG might be used in patients with partial CPS-I deficiency.
N-carbamylglutamate in organic acidemias
Neonatal hyperammonemia can be caused by some organic acidemias. Since the 1970s, hyperammonemia has been known to be a complication of propionic acidemia (PA) and methylmalonic acidemia (MMA), due to an inhibition of carbamoyl-phosphate synthesis, the first step of the urea cycle. In fact, in such disorders, propionyl-CoA accumulates and inhibits NAGS. The reduced synthesis of NAG, the natural stimulator of CPS-I, hinders the conversion of ammonia to urea in the liver.Citation21
Propionic acidemia
Propionic acid is an intermediate metabolite of branched-chain amino acids (valine, theonine, methionine), odd-chain fatty acids, and cholesterol degradation. Propionic acid is carboxylated to methylmalonic acid by a mitochondrial biotin-dependent enzyme: propionyl CoA carboxylase. It is composed of 2 subunits, α and β; the subunit α binds biotin. A failure of this enzyme determines the PA (MIM 606054). This inborn disease is inherited as an autosomal recessive trait. The genes involved are located in the following chromosomal regions 13q32 and 3q21–q22, and many mutations have been identified in different patients. Two forms are described based on the clinical manifestations: a severe form in which symptoms develop during the first days of life, and a milder form in which a patient may have an episode of severe ketoacidosis after a healthy period. The clinical findings are poor feeding, vomiting, dehydration, hypotonia, lethargy, severe ketoacidosis with rapid progress to coma, and death. Laboratory findings during the acute attack are a severe metabolic acidosis, hypoglycemia, and hyperammonemia. Propionic acid and methylcitric acid are markedly elevated in the blood and urine of infants with PA. To reach a definite diagnosis, a measurement of the enzyme activity in leukocytes or cultured fibroblasts is required. The treatment consists of hydration, correction of acidosis, with intravenous glucose, insulin, carnitine, sodium benzoate, and vitamin therapy, associated with a hypercaloric protein-free diet. Gebhardt et al described 2 patients with PA and hyperammonemia who were treated with NCG. Their blood ammonia levels were decreased even by the first dose of NCG and normalized in a 6-hour period. In both patients dialysis was avoided.Citation22 Also Jones and collaborators published 2 cases of patients with PA and high ammonia levels. The first newborn responded to administration of NCG optimally with a fall in plasma ammonia levels. The second patient did not respond to NCG therapy, but in this case the dose of NCG was lower (25 mg/kg rather than 250 mg/kg).Citation23 Other cases of PA treated with NCG have been described since these cases were described. In these studies the administration of NCG improved detoxification of ammonia.Citation24–Citation27 The effectiveness of NCG in decreasing ammonia levels in patients with PA was demonstrated by Tuchman et al. They investigated the in vivo synthesis of [13C]urea following administration of [1-13C]acetate, a stable isotope tracer, and NCG: in the patient with PA they observed an increase of 13CO2 in breath, of [13C] urea in plasma and production of absolute [13C]urea, although this was lower than in healthy subjects.Citation18 Similar results were published by Ah Mew et al:Citation28 they administered [13C]sodium acetate to 7 patients with PA and neonatal hyperammonemia, and measured the concentration of 13CO2 in blood and [13C]urea, before and after the start of NCG therapy. They demonstrated that NCG increases ureagenesis and decreases the blood ammonia and glutamine levels in PA patients.
Methylmalonic acidemia
Methylmalonic acid is normally derived from propionic acid as part of catabolism of branched-chain amino acids, cholesterol, and odd-chain fatty acids. The enzyme methylmalonyl CoA mutase converts the L-methylmalonic acid to succinic acid. This enzyme requires as a cofactor adenosylcobalamin, a metabolite of vitamin B12.Citation29 Deficiency of enzyme mutase or, less frequently, of adenosylcobalamin determines the accumulation of methylmalonic acid in plasma. Two forms of mutase apoenzyme deficiencies are identified, the mutation mut0 responsible for complete enzyme deficiency and mut− responsible for partial enzyme deficiency.Citation30 These patients are not responsive to B12-vitamin therapy, unlike the patients with a defect in the formation of adenosylcobalamin. The clinical findings of the patients with MMA due to mut0, mut−, and defect in metabolism of vitamin B12 are similar. Severe forms develop in the first days of life with feeding problems, vomiting, hypotonia, metabolic acidemia, and consensual tachypnea. Also this condition when not treated promptly may progress to coma and death. The milder forms evolve later with hypotonia, feeding failure, and developmental delay.Citation31 Laboratory findings are hyperammonemia and metabolic acidosis with increase of methylmalonic acid in blood. In addition propionic acid and its metabolites are found in the urine. Diagnosis can be confirmed by measuring mutase activity in cultured fibroblasts, or by identifying the mutant gene. Intravenous treatment must be immediately started with carnitine, glucose, insulin, arginine, sodium benzoate, and hypercaloric protein-free diet. It has been demonstrated that the administration of NCG decreases blood ammonia levels. Gebhardt et al described a case of a 6-day-old newborn affected by MMA and hyperammonemia (805 μmol/L). After the initial conventional treatment and peritoneal dialysis his plasma ammonia levels were 610 μmol/L. NCG therapy was started and blood ammonia levels dropped to 100 μmol/L 3 hours after the first NCG dose administration.Citation32 Also Levrat et al reported a case of MMA in which the hyperammonemia was resolved with insertion of NCG therapy.Citation33
Maple syrup urine disease
Branched-chain amino acids (leucine, isoleucine, and valine) are metabolized by a mitochondrial thiamine pyrophosphate (vitamin B1)-dependent complex enzyme system: branchedchain α-ketoacid dehydrogenase.Citation34 This enzyme is composed of 3 catalytic components, E1 (decarboxylase component, formed by 2 subunits E1α and E1β), E2 (acyltransferase component), and E3 (dehydrogenase component).Citation35 Deficiency of this enzyme determines maple syrup urine disease (MSUD, MIM 251000), so called for the typical sweet odor of maple syrup found in body fluids, especially urine. Based on clinical manifestation and response to vitamin B1 administration, 5 types of MSUD have been classified.Citation36 All these clinical phenotypes are inherited as an autosomal recessive trait. The gene for E1α, E1β, E2, and E3 subunits resides on chromosomes 19, 6, 1, and 7, respectively. There is no mutual relationship between clinical phenotypes and genotypes. The classic MSUD is the most severe form with major clinical findings. At birth the newborns are healthy; the clinical manifestations develop in the first week of life. These are poor feeding, vomiting, and in the following days lethargy and coma may appear. These patients are hypertonic, with muscular rigidity and severe opisthotonos. The neurological manifestations are convulsions, hypoglycemia, and cerebral edema may also be present.Citation37 In acute state, the neuroimaging may show cerebral edema most frequently in cerebellum, dorsal brainstem, cerebral peduncle, and internal capsule. In later ages of life hypomyelination and cerebral atrophy may be observed. Laboratory findings show increased levels of leucine, isoleucine, and valine in plasma; these amino acids and their respective ketoacids are elevated in urine and in cerebrospinal fluid.Citation38 The definitive diagnosis can be done by measuring the enzyme activity in leukocytes or cultured fibroblasts. The treatment of acute state is hydration with glucose infusion and a high caloric protein-free diet. In some patients peritoneal dialysis or hemodialysis are necessary. In 2009 for the first time, Kalkan Ucar et al used NCG in a patient with decompensated MSUD.Citation39 These authors demonstrated that the administration NCG together with conventional therapy decreases the blood levels of ammonia and it can be added to improve the acute metabolic decompensation in patients with MSUD.
Isovaleric acidemia
Isovaleric acidemia (MIM 243500) is a rare autosomal recessive inborn disease caused by deficiency of isovaleryl coenzyme A dehydrogenase (IVD). IVD is a mitochondrial flavoenzyme which catalyzes the conversion of isovaleryl-CoA to 3-methylcrotonyl-CoA in the leucine degradation. Two forms of this disease exist: an acute one in which the symptoms develop in early hours of life with vomiting, severe metabolic acidosis, lethargy, convulsion, and coma,Citation40 and a chronic intermittent form defined by episodes of vomiting, dehydration, metabolic acidosis, and alteration of mental status; these events develop in the first months or years of life. Characteristic “sweaty feet” may be present in acute attacks. If untreated, these patients may die or may develop severe central nervous system dysfunctions. Laboratory findings during the acute attacks show metabolic acidosis, neutropenia, and thombocytopenia. Hypocalcemia, hyperglycemia, and moderate to severe hyperammonemia may be present in some patients.Citation41 The diagnosis is confirmed by the increase of blood levels of the isovaleric acid and its metabolites and by the elevations of isovalerylglycine and 3-hydroxyisovaleric acid as well as other metabolites in urine.Citation42,Citation43 Mass screening in newborn is possible by tandem mass spectrometry (MS/MS) for the analysis of the acylcarnitine profile in blood spot. The gene of IVD has been mapped to chromosome 15q14–15q15. Also it is possible to measure the enzyme activity in leukocytes or cultured fibroblasts. Today many mutations of this gene have been identified.Citation44,Citation45 In acute attacks the treatment consists of hydration, correction of metabolic acidosis by infusing sodium bicarbonate, promotion of anabolism by increased caloric intake (infusing glucose), and reduced leucine intake. Treatment must be initiated promptly. The long-term treatment is a diet with lower protein intake (1.5 g/kg/day), and administration of L-carnitine (100 mg/kg/day) and glycine (150–250 mg/kg/day) to prevent the accumulation of isovaleric acid and its toxic metabolites.Citation46 Coude et alCitation47 demonstrated in rat hepatocytes that high levels of isovaleric acid inhibited ureagenesis due to a decrease in NAG levels, causing hyperammonemia. For this reason, in patients with isovaleric acidemia associated with hyperammonemia the administration of NCG may be added to conventional therapy. A clinical case report of IVA successfullytreated with carglumic acid was recently communicated by Kasapkara et al.Citation48
N-carbamylglutamate in hyperinsulinism hyperammonemia syndrome
The hyperinsulinism hyperammonemia syndrome (HI/ HA) is an inborn error of metabolism. It was described for the first time by Zammarchi et alCitation49 in 1996. HI/HA syndrome is caused by a mutation of the enzyme glutamate dehydrogenase (GDH) encoded by GLUDl.Citation50 These mutations result in higher activity of GDH. About 80% of cases are due to de novo mutations; however a dominant pattern has been demonstrated.Citation51 GDH is a mitochondrial matrix enzyme, expressed at high levels in liver, kidney, brain, and pancreatic β-cells. It is composed of 6 subunits and oxidizes the glutamate to α-ketoglutarate and ammonia using NAD and/or NADP as a cofactor.Citation52 The clinical manifestations of affected infants are variable episodes of hypoglycemia associated with hyperammonemia. Hypoglycemia may occur with fasting and in response to protein feeding.Citation53,Citation54 Patients with HI/HA syndrome show a large acute insulin response to leucine administration and the blood amino acid concentrations are normal. In these children blood ammonia levels are persistently elevated between 2 and 5 times over normal limit, but the hyperammonemia is not associated with lethargy, irritability, or coma. Stanley has supposed that overactivity of GDH in the brain decreases the levels of glutamate and glutamine, protecting the central nervous system from the neurotoxicity of the accumulation of these elements.Citation55 In this metabolic disorder the physiopathological basis of hyperammonemia is that the gain in GDH activity leads to an increase of ammonia levels and excessive oxidation of glutamate reduces the glutamate needed for the synthesis of NAG, slowing the urea cycle.Citation56 For hypoglycemia the conventional treatment is diazoxide (5–15 mg/kg/day), a KATP channel agonist. Some studies show that NCG is a potential therapy for hyperammonemia.Citation57,Citation58
N-carbamylglutamate in valproate-induced hyperammonemia?
Valproate-induced hyperammonemic encephalopathy is an unusual, but serious, adverse effect of valproic acid treatment that can lead to death, and is characterized by a decreasing level of consciousness, focal neurological deficits, cognitive slowing, vomiting, drowsiness, and lethargy. The mechanism by which hyperammonemia develops is still unclear. Hyperammonemia may result from the stimulation of kidney glutaminase in the renal cortex, from the depletion of mitochondrial acetyl CoA, and decreased production of NAG, but also from the accumulation of propionate, a valproate metabolite. The reduction of hepatic NAG, the obligatory activator of the first enzyme of the urea cycle, leads to the inhibition of hepatic mitochondrial CPS-I.Citation59
Two patients with valproate-induced hyperammonemia have been treated with carglumic acid.Citation60 NCG was effective in decreasing blood ammonia levels and its administration may be added to conventional therapy, as suggested by recent guidelines.Citation61
Side effects
The most common adverse reactions (occurring in more than 13% of patients) observed in patients receiving NCG, regardless of causality, are vomiting, abdominal pain, pyrexia, tonsilitis, anemia, ear infection, diarrhea, nasopharyngitis, and headache. In newborns, NCG is usually well tolerated without significant adverse effects. Tachycardia, profuse sweating, increased bronchial secretion, hyperthermia, and restlessness have been reported only at a dosage of 650 mg/kg/day.Citation62
Conclusion
We have described the beneficial effects of NCG to decrease blood ammonia levels and to improve ureagenesis in different conditions presenting with hyperammonemia. NCG is safe, fast acting and easy to administer. Dosing varies depending on the clinical condition and indication. Restarting the urea cycle, NCG increases ureagenesis and diminishes blood ammonia levels. It also improves the acute therapy and thus reduces the need for or the duration of peritoneal dialysis and hemodialysis. Favorable long-term outcomes in promptly treated patients with hyperammonemia suggest the need to start the NCG therapy as soon as possible, even before the exact diagnosis is set up.Citation26
Abbreviations
| ASL | = | argininosuccinate lyase |
| ASS | = | argininosuccinate synthase |
| CPS-I | = | carbamoyl-phosphate synthase I |
| HI/HA | = | hyperinsulinism hyperammonemia syndrome |
| MMA | = | methylmalonic acidemia |
| MSUD | = | maple syrup urine disease |
| NAG | = | N-acetylglutamate |
| NAGSl | = | N-acetylglutamate synthase |
| NCG | = | N-carbamylglutamate |
| OTC | = | ornithine transcarbamylase |
| PA | = | propionic acidemia |
Disclosure
The authors report no conflicts of interest in this work.
References
- LeonardJVWard PlattMPMorrisAAHypothesis: proposals for the management of a neonate at risk of hyperammonaemia due to a urea cycle disorderEur J Pediatr2008167330530917436013
- O’ConnorJEJordáAGrisolíaSAcute and chronic effects of carbamyl glutamate on blood urea and ammoniaEur J Pediatr198514331961973987713
- Ah MewNPayanIDaikhinYEffects of a single dose of N-carbamylglutamate on the rate of ureagenesisMol Gen Metab2009984325330
- KimSPaikWKCohenPPAmmonia intoxication in rats: protection by L-carbamylglutamate plus L-arginineProc Natl Acad Sci U S A19726912353035334509311
- RegleroARivasJMendelsonJWallaceRGrisoliaSDeacylation and transacetylation of acetyl glutamate and acetyl ornithine in rat liverFEBS Lett19778111317902767
- TuchmanMLeeBLichter-KoneckiUCross-sectional multicenter study of patients with urea cycle disorders in the United StatesMol Genet Metab200894439740218562231
- BachmannCKrähenbühlSColomboJPSchubigerGJaggiKHTönzON acetylglutamate synthetase deficiency: a disorder of ammonia detoxicationN Engl J Med198130495437453791
- HaberleJSchmidtEPauliSMutation analysis in patients with N acetylglutamate synthase deficiencyHum Mutat200321659359712754705
- CaldovicLMorizonoHPanglaoMGLate onset N-acetylglutamate synthase deficiency caused by hypomorphic allelesHum Mutat200525329329815714518
- CaldovicLMorizonoHTuchmanMMutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) geneHum Mutat200728875475917421020
- CaldovicLAh MewNShiDMorizonoHYudkoffMTuchmanMN-acetylglutamate synthase: structure, function and defectsMol Genet Metab2010100 Suppl 1S13S1920303810
- GesslerPBuchalPSchwenkHUWermuthBFavourable long-term outcome after immediate treatment of neonatal hyperammonemia due to N-acetylglutamate synthase deficiencyEur J Pediatr2010169219719919533169
- GuffonNVianey-SabanCBourgeoisJRabierDColomboJPGuibaudPA new neonatal case of N-acetylglutamate synthase deficiency treated by carbamylglutamateJ Inherit Metab Dis199518161657623444
- HinnieJColomboJPWermuthBDryburghFJN-Acetylglutamate synthetase deficiency responding to carbamylglutamateJ Inherit Metab Dis19972068398409427158
- MorrisAARichmondSWOddieSJPourfarzamMWorthingtonVLeonardJVN-acetylglutamate synthetase deficiency: favourable experience with carbamylglutamateJ Inherit Metab Dis19982188678689870213
- PleckoBErwaWWermuthBPartial N-acetylglutamate synthetase deficiency in a 13-year-old girl: diagnosis and response to treatment with N-carbamylglutamateEur J Pediatr1998157129969989877039
- CaldovicLMorizonoHDaikhinYRestoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamateJ Pediatr2004145455255415480384
- TuchmanMCaldovicLDaikhinYN-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkersPediatr Res200864221321718414145
- NordenströmAHalldinMHallbergBAlmJA trial with N- carbamylglutamate may not detect all patients with NAGS deficiency and neonatal onsetJ Inherit Metab Dis200730340017510757
- KuchlerGRabierDPoggi-TravertFTherapeutic use of carbamylglutamate in the case of carbamoyl-phosphate synthetase deficiencyJ Inherit Metab Dis19961922202228739970
- CoudeFXSweetmanLNyhanWInhibition by propionyl-coenzyme A of N-acetylglutamate synthetase in rat liver mitochondria. A possible explanation for hyperammonemia in propionic and methylmalonic acidemiaJ Clin Invest197964615441551500823
- GebhardtBDittrichSParbelSVlahoSMatsikaOBohlesHN-carbamylglutamate protects patients with decompensated propionic aciduria from hyperammonaemiaJ Inherit Metab Dis200528224124415877213
- JonesSReedCAVijaySWalterJHMorrisAAN-Carbamylglutamate for neonatal hyperammonaemia in propionic acidaemiaJ Inherit Metab Dis2008221 [Epub ahead of print]
- Fernández de MiguelSGimeno Díaz de AtauriATorres PeralRFernández CarriónFSerrano AyestaránON-carbamyl glutamate treatment in hyperammoniemia decompensated propionic acidaemiaAn Pediatr (Barc)200971657958019850540
- SchwahnBCPieterseLBissetWMGallowayPGRobinsonPHBiochemical efficacy of N-carbamylglutamate in neonatal severe hyperammonaemia due to propionic acidaemiaEur J Pediatr2010169113313419680687
- FilippiLGozziniEFioriniPMalvagiaSla MarcaGDonatiMAN-carbamylglutamate in emergency management of hyperammonemia in neonatal acute onset propionic and methylmalonic aciduriaNeonatology201097328629019887858
- SoyucenEDemirciEAydinAOutpatient treatment of propionic acidemia-associated hyperammonemia with N-carbamoyl-L-glutamate in an infantClin Ther201032471071320435240
- Ah MewNMcCarterRDaikhinYNissimIYudkoffMTuchmanMN-carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic acidemiaPediatrics20101261e208e21420566609
- AcquavivaCBenoistJFPereiraSMolecular basis of methylmalonyl-CoA mutase apoenzyme defect in 40 European patients affected by mut(o) and mut- forms of methylmalonic acidemia: identification of 29 novel mutations in the MUT geneHum Mutat200525216717615643616
- FowlerBLeonardJVBaumgartnerMRCauses of and diagnostic approach to methylmalonic aciduriasJ Inherit Metab Dis200831335036018563633
- ManoliIVendittiCPPagonRABirdTCDolanCRStephensKMethylmalonic acidemiaGeneReviewsSeattle (WA)University of Washington1993–2005
- GebhardtBVlahoSFischerDSewellABöhlesHN-carbamylglutamate enhances ammonia detoxification in a patient with decompensated methylmalonic aciduriaMol Genet Metab200379430330412948747
- LevratVForestIFouilhouxAAcquavivaCVianey-SabanCGuffonNCarglumic acid: an additional therapy in the treatment of organic acidurias with hyperammonemia?Orphanet J Rare Dis20083218234091
- DancisJHutzlerJLevitzMMetabolism of the white blood cells in maple-syrup-urine diseaseBiochim Biophys Acta19604334234313719556
- ChuangDTMaple syrup urine disease: it has come a long wayJ Pediatr19981323 Pt 2S17S239546032
- ChuangDTShihVEScriverCRBeaudetALSlyWSValleDDisorders of branched-chain amino acid and keto acid metabolismThe Metabolic and Molecular Basis of Inherited Disease7th edNew YorkMcGraw-Hill199512391277
- CromeLDuttonGRossCFMaple syrup urine diseaseJ Pathol Bacteriol19618137938413696537
- MackenzieDYWoolfLIMaple syrup urine disease; an inborn error of the metabolism of valine, leucine, and isoleucine associated with gross mental deficiencyBr Med J195915114909113608087
- Kalkan UcarSCokerMHabifSThe first use of N-carbamylglutamate in a patient with decompensated maple syrup urine diseaseMetab Brain Dis200924340941419688253
- LottITEricksonAMLevyHLDietary treatment of an infant with isovaleric acidemiaPediatrics19724946166185013425
- MendiolaJJrRobothamJLLiehrJGWilliamsJCNeonatal lethargy due to isovaleric acidemia and hyperammonemiaTex Med198480152546695347
- TanakaKBuddMAEfronMLIsselbacherKJIsovaleric acidemia: a new genetic defect of leucine metabolismProc Natl Acad Sci U S A19665612363425229850
- TanakaKIsselbacherKJThe isolation and identif ication of N- isovalerylglycine from urine of patients with isovaleric acidemiaJ Biol Chem196724212296629726027258
- VockleyJRoganPKAndersonBDExon skipping in IVD RNA processing in isovaleric acidemia caused by point mutations in the coding region of the IVD geneAm J Hum Genet200066235636710677295
- LeeYWLeeDHVockleyJKimNDLeeYKKiCSDifferent spectrum of mutations of isovaleryl-CoA dehydrogenase (IVD) gene in Korean patients with isovaleric acidemiaMol Genet Metab2007921–2717717576084
- VockleyJEnsenauerRIsovaleric acidemia: new aspects of genetic and phenotypic heterogeneityAm J Med Genet C Semin Med Genet2006142C29510316602101
- CoudeFXGrimberGParvyPRabierDRole of N-acetylglutamate and acetyl-CoA in the inhibition of ureagenesis by isovaleric acid in isolated rat hepatocytesBiochim Biophys Acta1983761113166639961
- KasapkaraCSEzguFSTumerLBiberogluGOkurIHasanogluAN-carbamylglutamate treatment for acute neonatal hyperammonaemia in isovaleric acidaemiaJ Inherit Metab Dis201033 Suppl 1S48
- ZammarchiEFilippiLNovembreEDonatiMABiochemical evaluation of a patient with a familial form of leucine-sensitive hypoglycemia and concomitant hyperammonemiaMetabolism19964589579608769351
- StanleyCALieuYKHsuBYHyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase geneN Engl J Med199833819135213579571255
- MacMullenCFangJHsuBYHyperinsulinism/hyperammonemia syndrome in children with regulatory mutations in the inhibitory guanosine triphosphate-binding domain of glutamate dehydrogenaseJ Clin Endocrinol Metab20018641782178711297618
- FahienLAMacDonaldMJKmiotekEHMertzRJFahienCMRegulation of insulin release by factors that also modify glutamate dehydrogenaseJ Biol Chem19882632713610136143047128
- PariniRColomboFLombardiAMMenniFBeccariaLHyperinsulinism plus hyperammonemiaJ Pediatr199813368008019842050
- HsuBYKellyAThorntonPSGreenbergCRDillingLAStanleyCAProtein-sensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndromeJ Pediatr2001138338338911241047
- StanleyCAHyperinsulinism/hyperammonemia syndrome: insights into the regulatory role of glutamate dehydrogenase in ammonia metabolismMol Genet Metab200481 Suppl 1S45S5115050973
- HuijmansJGDuranMde KlerkJBRoversMJScholteHRFunctional hyperactivity of hepatic glutamate dehydrogenase as a cause of the hyperinsulinism/hyperammonemia syndrome: effect of treatmentPediatrics2000106359660010969108
- PalladinoAAStanleyCAThe hyperinsulinism/hyperammonemia syndromeRev Endocr Metab Disord2010109 [Epub ahead of print]
- De LonlayPBenelliCFouqueFHyperinsulinism and hyperammonemia syndrome: report of twelve unrelated patientsPediatr Res200150335335711518822
- Segura-BrunaNRodriguez-CampelloAPuenteVRoquerJValproate- induced hyperammonemic encephalopathyActa Neurol Scand200611411716774619
- Pedron GinerCLopez MarinLQuijada FrailePValproate induced hyperammonaemic encephalopathy syndrome. Treatment with carglumic acidJ Inherit Metab Dis200831 Suppl 1S89
- ErgonProtocolo Hispano-Luso de diagnostico y tratamiento de las hiperamoniemias en pacientes neonatos y de màs de 30 dìas de vida2nd edMajadahonda, Madrid2009
- SchubigerGBachmannCBarbenPColomboJPTönzOSchüpbachDN-acetylglutamate synthetase deficiency: diagnosis, management and follow-up of a rare disorder of ammonia detoxicationEur J Pediatr199115053533562044610