
Abstract
Purpose
Endometrial cancer (EC) is a common gynaecologic malignancy with an increasing incidence rate and mortality in recent years. N6-methylandenosine (m6A)-related long noncoding RNA (lncRNA) plays a vital role in EC, emerging as one of the most abundant RNA modifications.
Materials and Methods
The Cancer Genome Atlas (TCGA) database and UCSC Xena were used to download data related to EC. Survival and univariate and multifactorial prognostic analyses were performed for m6A-related lncRNAs. The expression levels of the three lncRNAs were verified using q-PCR. A nomogram was used to create a clinical tool to assess overall survival. To investigate the relationship between m6A-related lncRNA and EC, we downloaded differential genes related to EC from the TCGA database and mined three m6A-related lncRNAs, namely SCARNA9, TRAF3IP2-AS1, and AL133243.2. The data were categorized into high- and low-risk groups based on m6A-associated lncRNA.
Results
Survival analysis revealed that the high-risk group had a lower survival rate. Survival analysis of three m6A-associated lncRNAs revealed that cases with high expression of SCARNA9 tended to have a poorer prognosis, whereas the opposite was true for TRAF3IP2-AS1, AL133243.2. Univariate and multifactorial prognostic analyses suggested statistical differences in patients’ age, FIGO stage, pathological grade, risk score, and prognosis of EC, which was confirmed by results of the separate prognostic factor analysis for the three lncRNAs. Risk status was validated as an independent prognostic indicator, and the prognostic nomogram combined patient age, pathological stage, and FIGO classification to assess 3–5-year survival. Cases from high- and low-risk groups were analysed for the tumour microenvironment and immune cell scores, and stromal cell scores were found to be lower in the high-risk group. Correlations were analysed using different databases for immune cell classification.
Conclusion
m6A-related lncRNAs may play a key role in the diagnosis and treatment of EC as targets of prognosis and the immune microenvironment.
Introduction
Endometrial cancer (EC) is one of the most common diseases of the female reproductive system, ranking the sixth most common gynaecologic malignancy globally, with nearly 382,000 new diagnoses and 89,900 deaths in 2018.Citation1 Epidemiological analysis of late age at menopause, postmenopausal hormone replacement therapy, and early age at menarche may serve as risk factors for the development of EC.Citation2 Most cases are diagnosed due to abnormal introitus bleeding and the prognosis is good in the early diagnosed cases, while the prognosis is poor in progressive cases, with a 5-year survival rate of only 15%.Citation3 Therefore, new treatment strategies and molecular indicators are crucial for the prognosis of endometrial cancer.
N6-methyladenosine modification (m6A) is the most abundant and prevalent RNA modification present in eukaryotic cells and contains three categories of m6A methyltransferases: writer, reader, and and eraser.Citation4 M6A is implicated in multiple aspects of cellular metabolism, including RNA splicing, transcription, translation, and degradation.Citation5 Studies have shown that m6A participates in the development of a multitude of tumours, including EC. IGF2BP1 acts as an m6a reader to recognize and stabilize PEG10 mRNA and enhance its expression, thereby accelerating cell proliferation and promoting the progression of EC.Citation6 FTO removes YTHDF2-mediated degradation of HOXB13 and increases its expression, thereby triggering the wnt pathway and facilitating invasion and metastasis of EC cells.Citation7 YTHDF3 is involved in multiple aspects of breast cancer brain metastasis by binding to its mRNA, promoting its translation, and regulating downstream genes.Citation8
lncRNAs are a class of non-coding RNAs with molecular mass greater than 200 and less than 2000, which were previously shown to be unable to encode proteins, however some have been found to have coding functions.Citation9 lncRNAs participate in the pathogenesis of many diseases, such as Mendelian disorders, cardiovascular diseases, and tumours.Citation10 lncRNA H19 promotes hepatocellular carcinoma bone metastasis by mediating PPP1CA/p38MAPK-dependent reduction in OPG expression.Citation11 LINC01969 is predominantly located in the cytoplasm and promotes the malignant biological behaviour of ovarian cancer by acting as a sponge of miR-144-5p adsorbed to regulate the expression of the downstream target gene LARP1.Citation12 Therefore, lncRNAs have essential roles in several aspects of tumour initiation and development.
The study of the tumour microenvironment (TME) has become increasingly prevalent. The tumour microenvironment consists of soluble molecules, tumour stromal cells, and extracellular matrix, of which the most investigated are immune cells and stromal cells.Citation13–Citation15 Several studies have indicated that the tumour microenvironment may be involved in multiple aspects of tumour cell invasion, metastasis, drug resistance, and immune escape.Citation16,Citation17 Furthermore, the study of immune-related lncRNAs has attracted increasing attention. lncRNA Xist modulated the expression of C/EBPa and KLF6 through competitive binding of miR-101 to mediate macrophage polarisation and thus affect the proliferation and migration of breast and ovarian cancer cells.Citation18 lncRNA LUCAT1 inhibits the transcription of interferon genes by interacting with STAT1 in the nucleus, thereby inhibiting the immune response of human cells.Citation19 lncRNA AFAP1-AS1 acts on KYSE410 cells through exosomal delivery of M2 type macrophages, down-regulating miR-26a, and promoting the expression of ATF2, thereby promoting cancer cell migration, invasion, and lung metastasis.Citation20
Currently, lncRNA studies are more common in EC, whereas m6A studies are scarce. In this study, we extracted three m6A-related lncRNAs based on data from EC patients in The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/) using bioinformatics and statistical analysis, and constructed prognostic models for m6A-associated lncRNAs to analyse the prognostic-related risk factors of EC. Endometrial patients were divided into high-and low-risk groups, and survival analysis and immune microenvironment analysis were performed. Finally, an accurate nomogram was constructed to validate and perform KEGG and GO enrichment analyses. These analyses suggest that these lncRNAs may serve as biomarkers for the diagnosis and prognosis of patients with EC.
Materials and Methods
Databases and m6A-Related Genes
Fragments per kilobase of transcript per million mapped reads (FPKM) data for EC were downloaded from the TCGA public database (https://portal.gdc.cancer.gov/),Citation21 which comprised 23 normal samples and 552 tumour samples. The clinical database of EC patients was downloaded from UCSC Xena (https://xenabrowser.net/).Citation21–Citation23 Then, the Perl software was used for data processing to generate a matrix of all differential genes in EC, which was then divided into lncRNA matrix and mRNA matrix. Additionally, 21 m6A-related genes were identified based on the current study, including readers (YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3, IGF2BP1, IGF2BP2, IGF2BP3, RBMX, HNRNPA2B1, and HNRNPC), writers (METTL3, METTL14, METTL16, RBM15, RBM15B, WTAP, VIRMA (KIA1499), and ZC3H13), and erasers (FTO and ALKBH5). By analysing the mRNA expression matrix and m6A-related genes, the expression matrix of m6A-related genes was extracted. The m6A-related lncRNA expression matrix and m6A-related lncRNA analysis were performed using the limma package of R software and BiocManage (corFilter=0.6, pvalueFilter=0.001).
Survival Analysis
Using the Perl programming language, the expression matrix of m6A-related lncRNAs was organised and analysed with reference to the clinical data of EC. Using the survival package, univariate and multivariate Cox regression analyses were performed on the clinical data of m6A-associated lncRNAs to obtain a series of m6A-associated lncRNAs with P-value, HR values, and risk scores. The risk score calculating formula is: Risk score = , where means
the coefficients,
is the FPKM value of each m6A-related lncRNAs.Citation24 Patients were divided into high and low-risk groups based on the risk scores, and survival analysis and heat mapping were conducted for the high and low-risk groups using the survivor, “survmine” and “pheatmap” packages in R software. Furthermore, survival analysis was performed individually on the m6A-associated lncRNAs associated with the construction model, and survival curves were plotted. To verify the independence of the high- and low-risk group model construction, all risk-related genes and m6A-associated lncRNAs were subjected to principal component analysis using the Limma and scatterplot3d packages in R software.
Independent Prognostic Analysis of m6A-Associated lncRNA and Validation of Clinical Characteristics
To assess the impact of certain clinical characteristics on the prognosis of m6A-related lncRNA, univariate independent prognostic analyses were performed in terms of age, pathological grade, and FIGO staging by using the “survival” packages in R software, respectively. To validate the accuracy of the model, the multivariate independent prognostic analysis was performed and ROC curves were plotted for the above clinical characteristics with the “survROC” package. To authenticate the role of the clinical characteristics on prognosis from the different perspectives, we used multivariate COX regression to plot nomogram by using the “rms”, “foreign”, and “survival” packages in R software.
Tumour Immune Microenvironment Analysis
The immune cells and stromal cells in the tumour microenvironment (TME) were scored according to the ESTIMATE algorithm using the limma package and the ESTIMATE package of R language. The tumour microenvironment scores and clinical data were collated and combined for analysis using Perl programs to obtain immune and stromal scores in different risk states. By utilising the R software, box plots were drafted based on the immune score and stromal scores of patients in the high and low-risk group. The ESTIMATE algorithm in the R software ESTIMATE package was used to estimate the ratio of the immune matrix component in the TME for each sample, presented in the form of three scores: ESTIMATE score, stromal score, and immune score. This means that the higher the corresponding score, the greater the ratio of the corresponding component in the TME. Analysing and collating the risk scores of m6A-related genes and immune cells, correlation analysis was performed using the scales, “ggplot2” and “ggtext” packages in R software to plot bubble plots. The limma package, corrplot package, and BioManager package in R software were used for co-expression analysis of all differential gene matrices of endometrial EC with prognosis-associated m6A-related lncRNA matrices, where the asterisks (*) represents statistically significant differences.
Functional Annotation
The expression matrix of all genes and clinical data of different risk states were collated, followed by gene enrichment analysis (GO analysis) using the GSEA (4.0.2) software.
Samples and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
We collected 20 EC tissue and 11 normal endometrial tissue specimens. These specimens were obtained from surgical patients at Wuhan Union Hospital and were performed in strict accordance with the relevant regulations of medical ethics (Ethics Committee of Tongji Medical College of Huazhong University of Science and Technology), and informed consent was obtained from the participating patients.
To further verify the expression levels of m6A-related lncRNAs in the tissue samples, we extracted total RNA using TRIzol (RNAiso Plus, Takara, Japan) and cDNA was obtained using a reverse transcription kit (ABclonal, Wuhan, China). Expression of three m6A-related lncRNAs was calculated using 2-CT values, and GAPDH mRNA expression was used as a reference. The primer sequences used in this study were as follows: lncSCARNA9 forward 5ʹ-AGTCTTTCCAGTCTACCTGATGC-3ʹ and reverse 5ʹ-CATTGCCCAGAAATGATTAGGCT-3ʹ; lncTRAF3IP2 -AS1 forward 5ʹ-ACTGGTTGACAGAGCACCAAC-3ʹ, and reverse 5ʹ-AAATCCCATCCGTCCTTGCCT; lncAL133243.2 forward 5ʹ- AGTCCACCATTGCTCAACCGA-3ʹ; and reverse 5ʹ-ATCGGCCTTACATCTCCTGGC-3ʹ.
Bioinformatic Analysis
Perl programming language (Strawberry-Perl-5.30.0.1) was used to perform data collation and consolidation, and the databases were analysed using different packages in R software (R-3.6.1), such as limma, survival, ggplot2, and rms. The Mann–Whitney U-test was employed to explore the mRNA levels of m6A methylation-associated lncRNAs. Differences between the two subgroups were assessed with Student’s t-test. Survival curves were plotted using the Kaplan-Meier method. Univariate and multivariate analyses were performed with Cox regression models to explore the independent prognostic value of risk scores in combination with other clinical characteristics. The Q-PCR data were collated and analysed using GraphPad software. A value of p < 0.05 was considered statistically significant.
Results
Univariate and Multifactorial Cox Regression Analysis of Prognosis and Co-Expression Analysis of m6A-Related Genes with lncRNA
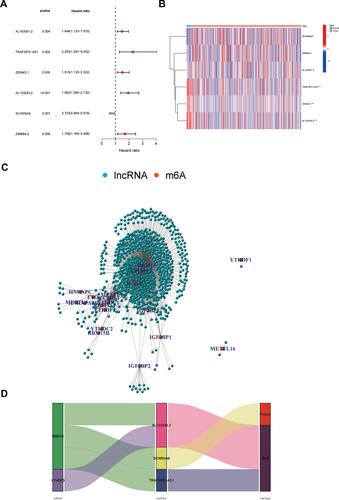
To determine whether m6A-associated lncRNAs are associated with the prognosis of EC patients, we identified six m6A-associated lncRNAs (TRAF3IP2-AS1, Z83843.1, AL133243.2, AL163051.2, Z98884.2, and SCARNA9) using univariate Cox regression analysis ( and ). And the ROC curves (Supplementary Figure 1A) demonstrated that differential genes of EC harbored a promising ability to predict OS in the TCGA cohort (1-year AUC = 0.585, 3-year AUC = 0.639, 5-year OS = 0.644). SCARNA9 was negatively correlated with the prognosis of EC, while the remaining lncRNAs were positively correlated; subsequently, we performed co-expression analysis of m6A-related genes with lncRNAs which were differentially expressed in EC. demonstrates that the variably expressed lncRNAs in EC were mainly concentrated in three m6A methylation enzymes, RBM15, YTHDF3, and WTAP. Multifactorial Cox regression analysis identified three m6A-associated lncRNAs (TRAF3IP2-AS1, AL133243.2, and SCARNA9) associated with the prognosis of EC (). Finally, the Sankey diagram was plotted to visualize the co-expression relationship between the three m6A-related lncRNAs and m6A-related genes based on risk scores ().
Table 1 Multivariate Cox Regression Analysis of the m6A-Related lncRNAs
Figure 1 Univariate and multifactorial Cox regression analysis of prognosis and co-expression analysis of m6A-related genes with lncRNA. (A) Forest plot of m6A-related lncRNAs. HR>1(red)represents risk-associated lncRNAs, HR<1 (green) indicates protective lncRNA. (B) Heatmap of m6A-related prognostic lncRNAs. *p < 0.05, ***p < 0.001. (C) Network of the correlations between m6A-related genes and m6A-related prognostic lncRNAs. (D) Co-expression Sankey diagram. The left column represents m6A-related genes, the middle column represents m6A-related lncRNAs, and the right column represents risk types.
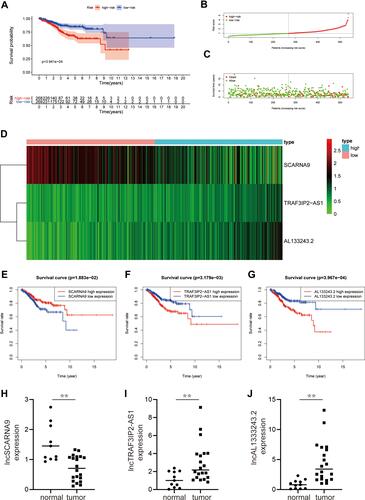
Survival Analysis of the Three m6A-Related Prognostic lncRNA
We classified patients with EC into high-risk and low-risk groups based on the median risk scores. The risk score formula was calculated as follows: risk score = (expression level of TRAF3IP2-AS1 multiplied by 0.695) + (expression level of AL133243.2 multiplied by 0.673) + (expression level of SCARNA9 multiplied by −0.346). To compare the overall survival (OS) rates of patients with different risk score statuses and to analyse the effect of different lncRNAs on the prognosis of patients, survival curves were plotted (). As illustrated in , EC patients with lower risk scores had higher overall survival rates; however, the OS rates of the patients in the high-risk group were diametrically opposite. and show the risk scores and the distribution of patient survival status associated with the different risk statuses and it shows that patients in the high-risk status group had higher risk scores and dead status. The heat map in illustrated the expression of three m6A-related (SCARNA9, TRAF3IP2-AS1, and AL133243.2) prognostic lncRNAs at different risk states. The expression of SCARNA9 was higher in the low-risk group than in the high-risk group, while the opposite trend was observed for TRAF3IP2-AS1 and AL133243.2. Then, we performed survival analysis of these three lncRNAs separately and found that low expression of TRAF3IP2-AS1 and AL133243.2, and high expression of SCARNA9 was associated with excellent prognosis (–). To verify the difference in expression of m6A-related lncRNAs between EC tissue and normal endometrium, q-PCR was performed. The results (–) suggested that the expression level of lncSCARNA9 was higher in normal endometrial tissues than in EC tissues, while the opposite trend was true for lncTRAF3IP2-AS1 and lncAL133243.2. The lncRNA expression levels were associated with the same prognostic trend.
Figure 2 Survival analysis of the three m6A-related prognostic lncRNA. (A) A survival curve based on risk scores of EC. (B and C) Risk score distribution and survival status of patients with EC. (D) Hierarchical clustering of three m6A-related lncRNAs expression levels based on risk scores. (E–G) KM curves illustrating the relationship between different expression levels of three m6A-related lncRNAs and overall survival. (H–J) The expression level of lncRNAs (SCARNA9, TRAF3IP2-AS1, and AL133243.2) were demonstrated by q-PCR. **p < 0.01.
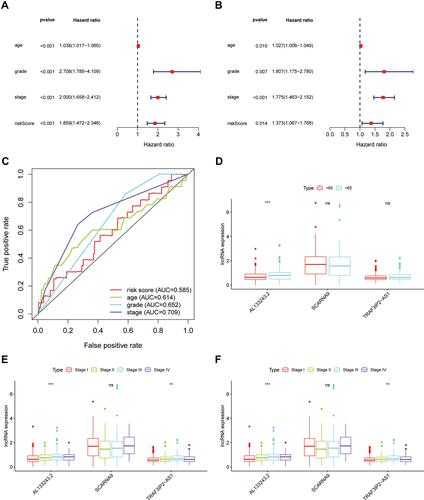
Hierarchical Analysis of m6A-Related lncRNA
To verify the influence of some clinical characteristics such as age, pathological grade, and FIGO stage on the prognosis of patients with EC, we examined univariate and multivariate COX analyses and plotted multi-index ROC curves to verify their reliability. We demonstrated that age, pathological stage, and FIGO classification were all risk factors for the patient’s prognosis as shown in –. M6A-related lncRNAs were significantly correlated with OS (HR,1.859; 95% CI:1.472–2.346, p value<0.001) by univariate Cox analysis and verified by multivariate Cox analyses. Moreover, the ROC curves suggested that the AUC was greater than 0.5, indicating a high degree of authenticity. Furthermore, we analysed the correlation of each m6A-related lncRNA with age, pathological stage, and FIGO classification separately; patients with a high expression level of AL133243.2, were typically accompanied by age >65 years, high pathological grade, and late FIGO staging. SCARNA9 expression was unrelated to age and FIGO stage, and negatively correlated with pathological grade. The expression of TRAF3IP2-AS1 increased in patients with more advanced FIGO staging and pathological grade (–).
Figure 3 Hierarchical Analysis of m6A-related lncRNA. (A and B) Univariate and multivariate analyses revealed that risk score was an independent prognostic predictor. (C) Multi-index ROC curve for the risk score, age, grade and stage, accompanied by AUCs of 0.585,0.614, 0.652, and 0.792, respectively. (D–F) Patients with different clinicopathological features (including age, pathologic grade and FIGO stage) had different expression levels of the m6A-related lncRNAs. **p < 0.01, ***p < 0.001, ns, p > 0.05.
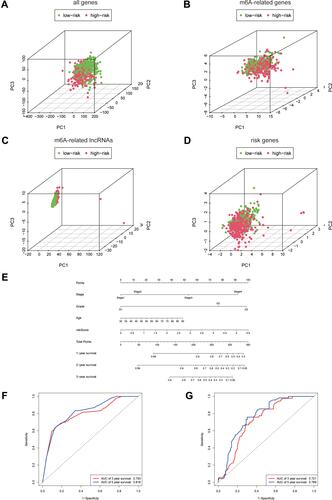
Principal Component Analysis and Construction of the m6A-Related Based Nomogram
The principal component analysis (PCA) at different levels was used to validate whether the risk status was distinguished among the different influencing factors. As shown in –, the results suggested that the distributions of the high-risk and low-risk groups were relatively independent. Then, to establish a clinical quantitative tool applicable to the assessment of OS (one-year survival, two-year survival, and three-year survival) in patients with EC, we constructed a nomogram based on the m6A-related risk scores, FIGO staging, pathological grading, and age in the TCGA database (). The EC patients were randomly divided into the training (70%) and validation groups (30%). We then plotted ROC curves to verify the accuracy of the model. The outcomes revealed that the AUC values of the 3-year and 5-year survival rates were 0.793 and 0.816 for the training group () and 0.721 and 0.769 for the validation group (), respectively, indicating the high stability of the nomogram model.
Figure 4 Principal Component Analysis and construction of the m6A-related based Nomogram. (A–D) Principal component analysis of low and high-risk groups based on all differential genes, m6A-related genes, m6A-related lncRNAs and risk-associated genes. (E) Nomogram based on risk score, age, pathologic grade and FIGO stage. (F and G) Time-dependent receiver operating characteristic (ROC) curves of training group and validation group for the nomogram (for predicting 3, and 5-year OS).
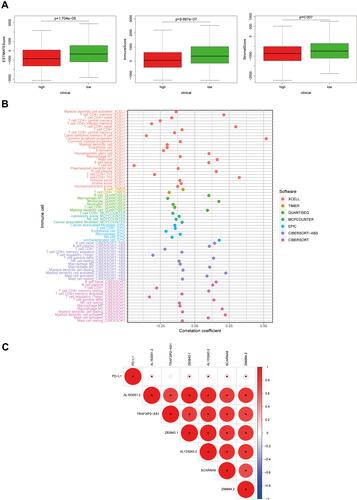
Tumour Immune Microenvironment Score and Immune Cell Infiltration Analysis
A correlation between m6A and tumour-infiltrating immune cells has recently been demonstrated. For instance, METTL3 activated T cells and enhanced the activation of the TLR4/NF-κB signalling pathway by mediating the methylation of TRL4, CD80, and CD40 signalling ligand Tirap transcripts, thus enhancing their translation in dendritic cells, which in turn stimulates cytokine production.Citation11 Persistent neoantigen-specific immunity is modulated by mRNA m6A methylation via the m6A-binding protein YTHDF15.Citation12 This research evaluated the correlation of m6A-related genes with tumour-infiltrating immune cells in EC. First, we assessed the stromal and immune scores of patients with EC based on risk status, and the results revealed that both the stromal and immune scores were lower in the high-risk group than in the low-risk group, which was statistically different (). Therefore, we assessed the relationship between different risk scores and tumour-infiltrating immune cells. We collected and collated the risk status of different immune cells from different algorithms (including XCELL, TIMER, QUANTISEQ, MCPCOUNTER, EPIC, CIBERSORT-ABS, and CIBERSORT), and the risk status of lncRNAs associated with m6A was collated and merged to obtain the bubble graph (). The results suggested that CD8+ T cells, NK cells, and M0 macrophages were negatively correlated in different databases, while M2 macrophages were positively correlated. To further clarify the correlation between prognosis-related lncRNAs and immune genes, we verified the co-expression of PD-L1 with six lncRNAs. The results indicated that PD-L1 was positively correlated with Z83843.1, AL133243.2, AL163051.2, Z98884.2, and SCARNA9, but not with TRAF3IP2-AS1 (). Additional immunoassay sites, such as PD-1, cytotoxic T lymphocyte associated antigen (CTLA-4), lymphocyte-activation gene 3 (LAG-3), T cell immunoglobulin domain and mucin domain-3 (TIM-3), T-cell immune receptor with immunoglobulin and ITIM domain (TIGIT), and killer cell immunoglobulin like receptor, three Ig domains and long cytoplasmic tail 1 (KIR3DL1), were verified and their results were consistent with the PD-L1 results described above (Supplementary Figure 1B).
Figure 5 Tumor immune microenvironment score and immune cell infiltration analysis. (A) ESTIMATE score, stromal score, and immune score of different groups were evaluated by ESTIMATE. (B) Immune cell infiltration analysis based on m6A-related lncRNAs risk scores. (C) Co-expression analysis of m6A-related prognostic lncRNAs and immune gene PD-L1.
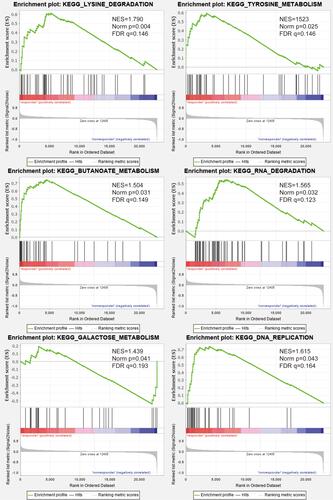
Pathway and Process Enrichment Analysis
To investigate the differences in signalling pathways, molecular mechanisms, and biological processes of m6A-related lncRNAs between the high-and low-risk groups, we performed the KEGG enrichment analysis. The enrichment analysis () indicated that the high-risk group was mainly enriched in metabolic and RNA-related pathways, such as tyrosine metabolism, galactose metabolism, RNA polymerase, and DNA replication. Related genes enriched in the above pathways were provided in the Supplementary Table 1. These results may inform us about the biological pathways associated with the m6A-related lncRNAs.
Figure 6 Functional annotation. Gene set enrichment analysis (GSEA) indicated significant enrichment of m6A‐related phenotype in the high-risk patients.
Discussion
In the present study, 552 cases of EC from the TCGA database were analysed in order to investigate the relevance of m6A-related lncRNAs in the prognosis and immune microenvironment of EC. Univariate Cox regression analysis identified six m6A-associated lncRNAs affecting EC prognosis, and further multivariate Cox regression analysis yielded three lncRNAs, SCARNA9, TRAF3IP2-AS1, and AL133243.2. Moreover, the correlation of these three lncRNAs with clinical characteristics and expression levels was performed separately. Based on the median risk scores, we categorised patients with EC into high-risk and low-risk groups, and the overall survival rate of the high-risk group was lower than that of the low-risk group. Combining the age, pathological grading, FIGO stage, and risk score of the cases from the TCGA databases, we constructed a nomogram model and used the ROC curve to test its excellent accuracy and stability to predict the overall survival rate of EC patients. Immunisation scores of high-and low-risk groups were compared according to risk scores as variables, and the association of m6A-related lncRNAs with immune cell types and immune gene mutation loci was analysed using different algorithms.
M6A affects disease development by influencing the expression of target genes, and is involved in almost all processes of mRNA, including shearing, translation, degradation, export, and folding.Citation25,Citation26 An increasing number of studies have shown that long non-coding RNAs are involved in tumorigenesis and progression as primary regulators at the level of the m6A epigenetic modifications.Citation27 Knockdown of ALKBH5 inhibits the malignant biological behaviour of colon cancer cells through NEAT1 demethylation.Citation28 Linc00460 binds to the 3ʹUTR region of HMGA1 through interaction with IGF2BP2 and DHX9 to enhance the stability of HMGA1 mRNA and induce proliferation, invasion, and migration of colon cancer cells.Citation29 In cervical cancer cells, IGF2BP3 enhances the stability of lnRNA KCNMB2-AS1 by binding to it, thereby promoting tumour cell proliferation.Citation30 M6A-modified NEAT1-1 can facilitate the development of bone metastasis in prostate cancer cells. Furthermore, the m6A level of NEAT1-1 can be used as a predictor of death in patients with prostate cancer bone metastases.Citation31 ETTL3 upregulates the expression of LINC00958 by promoting its transcriptional stability, while LINC00958 acts as miR-378a-3p endogenous competing RNA that promotes YY1 and thus breast cancer progression.Citation32 Collectively, these findings suggest that m6A, as a regulator of lncRNA, plays an essential role in the initiation and development of cancers. Hence, we should focus on the interactions between m6A modifications and lncRNAs to provide new insights into the diagnosis and treatment of tumours.
We analysed the prognostic association of six m6A-related lncRNAs with 552 cases of EC, of which three were identified to be highly relevant by multivariate Cox regression analysis. Microarray analysis in cervical cancer revealed that SCARNA9 expression is down-regulated in cancer tissues.Citation33 An analysis demonstrated that SCARNA9, an autophagy gene-related lncRNA, is associated with the prognosis of EC.Citation34 In glioma cells, TRAF3IP2-AS1 was identified using the lncRNA-mediated ceRNA network as one of the pivotal genes that may influence glioma initiation and development through signalling pathways such as cytokine-cytokine interactions and mTOR.Citation35 In bladder cancer, autophagy gene-associated lncRNA Z83843.1 is tightly connected with the overall survival of patients and can be considered as an independent prognostic predictor.Citation36 Moreover, a study showed that in lung adenocarcinoma, Z83843.1 was intimately associated with the metastasis of cancer cells.Citation37 Among the six lncRNAs analysed in this study, some have been reported to be associated with tumours, however, few have been reported in EC, and almost none were associated with m6A. Therefore, our bioinformatics analysis helps us to identify which m6A-related lncRNAs can be used as prognostic, immune microenvironmental targets in EC, and provide new insights for subsequent studies of tumour progression in EC.
However, in this study, we used the information of EC cases from the TCGA database and clinical data to analyse and validate them using different analytical methods. Nevertheless, some limitations exist in this study, as this work is based solely on bioinformatics analysis, and the interaction between these lncRNAs and m6A was not validated in vivo and in vitro.
Conclusion
The above analyses revealed that these lncRNAs were involved in endometriogenesis development through the regulation of m6A-related enzymes. Therefore, these lncRNAs may play a key role in the diagnosis and treatment of endometrial cancer as targets of prognosis and immune microenvironment.
Data Sharing Statement
We obtained RNA-seq data from TCGA (http://cancergenome.nih.gov/) and clinical information from the UCSC Xena (https://xenabrowser.net). All gene sets were downloaded from GSEA (http://software.broadinstitute.org/gsea/index.jsp).
Ethics Approval and Consent to Participate
Informed consent for all patients was approved by the Ethics Committee of Tongji Medical College of Huazhong University of Science and Technology (No.2021-S046), and the research was supported by the Union Hospital affiliated to Tongji Medical College of Huazhong University of Science and Technology.
Author Contributions
Rui Shi conceived the completed experiments and wrote the paper, Ziwei Wang and Jun Zhang contributed to the data collation, Zhicheng Yu, Lanfen An, and Sitian Wei participated in reading and commenting on the paper, and Dilu Feng and Hongbo Wang designed and supervised the paper. All authors read and approved the final manuscript. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Acknowledgment
The authors are very grateful for the valuable data provided by GSEA and TCGA databases. Furthermore, the authors are grateful for the financial support of the instructor and the technical and material support of the laboratory staff.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Funding
References
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
- Faber MT, Sperling CD, Bennetsen AKK, Aalborg GL, Kjaer SKA. Danish nationwide study of risk factors associated with Type I and Type II endometrial cancer. Gynecol Oncol. 2021;161(2):553–558. doi:10.1016/j.ygyno.2021.02.010
- O’Flynn H, Ryan NAJ, Narine N, Shelton D, Rana D, Crosbie EJ. Diagnostic accuracy of cytology for the detection of endometrial cancer in urine and vaginal samples. Nat Commun. 2021;12(1):952. doi:10.1038/s41467-021-21257-6
- Jiang X, Liu B, Nie Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6(1):74.
- Huang J, Chen Z, Chen X, Chen J, Cheng Z, Wang Z. The role of RNA N (6)-methyladenosine methyltransferase in cancers. Mol Ther Nucleic Acids. 2021;23:887–896. doi:10.1016/j.omtn.2020.12.021
- Zhang L, Wan Y, Zhang Z, et al. IGF2BP1 overexpression stabilizes PEG10 mRNA in an m6A-dependent manner and promotes endometrial cancer progression. Theranostics. 2021;11(3):1100–1114. doi:10.7150/thno.49345
- Zhang L, Wan Y, Zhang Z, et al. FTO demethylates m6A modifications in HOXB13 mRNA and promotes endometrial cancer metastasis by activating the WNT signalling pathway. RNA Biol. 2020;1–14.
- Chang G, Shi L, Ye Y, et al. YTHDF3 Induces the Translation of m(6)A-Enriched Gene Transcripts to Promote Breast Cancer Brain Metastasis. Cancer Cell. 2020;38(6):857–71 e7. doi:10.1016/j.ccell.2020.10.004
- Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–641. doi:10.1016/j.cell.2009.02.006
- Maass PG, Luft FC, Bahring S. Long non-coding RNA in health and disease. J Mol Med. 2014;92(4):337–346. doi:10.1007/s00109-014-1131-8
- Wang H, Hu X, Huang M, et al. Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat Commun. 2019;10(1):1898. doi:10.1038/s41467-019-09903-6
- Han D, Liu J, Chen C, et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019;566(7743):270–274. doi:10.1038/s41586-019-0916-x
- Thomas D, Radhakrishnan P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer. 2019;18(1):14. doi:10.1186/s12943-018-0927-5
- Wormann SM, Diakopoulos KN, Lesina M, Algul H. The immune network in pancreatic cancer development and progression. Oncogene. 2014;33(23):2956–2967. doi:10.1038/onc.2013.257
- Huang H, Zhou W, Chen R, Xiang B, Zhou S, Lan L. CXCL10 is a Tumor Microenvironment and Immune Infiltration Related Prognostic Biomarker in Pancreatic Adenocarcinoma. Front Mol Biosci. 2021;8:611508. doi:10.3389/fmolb.2021.611508
- Chen F, Zhuang X, Lin L, et al. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med. 2015;13:45. doi:10.1186/s12916-015-0278-7
- Park JH, van Wyk H, Roxburgh CSD, Horgan PG, Edwards J, McMillan DC. Tumour invasiveness, the local and systemic environment and the basis of staging systems in colorectal cancer. Br J Cancer. 2017;116(11):1444–1450. doi:10.1038/bjc.2017.108
- Zhao Y, Yu Z, Ma R, et al. lncRNA-Xist/miR-101-3p/KLF6/C/EBPalpha axis promotes TAM polarization to regulate cancer cell proliferation and migration. Mol Ther Nucleic Acids. 2021;23:536–551. doi:10.1016/j.omtn.2020.12.005
- Agarwal S, Vierbuchen T, Ghosh S, et al. The long non-coding RNA LUCAT1 is a negative feedback regulator of interferon responses in humans. Nat Commun. 2020;11(1):6348. doi:10.1038/s41467-020-20165-5
- Mi X, Xu R, Hong S, Xu T, Zhang W, Liu M. M2 Macrophage-Derived Exosomal lncRNA AFAP1-AS1 and MicroRNA-26a Affect Cell Migration and Metastasis in Esophageal Cancer. Mol Ther Nucleic Acids. 2020;22:779–790. doi:10.1016/j.omtn.2020.09.035
- Chang Y, Wang X, Xu Y, et al. Comprehensive characterization of cancer-testis genes in testicular germ cell tumor. Cancer Med. 2019;8(7):3511–3519. doi:10.1002/cam4.2223
- Ding B, Lou W, Xu L, Li R, Fan W. Analysis the prognostic values of solute carrier (SLC) family 39 genes in gastric cancer. Am J Transl Res. 2019;11(1):486–498.
- Nasif D, Campoy E, Laurito S, et al. Epigenetic regulation of ID4 in breast cancer: tumor suppressor or oncogene? Clin Epigenetics. 2018;10(1):111. doi:10.1186/s13148-018-0542-8
- Yang B, Shen J, Xu L, et al. Genome-Wide Identification of a Novel Eight-lncRNA Signature to Improve Prognostic Prediction in Head and Neck Squamous Cell Carcinoma. Front Oncol. 2019;9:898. doi:10.3389/fonc.2019.00898
- Liu Q, Gregory RI. RNAmod: an integrated system for the annotation of mRNA modifications. Nucleic Acids Res. 2019;47(W1):W548–W55. doi:10.1093/nar/gkz479
- Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45(10):6051–6063. doi:10.1093/nar/gkx141
- Sun T, Wu Z, Wang X, et al. LNC942 promoting METTL14-mediated m(6)A methylation in breast cancer cell proliferation and progression. Oncogene. 2020;39(31):5358–5372. doi:10.1038/s41388-020-1338-9
- Guo T, Liu DF, Peng SH, Xu AM. ALKBH5 promotes colon cancer progression by decreasing methylation of the lncRNA NEAT1. Am J Transl Res. 2020;12(8):4542–4549.
- Hou P, Meng S, Li M, et al. LINC00460/DHX9/IGF2BP2 complex promotes colorectal cancer proliferation and metastasis by mediating HMGA1 mRNA stability depending on m6A modification. J Exp Clin Cancer Res. 2021;40(1):52. doi:10.1186/s13046-021-01857-2
- Zhang Y, Wang D, Wu D, Zhang D, Sun M. Long Noncoding RNA KCNMB2-AS1 Stabilized by N(6)-Methyladenosine Modification Promotes Cervical Cancer Growth Through Acting as a Competing Endogenous RNA. Cell Transplant. 2020;29:963689720964382. doi:10.1177/0963689720964382
- Wen S, Wei Y, Zen C, Xiong W, Niu Y, Zhao Y. Long non-coding RNA NEAT1 promotes bone metastasis of prostate cancer through N6-methyladenosine. Mol Cancer. 2020;19(1):171. doi:10.1186/s12943-020-01293-4
- Rong D, Dong Q, Qu H, et al. m(6)A-induced LINC00958 promotes breast cancer tumorigenesis via the miR-378a-3p/YY1 axis. Cell Death Discov. 2021;7(1):27. doi:10.1038/s41420-020-00382-z
- Roychowdhury A, Samadder S, Das P, et al. Deregulation of H19 is associated with cervical carcinoma. Genomics. 2020;112(1):961–970. doi:10.1016/j.ygeno.2019.06.012
- Wang Z, Zhang J, Liu Y, Zhao R, Zhou X, Wang H. An Integrated Autophagy-Related Long Noncoding RNA Signature as a Prognostic Biomarker for Human Endometrial Cancer: a Bioinformatics-Based Approach. Biomed Res Int. 2020;2020:5717498. doi:10.1155/2020/5717498
- Zan XY, Li L. Construction of lncRNA-mediated ceRNA network to reveal clinically relevant lncRNA biomarkers in glioblastomas. Oncol Lett. 2019;17(5):4369–4374.
- Sun Z, Jing C, Xiao C, Li T. An autophagy-related long non-coding RNA prognostic signature accurately predicts survival outcomes in bladder urothelial carcinoma patients. Aging. 2020;12(15):15624–15637. doi:10.18632/aging.103718
- Yao X, Zhang H, Tang S, Zheng X, Jiang L. Bioinformatics Analysis to Reveal Potential Differentially Expressed Long Non-Coding RNAs and Genes Associated with Tumour Metastasis in Lung Adenocarcinoma. Onco Targets Ther. 2020;13:3197–3207. doi:10.2147/OTT.S242745