
Abstract
Purpose
To construct a prognosis model of melanoma based on pyroptosis related genes.
Methods
Melanoma RNA-sequencing data was downloaded from TCGA. First, the lnRNAs related to pyroptosis were obtained through Pearson correlation analysis. Then, the prognosis model of pyroptosis related genes was constructed by Cox regression and Lasso regression. Melanoma patients were divided into high-risk and low-risk groups by risk score, and the differences in prognosis and immune microenvironment between the two groups were explored.
Results
We found that the high-risk group had a significantly poorer prognosis, and different groups differed in immune infiltration, m6A methylation, and immune checkpoint.
Conclusion
Our prognostic model can provide a reference for the study of pyroptosis in melanoma cells and provide a new idea for melanoma treatment.
Introduction
Melanoma is a highly malignant skin tumor with strong invasiveness and poor prognosis.Citation1 The National Cancer Institute estimates that there will be about 100,000 new cases of melanoma and 6850 deaths in the United States in 2020.Citation2 Risk factors for melanoma include light skin color, UV exposure, nevi, genetic predisposition, and a family history of melanoma.Citation2 Since the beginning of the 21st century, research on the treatment of melanoma, especially the discovery of immune checkpoint inhibitors and BRAF inhibitors, has greatly improved the survival rate of melanoma patients.Citation3,Citation4 However, although the mortality of melanoma patients is on the decline, the morbidity of melanoma is still increasing year by year.Citation5 At the same time, drug resistance of PD-1/PD-L1 inhibitors and BRAF mutation inhibitors has also gradually emerged, which makes the exploration of new biomarkers urgent.Citation6
Pyroptosis, an inflammatory form of programmed cell death, shares many similarities with apoptosis, but there are also significant differences.Citation7 When cell pyroptosis occurs, there will be many pores on the cell membrane so that the cell will have osmotic swelling and membrane rupture, and the inflammatory substances will be discharged, causing an obvious inflammatory reaction.Citation7 This is the morphological characteristic of cell pyroptosis. As early as 1992, Zychlinsky et al observed that Shigella flexneri can induce this death in host macrophages, but due to limited knowledge at the time, it was still considered to be apoptosis.Citation8 It was not until 2001 that Cookson et al found that this death was different from apoptosis and had Caspase-1-dependent characteristics, which induced them to name it “pyroptosis” for the first time.Citation9 “Pyro” means “fire” and “ptosis” means “fall”, which vividly describes the characteristics of this type of death. According to the differences in the mechanism of cell pyroptosis, it can be divided into the classical pyroptosis pathway mediated by Caspase-1 and the non-classical pyroptosis pathway mediated by Caspase-4/5/11.Citation10 Activated caspase cleaves Gasdermin protein to form the N-terminal domain (GSDM-N) and C-terminal domain (GSDM-C).Citation11 GSDM-N has pore-forming activity and is “oligomerized” to form pores of varying sizes in the cell membrane, mediating osmotic swelling of the cell and the expulsion of inflammatory molecules such as IL-1β and IL-18.Citation7 Pyroptosis is a double-edged sword in cancer. On the one hand, we can induce Pyroptosis of tumor cells to inhibit tumor growth. On the other hand, the inflammatory substances released in the process of cell pyroptosis form the inflammatory microenvironment of the tumor and promote tumor growth by activating different signaling pathways.Citation12 The correlation between pyroptosis and cancer still needs further research to clarify. Among them, the correlation between pyroptosis and melanoma is still unclear.
Now it’s been 60 years since Jacob and Monod discovered mRNA.Citation13 Their momentous discovery laid the foundation for the “genetic central dogma”.Citation14 To this day, the “genetic central dogma” remains the classic law of genetics. But in 2001, the Human Genome Project found that only 1.5% of nucleic acid sequences are transcribed into mRNA to encode proteins, while the remaining 98.5% are often considered “junk sequences”.Citation15 With the development of research, people found that these “junk sequences” can be transcribed into a variety of non-coding RNAs. LncRNA is one of them, which usually refers to non-coding RNA transcripts with more than 200 nucleotide sequences, and plays an important role in homeostasis regulation and the occurrence and development of tumors.Citation16 At present, many studies have confirmed the relationship between lncRNA and cell apoptosis, but the specific role network is still unclear.
In this study, we explored pyroptosis-related lncRNAs in melanoma and constructed a prognostic model, which showed that patients with a high-risk score had a worse prognosis. Our conclusions can provide some new ideas for the prognosis and treatment of melanoma.
Methods
Information Extraction of Datasets
Melanoma RNA sequencing data downloaded from TCGA (https://portal.gdc.cancer.gov/) in the database. Inclusion criteria were: (1) patients diagnosed with melanoma; (2) Patients had detailed lncRNA and clinical information. A total of 457 melanoma patients met the inclusion criteria, and their lncRNA and clinical information were downloaded (those with a follow-up of fewer than 30 days were excluded).
Identification of lncRNAs Associated with Cell Pyroptosis
Thirty-three apoptosis-related genes were obtained from previous literature. The Pearson correlation method was used to identify lncRNAs related to cell pyroptosis. The correlation coefficients R2>0.3 and P<0.001 were considered to be pyroptosis-related lncRNAs. We used Cytoscape software to visualize lncRNA co-expression networks.
Identification of Prognostic Pyroptosis-Related lncRNAs
First, we performed univariate Cox regression on pyroptosis-related lncRNAs. We used the package “forestplot” in R software for COX regression analysis. Then Lasso regression was performed for lncRNAs with P<0.05, and multivariate Cox regression was performed for the results obtained. According to the final results, risk scores were calculated. Using the regression coefficient (β), the scoring formula is: . Using a median risk score, patients were divided into high-risk and low-risk groups.
Construction of the Prognostic Model
After patients were divided into high-risk and low-risk groups, Kaplan-Meier survival analysis was performed for both groups to identify the prognostic value of the model. The accuracy of the established prognosis model was verified by the calibration curve and ROC curve. We used the “plotROC” package in R software to draw ROC curves.
Clinical Value of the Established Prognostic Model
We used univariate and multivariate Cox regressions, respectively, to test whether risk score and clinical characteristics (age, sex, stage) were effective prognostic indicators for melanoma patients. Nomograms were used to demonstrate clinical characteristics and risk scores concerning 1, 3, and 5-years survival.
Correlation Analysis of Immune Checkpoint and M6A-Methylation
We then collected 47 immune checkpoint-related genes (ICPs) and 25 M6A-methylated related genes by reading the published literature and investigated the expression of these genes in the high- and low-risk groups in two data sets. Differentially expressed ICP and ICD genes were retained (P <0.05) and displayed as a bar chart. Rank sum test was used for comparison between groups.
Model to Predict Potential Drug Candidates for Melanoma
By using the “pRRophetic” package and the expression matrix of melanoma patients, we predicted the minimum drug inhibition concentration (IC50) of drugs in melanoma patients, and finally obtained some drugs that are related to the model and may become candidates for the treatment of melanoma.
Gene Set Enrichment Analysis
Gene Set Enrichment Analysis (GSEA) is a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (eg phenotypes). We used GSEA analysis to explore KEGG pathways differences in gene function between the high-risk and low-risk groups.
Results
The Flow Chart
The flow chart is shown in . First, we downloaded melanoma data from TCGA database. Then, we constructed a prognostic model based on 9 pyroptosis related lncRNAs. Subsequently, we investigated differences in immune infiltration and m6A methylation between the high-risk and low-risk groups. Finally, we performed GSEA analysis.
Figure 1 The flow chart of our study.
Nine Pyroptosis-Related lncRNAs Were Identified to Calculate the Risk Score
Firstly, the lncRNA closely associated with pyroptosis-related genes in melanoma was identified by Pearson-correlation test. Cox regression and Lasso regression were performed on the pyroptosis-related lncRNAs of melanoma in the TCGA database, and 9 pyroptosis-related lncRNAs with prognostic value were finally obtained (). The risk score was calculated by these 9 lncRNAs. Based on the median risk score, 457 melanoma patients in TCGA were divided into high-risk and low-risk groups ().
Table 1 9 Pyroptosis-Related lncRNAs with Prognostic Value
Figure 2 (A) The risk score was calculated by these 9 lncRNAs. Based on the median risk score, 457 melanoma patients in TCGA were divided into high-risk and low-risk groups. (B) Prognostic differences between the high-risk and low-risk groups were investigated. High risk group was found to indicate poor prognosis (P <0.001). (C) In the form of dot plot, we can intuitively see that the survival time of patients in the high-risk group is concentrated at the bottom, that is, the survival time is shorter. (D) Heat map reflects the relationship between the expression status of 9 pyroptosis-related lncRNAs and clinical features such as TNM stage, sex and age.
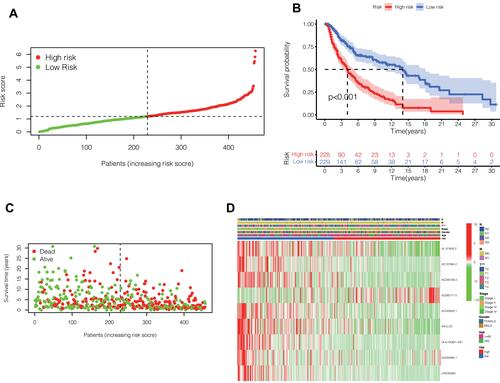
The Group Model Based on Risk Score Can Effectively Predict the Prognosis of Melanoma Patients
After classifying melanoma patients into high-risk and low-risk groups, we performed a survival analysis. As can be seen from the , the survival rate of patients in the high-risk group decreased significantly, and the difference was statistically significant (P <0.001). In , in the form of dot plot, we can intuitively see that the survival time of patients in the high-risk group is concentrated at the bottom, that is, the survival time is shorter. heat map reflects the relationship between the expression status of 9 pyroptosis-related lncRNAs and clinical features such as TNM stage, sex and age. Heatmap showed significant differences in T staging between patients in the high- and low-risk groups. There was no significant difference in other clinical features.
Risk Score is an Independent Factor Affecting Patient Survival with High Accuracy
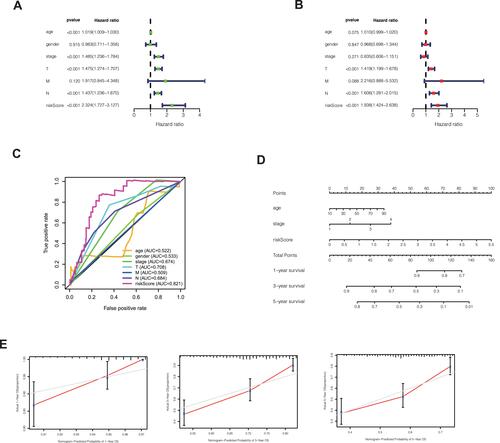
In order to explore whether risk score is an independent influencing factor for melanoma patients, univariate and multivariate Cox regression were performed for risk score, age, gender, TNM stage and other factors. The clinical characteristics of the patients in the database are as follows (). Univariate Cox regression showed that age, stage, T stage, N stage and risk score were independent influencing factors for the prognosis of melanoma patients (). Multivariate Cox regression showed that T stage, N stage and risk score were independent influencing factors for the prognosis of melanoma patients (). The ROC curve showed that the AUC value of the risk score was 0.821, demonstrating its sensitivity (). Nomogram can see that the risk score is associated with 1, 3, and 5 year survival (). Calibration shows that Nomogram can accurately predict 1, 3, and 5-year survival of patients with melanoma ().
Table 2 The Clinical Characteristics of the Patients in the Database
Figure 3 (A) Univariate Cox regression showed that age, stage, T stage, N stage and risk score were independent influencing factors for the prognosis of melanoma patients. (B) Multivariate Cox regression showed that T stage, N stage and risk score were independent influencing factors for the prognosis of melanoma patients. (C) Among the ROC curves of patient prognosis and clinical characteristics, the risk score AUC was the highest (0.821). (D) Nomogram was used to predict 1, 3, 5-year survival. (E) Calibration shows that Nomogram can accurately predict 1, 3, and 5-year survival of patients with melanoma.
Correlation Analysis of Immune Checkpoint and M6A Methylation in Different Groups
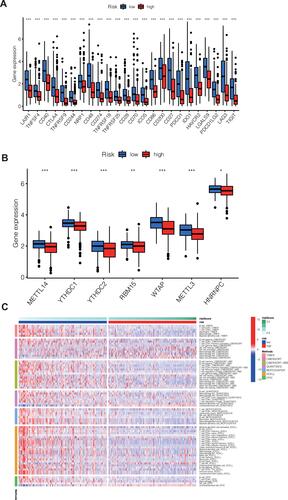
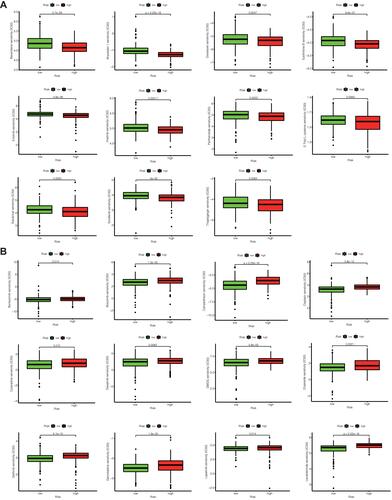
Many immune checkpoint genes were expressed differently in samples in the two groups and tended to be higher in the low-risk group (***P<0.001, ). Among the samples from the high-risk and low-risk groups, the expression of 7 M6A-methylation related genes differed, and all of them showed a tendency to be upregulated in the low-risk group (***P<0.001; * P < 0.05; ).
Figure 4 (A) Immune checkpoint genes that were differentially expressed in the high-risk and low-risk groups were selected and presented in a boxplot. (***p < 0.001) (B) Methylation-related genes that were differentially expressed in the high-risk and low-risk groups were selected and presented in a boxplot. (*p < 0.05, **p < 0.01, ***p < 0.001) (C) Heat maps of immune cells that differ between the high-risk and low-risk groups.
Analysis of Immune Cell Infiltration Levels in Different Groups
The levels of immune infiltration in high-risk and low-risk groups were calculated by algorithm such as TIMER, CIBERSORT, Quantiseq and other methods. The heat map shows the level of each immune cell infiltration in the tumor microenvironment of the two groups of patients under different algorithms (). We can see that the low-risk group tended to have higher levels of immune cells in the tumor microenvironment.
Analysis of Sensitivity Difference of Antitumor Drugs in Different Groups
The study of the sensitivity of different groups of patients to anti-tumor drugs can provide help for the formulation of future treatment regimens. The high-risk group was more sensitive to certain drugs, such as Bexarotene, Bryostatin and Docetaxel (). The low-risk group was more sensitive to Bortezomib, Bosutinib, Camptothecin, etc (). This has guiding significance for us to select drugs according to the group situation.
Figure 5 (A) The high-risk group was more sensitive to certain drugs, such as Bexarotene, Bryostatin and Docetaxel. (B) The low-risk group was more sensitive to Bortezomib, Bosutinib, Camptothecin, etc.
Gene Set Enrichment Analysis
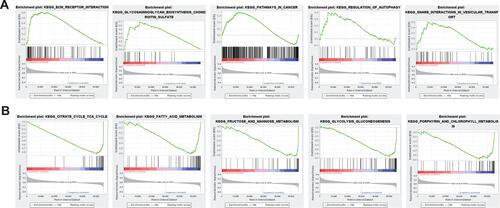
To explore the functional differences in gene expression between the high-risk and low-risk groups, we performed GSEA analysis. The results showed that gene expression in high-risk group was mainly related to ECM- receptor interaction, cancer signaling pathway and autophagy ( and ).
Figure 6 (A) GSEA analysis in high-risk group:Gene expression in high-risk group was mainly related to ECM- receptor interaction, cancer signaling pathway and autophagy. (B) GSEA analysis in low-risk group.
Discussion
The mode of cell death has always been the focus of research. Since cancer cells have the ability to proliferate indefinitely, inducing cancer cell death has become a potential therapeutic direction.Citation17 Programmed cell death (PCD) includes apoptosis, pyroptosis, and ferroptosis. Among them, pyroptosis has great potential in the treatment of cancer.Citation18 Cell pyroptosis plays a dual role in cancer. On the one hand, we can reduce tumor load by inducing cancer cell pyroptosis; on the other hand, cell pyroptosis of cancer cells can lead to the expulsion of inflammatory substances, thus constructing the inflammatory microenvironment of tumor and providing conditions for tumor growth.Citation7 Currently, the role of cell apoptosis in the physiology of cancer, including melanoma, remains unclear.
LncRNA is a class of non-coding RNAs with nucleotide numbers greater than 200. Known studies have revealed that lncRNA plays an important role in inflammation, tumor formation, and the acquisition of drug resistance.Citation19 Many studies have confirmed that lncRNAs are involved in cell apoptosis, but the mechanism of their role in cell apoptosis in melanoma is poorly understood.
In this study, we explored the lncRNAs related to cell pyroptosis in melanoma and identified those lncRNAs with prognostic values. Through Cox regression and Lasso regression, we constructed a prognostic model consisting of 9 lncRNAs. By calculating the risk scores, 457 patients were assigned to a high-risk or low-risk group. Compared with the low-risk group, the survival rate was significantly lower in the high-risk group, with a statistically significant difference (P <0.001). ROC curves and calibration curves proved the accuracy of the prognostic model. And not only that, we found that the expression levels of immune checkpoint-related genes and M6A methylation-related genes were higher in the low-risk group.
For melanoma, a tumor with a poor prognosis, the establishment of an effective prognostic model and the exploration of its mechanism of action can improve the outcome of treatment. At present, immune checkpoint inhibitors and targeted inhibitors have achieved good therapeutic effects in melanoma. But these treatments can gradually build up resistance and bring regret to patients.Citation20 Cell pyroptosis is a promising new direction for the treatment of melanoma. GSDME, a key protein in the pyroptosis pathway, is not expressed in many cancer cells but is highly expressed in melanoma, which becomes the Achilles’ heel of melanoma.Citation21 Induction of melanoma pyroptosis may play a potential role in improving the prognosis of advanced melanoma.Citation22 Yu et al found that autophagy in melanoma is closely related to pyroptosis, a process mediated by EEF-2K.Citation23 Erkes et al also found that BRAF and MEK inhibitors can change the tumor microenvironment by inducing melanoma cell pyroptosis, thus improving the prognosis.Citation24 Therefore, it can be seen that there is a close correlation between cell apoptosis and melanoma, which has a great exploration prospect.
Melanoma is a highly immune-related tumor, and the discovery of immune checkpoint inhibitors is a milestone.Citation25 Clinical use of CTLA-4 and PD-1 inhibitors has benefited many melanoma patients, greatly improving disease-free survival and overall survival in melanoma patients.Citation26 Exploring the crosstalk between pyroptosis and immune checkpoints can further help us understand the clinical significance of apoptosis. According to our results, the expression of immune checkpoint-related genes was higher in the low-risk group, suggesting that immune checkpoint inhibitor therapy may be more beneficial in the low-risk group.
Epigenetics exists in a wide range of organisms, and posttranscriptional modification is a common regulatory pathway.Citation27 M6A-methylation is the most common post-transcriptional modification in eukaryotes.Citation28 This process is mediated by methylase (writer), demethylase (eraser), and methylated reading protein (reader), and is involved in the homeostasis regulation of organisms and the occurrence and development of diseases.Citation29 The regulation of M6A methylation in cells has also become a new idea for the treatment of diseases, especially tumors.Citation29 Through the correlation analysis of pyroptosis-related lncRNAs and M6A-methylation-related genes, we found that the methylation level in the low-risk group tended to be higher, which provided ideas for the future treatment of M6A methylation in melanoma. At the same time, it indirectly indicates that there is a certain degree of crosstalk between pyrosis and M6A methylation.
All in all, our study established a prognostic model of pyroptosis-associated lncRNA in melanoma patients and studied its mechanism of action. This is not only meaningful for guiding prognosis but also can facilitate the study of tumor physiology of melanoma, such as immune checkpoints and M6A methylation. However, our study has some limitations. We lack in vivo and in vitro experiments to verify the expression and function of lncRNAs in the prognostic model, and we will improve this aspect in the future.
Conclusion
Our study established a prognostic model of pyroptosis-related lncRNAs in melanoma patients to provide a reference for the study of pyroptosis in melanoma cells, and provide a new idea for melanoma treatment. But our study has some limitations due to the lack of validation.
Ethics Approval
All procedures performed were in accordance with the declaration of the ethical standards of the institutional research committee and with the 1964 Helsinki 387 Declaration and its later amendments. The ethics committee has approved this study of the First Affiliated Hospital of Nanjing Medical University (No. 2021-SR-418).
Acknowledgments
We are very grateful for data provided by databases such as TCGA, GEO. Jiaheng Xie, Haobo Li, Liang Chen, and Yuan Cao are co-first authors for this study.
Disclosure
All authors declare that no conflict of interest exists.
Additional information
Funding
References
- Pavri SN, Clune J, Ariyan S, Narayan D. Malignant melanoma: beyond the basics. Plast Reconstr Surg. 2016;138(2):330e–340e. doi:10.1097/PRS.0000000000002367
- Rastrelli M, Tropea S, Rossi CR, Alaibac M. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014;28(6):1005–1011.
- Xie J, Zhu Z, Cao Y, Ruan S, Wang M, Shi J. Solute carrier transporter superfamily member SLC16A1 is a potential prognostic biomarker and associated with immune infiltration in skin cutaneous melanoma. Channels. 2021;15(1):483–495. doi:10.1080/19336950.2021.1953322
- Xie J, Ruan S, Zhu Z, et al. Database mining analysis revealed the role of the putative H+/sugar transporter solute carrier family 45 in skin cutaneous melanoma. Channels. 2021;15(1):496–506. doi:10.1080/19336950.2021.1956226
- Eggermont AMM, Crittenden M, Wargo J. Combination immunotherapy development in melanoma. Am Soc Clin Oncol Educ Book. 2018;38(38):197–207. doi:10.1200/EDBK_201131
- Winder M, Virós A. Mechanisms of drug resistance in melanoma. Handb Exp Pharmacol. 2018;249:91–108.
- Fang Y, Tian S, Pan Y, et al. Pyroptosis: a new frontier in cancer. Biomed Pharmacother. 2020;121:109595. doi:10.1016/j.biopha.2019.109595
- Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358(6382):167–169. doi:10.1038/358167a0
- Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9(3):113–114. doi:10.1016/s0966-842x(00)01936-3
- Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 2019;26(1):99–114. doi:10.1038/s41418-018-0212-6
- Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103. doi:10.1038/nature22393
- Ruan J, Wang S, Wang J. Mechanism and regulation of pyroptosis-mediated in cancer cell death. Chem Biol Interact. 2020;323:109052. doi:10.1016/j.cbi.2020.109052
- Jathar S, Kumar V, Srivastava J, Tripathi V. Technological developments in lncRNA biology. Adv Exp Med Biol. 2017;1008:283–323.
- Crick F. Central dogma of molecular biology. Nature. 1970;227(5258):561–563. doi:10.1038/227561a0
- Qian X, Zhao J, Yeung PY, Zhang QC, Kwok CK. Revealing lncRNA structures and interactions by sequencing-based approaches. Trends Biochem Sci. 2019;44(1):33–52. doi:10.1016/j.tibs.2018.09.012
- Robinson EK, Covarrubias S, Carpenter S. The how and why of lncRNA function: an innate immune perspective. Biochim Biophys Acta Gene Regul Mech. 2020;1863(4):194419. doi:10.1016/j.bbagrm.2019.194419
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31(1):51–72. doi:10.1146/annurev-immunol-032712-100008
- Yang F, He Y, Zhai Z, Sun E. Programmed cell death pathways in the pathogenesis of systemic Lupus Erythematosus. J Immunol Res. 2019;2019:3638562. doi:10.1155/2019/3638562
- Yang G, Lu X, Yuan L. LncRNA: a link between RNA and cancer. Biochim Biophys Acta. 2014;1839(11):1097–1109. doi:10.1016/j.bbagrm.2014.08.012
- Luís R, Brito C, Pojo M. Melanoma metabolism: cell survival and resistance to therapy. Adv Exp Med Biol. 2020;1219:203–223.
- Zhou B, Zhang JY, Liu XS, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28(12):1171–1185. doi:10.1038/s41422-018-0090-y
- Zhao P, Wang M, Chen M, et al. Programming cell pyroptosis with biomimetic nanoparticles for solid tumor immunotherapy. Biomaterials. 2020;254:120142. doi:10.1016/j.biomaterials.2020.120142
- Yu P, Wang HY, Tian M, et al. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta Pharmacol Sin. 2019;40(9):1237–1244. doi:10.1038/s41401-019-0222-z
- Erkes DA, Cai W, Sanchez IM, et al. Mutant BRAF and MEK inhibitors regulate the Tumor immune microenvironment via pyroptosis. Cancer Discov. 2020;10(2):254–269. doi:10.1158/2159-8290.CD-19-0672
- Atherton MJ, Morris JS, McDermott MR, Lichty BD. Cancer immunology and canine malignant melanoma: a comparative review. Vet Immunol Immunopathol. 2016;169:15–26. doi:10.1016/j.vetimm.2015.11.003
- Lin WM, Fisher DE. Signaling and immune regulation in melanoma development and responses to therapy. Annu Rev Pathol. 2017;12(1):75–102. doi:10.1146/annurev-pathol-052016-100208
- Li J, Xie J, Wu D, et al. A pan-cancer analysis revealed the role of the SLC16 family in cancer. Channels. 2021;15(1):528–540. doi:10.1080/19336950.2021.1965422
- Ma S, Chen C, Ji X, et al. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. 2019;12(1):121. doi:10.1186/s13045-019-0805-7
- Qin Y, Li L, Luo E, et al. Role of m6A RNA methylation in cardiovascular disease. Int J Mol Med. 2020;46(6):1958–1972. doi:10.3892/ijmm.2020.4746