
Abstract
Aim
The aim of the present study was to identify the association between tumor grade and liquid–liquid phase separation (LLPS)-related genes, and to generate a LLPS-related gene-based risk index (LLPSRI) as a prognostic tool for hepatocellular carcinoma (HCC).
Methods
Weighted gene correlation network analysis was performed to test whether the LLPS-related gene modules were associated with tumor grade of HCC. The candidate modules were subjected to functional enrichment analysis. We generated a LLPSRI using the expression profiles of the hub genes among the candidate modules in order to identify patients at high risk. Then, the biological characteristics of the high-risk patients were revealed using gene set enrichment analysis. Additionally, an independent external data set was used to validate the LLPSRI.
Results
Four gene modules showed a significant positive correlation with tumor grade and involved various cancer-related pathways. Among the hub genes, six were selected to generate the LLPSRI, which was significantly associated with prognosis of HCC patients. The LLPSRI could successfully divide patients with HCC into high- and low-risk groups, and patients in the high-risk group showed shorter overall survival than those in the low-risk group. E2F, MYC, and mTORC1 signaling may be important determinants of survival in the high-risk group. The prognostic value of the LLPSRI was validated with the independent external data set.
Conclusion
We identified LLPS-related gene modules that are associated with HCC tumor grade. The LLPSRI may be useful as a prognostic marker of HCC, and it may reliably stratify patients into groups at low or high risk of worse survival. Our analysis also suggests that certain biological characteristics of HCC may be associated with high risk of worse survival.
Introduction
Hepatocellular carcinoma (HCC) is the most common type of malignant tumor, and it is a global health challenge whose incidence is growing worldwide.Citation1,Citation2 Infection with hepatitis B virus (HBV) or hepatitis C virus (HCV) as well as cirrhosis are considered risk factors for HCC development.Citation3 Our understanding of the pathophysiology of HCC has improved in recent decades, but this knowledge has yet to be translated into clinical practice. Novel concepts are required to reveal the complex mechanisms underlying this disease. In recent years, liquid–liquid phase separation (LLPS) has emerged as a new concept to explain how cells perform their vital functions.Citation4 LLPS refers to the generation of membraneless intracellular condensates where essential processes occur, including chromatin organization, X-chromosome inactivation, transcription, DNA damage response, autophagy, and tumorigenesis.Citation5–Citation7 Aberrant LLPS is associated with various diseases, including cancer.Citation8–Citation11 For example, LLPS underlies the formation of nucleoli, whose morphology is considered an indicator of tumor malignancy and differentiation.Citation12 Numerous genes encoding proteins or RNA have been implicated in LLPS.Citation9 The field of LLPS-mediated regulation of biological processes is still in an early stage, and much more needs to be learned about the molecular mechanisms involved and what they mean for cells and organisms.
At the same time, HCC is a highly heterogeneous disease,Citation3 and certain patients with early-stage disease may be at higher risk of worse survival. A “one-size-fits-all” method to predict patients prognosis is unreliable, so novel prognostic models are urgently needed. Considering that LLPS is associated with HCC development, we hypothesized that the expression pattern of LLPS-related genes may be associated with prognosis of HCC. In the present study, we identified modules of LLPS-related genes that are associated with tumor grade, and from a subset of hub genes we created an index to predict prognosis of patients with HCC.
Materials and Methods
Data Processing
The TCGA-LIHC data set, including RNA sequencing (RNA-seq) expression profiles (displayed as read counts) and clinical information of HCC patients in The Cancer Genome Atlas (https://www.cancer.gov/), were downloaded to perform weighted gene correlation network analysis (WGCNA)Citation13 and to generate an LLPS-related gene-based risk index (LLPSRI) in HCC. The RNA-seq profiles were normalized using the voom method in the limma package.Citation14 To be included in our study, both gene expression profiles and clinical information had to be available, and the tumor had to be HCC. Data for healthy liver tissues, samples from patients with combined HCC and cholangiocarcinoma, and HCC samples of unknown tumor grade were excluded from our analysis. Another processed HCC data set (LIRI-JP) from the JP Project of the International Cancer Genome Consortium (https://dcc.icgc.org/) was downloaded from the HCCDB databaseCitation15 and used to validate the LLPSRI. The LLPS-related genes were downloaded from PhaSepDB (http://db.phasep.pro/).Citation16 The workflow of our study is shown in . Our institution waived the requirement for ethical approval for our study because it retrospectively meta-analyzed data already deposited in public databases.
Figure 1 Workflow of the present study.
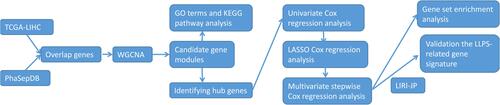
Identification of Modules Associated with Tumor Grade Using WGCNA
A total of 2158 LLPS-related genes were found in the RNA-seq expression profiles of the TCGA-LIHC data set. We extracted the expression profiles of LLPS-related genes in order to perform WGCNA using the WGCNA package. First, hierarchical clustering analysis was performed, and the samples with significantly different gene expression patterns were removed. Then, the soft thresholding power value was screened during module construction. A suitable power value was identified if the degree of independence was > 0.9. The minimum module size was set to 30, and the individual module was assigned a unique color. If a module showed a significant positive correlation with tumor grade (P < 0.05), it was considered a candidate module.
Functional Enrichment Analysis of Candidate Modules
The clusterProfiler packageCitation17 was used to perform functional enrichment analysis to reveal the potential biological functions of candidate modules, based on Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Results were considered significant if they were associated with P < 0.01 after adjustment using the Benjamini & Hochberg method.
Hub Genes and the LLPSRI
In WGCNA, module membership (MM) was defined as the association between a gene and its module. In the present study, a gene with MM > 0.8 was considered a hub gene.Citation18 The expression profiles of the hub genes were used to generate the LLPSRI. Briefly, univariate Cox regression analysis was carried out to identify overall survival (OS)-associated genes. A gene with P < 0.05 was considered significant. The gene expression profiles of the OS-associated genes were used to perform least absolute shrinkage and selection operator (LASSO) Cox regression analysis using the “glmnet” package.Citation19 The features with non-zero coefficients were selected in 10-fold cross-validation for multivariate stepwise Cox regression, and the LLPSRI was defined as
LLPSRI=Exprgene1*Coefgene1+Exprgene2*Coefgene2+Exprgene3*Coefgene3+...
where “Expr” represents the expression value of the candidate genes in the multivariate Cox regression analysis, and “Coef” is the corresponding estimated regression coefficient. Then, patients with HCC were divided into low- or high-risk groups based on the median LLPSRI. OS was compared between the low- and high-risk groups using the log-rank method.
Gene Set Enrichment Analysis (GSEA)
We performed GSEACitation20,Citation21 to explore the biological characteristics of high-risk patients. The hallmark gene set and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway gene sets from the Molecular Signatures Database (version 7.2)Citation20,Citation22 were used as the reference gene set. GSEA was carried out using the GSEA software written in JAVA (http://www.broadinstitute.org/gsea). We set the cutoff criteria as a false discovery rate value < 0.05.
Validation of the LLPSRI
As we did with patients in the TCGA-LIHC dataset, we assigned each patient in the JP-ICGC an LLPSRI according to the above formula. Then, patients with HCC were divided into low- or high-risk groups based on the median PRS. OS was compared between the low- and high-risk groups using the log-rank method.
Results
Multiple LLPS-Related Gene Modules are Associated with HCC Tumor Grade
Four samples of HCC were removed as they showed significantly different expression patterns from the other samples (). In the end, 352 HCC samples were used for WGCNA. The power was 4, which was the lowest value for the scale with an independence degree of up to 0.90 (). WGCNA identified nine modules (), among which the black, turquoise, brown, and yellow modules showed a significant positive correlation with tumor grade and were therefore selected as candidate modules for subsequent analysis (). The green module, in contrast, negatively correlated with tumor grade. Gene significance (GS) was defined by WGCNA as the correlation of a gene with tumor grade. GS and MM correlated significantly with each other in these four candidate modules (-).
Figure 2 Weighted correlation network analysis. (A) Four samples show significantly different expression patterns. (B) Network topologies at different soft-thresholding powers, indicated by the red numbers. (C) Dendrogram of genes clustered based on a dissimilarity measure (1-Topological Overlap Matrix). Each color represents a module in the gene co-expression network constructed by WGCNA. (D) Heatmap of the correlation between module eigengenes and tumor grade. (E–H) Gene significance and module membership correlated significantly with each other in the brown, black, turquoise, and yellow modules.
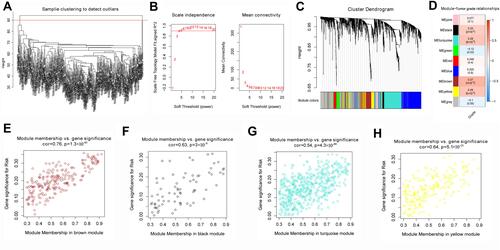
Tumor Grade-Related Gene Modules are Involved in Various Pathways
Analysis of GO indicated the genes of these four candidate modules were involved mainly in GOs related to nucleic acid metabolism (). KEGG pathway enrichment analysis showed that the genes were involved tumor-related pathways, such as the p53 signaling pathway, cell cycle, and FoxO signaling pathway ().
Figure 3 Functional enrichment analysis. (A) Gene Ontology analysis and (B) Kyoto Encyclopedia of Genes and Genomes pathway analysis of the four modules.
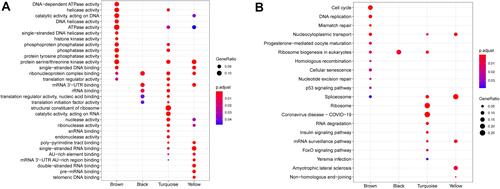
The LLPSRI May Be a Prognostic Indicator in HCC
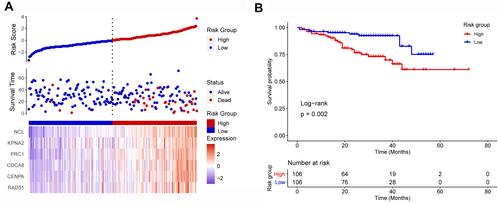
Forty-three LLPS-related genes were considered as OS-associated genes in univariate Cox analysis (), and LASSO analysis showed nine of them to have non-zero coefficients ( and ). A subset of six genes (CDCA8, CENPA, KPNA2, NCL, PRC1, and RAD51) was selected by multivariate stepwise Cox regression to generate the LLPSRI (). The LLPRRI was significantly associated with OS [hazard ratio (HR) 2.718, 95% confidence interval (CI) 2.160–3.420, P < 0.001]. The patients were divided into high- and low-risk groups according to the median LLPSRI (), and patients in the high-risk group had shorter OS than those in the low-risk group (). In fact, the LLPSRI was associated with OS at an even greater significance level than some clinicopathological characteristics, including sex, age, American Joint Committee on Cancer stage, and vascular invasion ().
Table 1 The Liquid-Liquid Phase Separation-Related Overall Survival-Associated Genes and Coefficients
Figure 4 The liquid–liquid phase separation-related gene-based risk index (LLPSRI) in the TCGA-LIHC data set. (A) Nine genes were identified as optimal features in LASSO analysis. (B) Risk factor correlation diagram. Top panel: risk predictions for each patient. The horizontal dotted line represents the median risk value, which distinguished a group at low risk (blue) and a group at high risk (red). Middle panel: patients ranked by predicted risk value, showing slightly longer survival time among patients at low risk than among those at high risk. Blue dots represent living patients; red dots, deceased patients. Bottom panel: expression of candidate genes. (C) The high-risk group showed worse overall survival than the low-risk group. (D) The LLPSRI is a prognostic factor independent of routine clinicopathological characteristics.
Biological Characteristics of Patients in the High-Risk Group
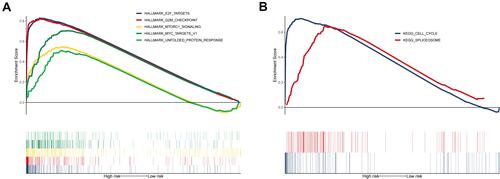
Five hallmark gene sets, hallmark E2F targets, hallmark G2M checkpoint, hallmark MTORC1 signaling, hallmark MYC targets v1, and hallmark unfold protein response, were significantly enriched in the samples of high-risk group (). Two pathways, cell cycle and spliceosome, were significantly enriched in the samples of high-risk group ().
Figure 5 Gene set enrichment analysis. (A) Five hallmark and (B) two Kyoto Encyclopedia of Genes and Genomes gene sets were significantly enriched in the high-risk samples.
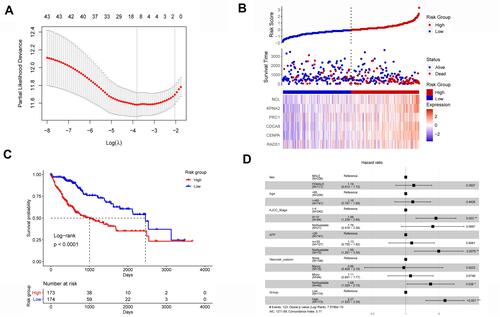
Validation of the LLPSRI Using an Independent Data Set
All HCC patients in the LIRI-JP data set were assigned LLPSRIs using the above formula. The LLPRRI was significantly associated with OS (HR 2.939, 95% CI 1.886–4.578, P < 0.001). As we did with patients in the TCGA-LIHC data set, we divided the LIRI-JP patients into high- and low-risk groups according to the median LLPSRI (). Patients in the high-risk group had shorter OS than those in the low-risk group ().
Figure 6 Validation of the prognostic value of the liquid–liquid phase separation-related gene-based risk index using an external data set. (A) Patients with HCC were divided into high- and low-risk groups, whose risk score, survival and gene expression were compared. (B) The overall survival rate was higher among low-risk patients than among high-risk patients.
Discussion
Many gene-based signatures have been proposed for prognostic stratification of HCC.Citation23–Citation25 Lin et al proposed a twenty gene-based prognostic stratification in HCC.Citation18 Lv et al proposed a seven gene-based risk index for predicting early recurrence in HCC.Citation26 Huang et al reported a RNA binding protein-related prognostic signature for HCC.Citation27 These studies have usually not started from subsets of genes with particular biological significance. The present study, in contrast, started from the newly introduced concept of LLPS as important in health and disease. Focusing on genes related to LLPS, we defined several gene modules that were associated with HCC tumor grade. Our LLPSRI was generated using the hub genes in the modules and was found to be associated with OS. Furthermore, its prognostic value was validated with a second, independent data set. These results suggest that LLPS may be a useful concept for explaining the pathogenesis of HCC. The LLPSRI may provide a valuable reference for the personalized management of HCC patients.
According to our GSEA, the target genes of transcription factors E2F and MYC are significantly enriched in high-risk samples, so the networks regulated by these two transcription factors are likely related to LLPS. Previous studies suggested critical roles for E2F and MYC in hepatocarcinogenesis,Citation28–Citation30 which the present analysis supports in the case of HCC. Our results justify further work to elucidate how E2F and MYC are involved in LLPS. In addition, we found that high-risk patients may show greater mTORC1 signaling, which is considered a major tumor-initiating pathway in HCC.Citation31 This suggests that mTOR inhibitors may benefit our HCC patients in the high-risk group, but not those in the low-risk group. Thus, the LLPSRI may be useful for screening patients for potential response when conducting prospective trials of mTOR inhibitors.
Our LLPSRI consists of six LLPS-related genes, some of which have previously been associated with HCC. Aberrant expression of KPNA2 correlates with early recurrence and poor prognosis in patients with small HCC.Citation32 PRC1 promotes early recurrence of HCC in association with the Wnt/β-catenin signalling pathway.Citation33 Silencing CDCA8 can suppress HCC growth and stemness.Citation34 RAD51 appears to influence the onset, progression and drug resistance of various cancers.Citation35 NCL and CENPA should be explored further to confirm and clarify their involvement in HCC.
The present study may provide new insights into the associations of LLPS with HCC tumor grade and prognosis, but several limitations should be noted. Most importantly, our analysis is based exclusively on bioinformatics, so our findings should be verified and extended in experimental studies, ultimately prospective studies with patients. Second, some of the genes that we identified here have yet to be linked to HCC. Indeed, whether the LLPSRI is linked to prognosis through causality or simple correlation needs to be explored.
Conclusion
We identified LLPS-related gene modules that are associated with HCC tumor grade. The LLPSRI may be useful as a prognostic marker of HCC, and it may reliably stratify patients into groups at low or high risk of worse survival. Our bioinformatic analysis also identified some biological characteristics of HCC that may be linked to worse survival. In these ways, our study generates numerous testable hypothesis to help guide future research into the mechanisms and personalized treatment of HCC.
Data Sharing Statement
The raw analyses from this study can be obtained from the corresponding author upon reasonable request.
Disclosure
Zhao-Shan Fang and Zhi Zhang are co-first authors for this study. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Funding
References
- Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi:10.1038/nrdp.2016.18
- Villanueva A. Hepatocellular Carcinoma. N Engl J Med. 2019;380(15):1450–1462. doi:10.1056/NEJMra1713263
- Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
- Brangwynne CP, Eckmann CR, Courson DS, et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324(5935):1729–1732. doi:10.1126/science.1172046
- Shin Y, Brangwynne CP. Liquid phase condensation in cell physiology and disease. Science. 2017;357(6357). doi:10.1126/science.aaf4382
- Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18(5):285–298. doi:10.1038/nrm.2017.7
- Nozawa RS, Yamamoto T, Takahashi M, et al. Nuclear microenvironment in cancer: control through liquid-liquid phase separation. Cancer Sci. 2020;111(9):3155–3163. doi:10.1111/cas.14551
- Alberti S, Carra S. Quality control of membraneless organelles. J Mol Biol. 2018;430(23):4711–4729. doi:10.1016/j.jmb.2018.05.013
- Aguzzi A, Altmeyer M. Phase separation: linking cellular compartmentalization to disease. Trends Cell Biol. 2016;26(7):547–558. doi:10.1016/j.tcb.2016.03.004
- Alberti S, Hyman AA. Are aberrant phase transitions a driver of cellular aging? Bioessays. 2016;38(10):959–968. doi:10.1002/bies.201600042
- Alberti S, Dormann D. Liquid-liquid phase separation in disease. Annu Rev Genet. 2019;53:171–194. doi:10.1146/annurev-genet-112618-043527
- Shimazui T, Koiso K, Uchiyama Y. Morphometry of nucleoli as an indicator for grade of malignancy of bladder tumors. Virchows Arch B Cell Pathol Incl Mol Pathol. 1990;59(3):179–183. doi:10.1007/BF02899403
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi:10.1186/1471-2105-9-559
- Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
- Lian Q, Wang S, Zhang G, et al. HCCDB: a database of Hepatocellular carcinoma expression atlas. Genomics Proteomics Bioinformatics. 2018;16(4):269–275. doi:10.1016/j.gpb.2018.07.003
- You K, Huang Q, Yu C, et al. PhaSepDB: a database of liquid-liquid phase separation related proteins. Nucleic Acids Res. 2020;48(D1):D354–D359. doi:10.1093/nar/gkz847
- Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
- Lin Y, Liang R, Ye J, et al. A twenty gene-based gene set variation score reflects the pathological progression from cirrhosis to hepatocellular carcinoma. Aging (Albany NY). 2019;11(23):11157–11169. doi:10.18632/aging.102518
- Friedman JH, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate. Descent. 2010;33(1):22. doi:10.18637/jss.v033.i01
- Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
- Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. doi:10.1038/ng1180
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–1740. doi:10.1093/bioinformatics/btr260
- Gu JX, Zhang X, Miao RC, et al. Six-long non-coding RNA signature predicts recurrence-free survival in hepatocellular carcinoma. World J Gastroenterol. 2019;25(2):220–232. doi:10.3748/wjg.v25.i2.220
- Wang K, Chen X, Jin C, et al. A novel immune-related genes prognosis biomarker for hepatocellular carcinoma. Aging (Albany NY). 2020;13(1):675–693. doi:10.18632/aging.202173
- Lei J, Zhang D, Yao C, Ding S, Lu Z. Development of a predictive immune-related gene signature associated with hepatocellular carcinoma patient prognosis. Cancer Control. 2020;27(1):1073274820977114. doi:10.1177/1073274820977114
- Lv Y, Wei W, Huang Z, et al. Long non-coding RNA expression profile can predict early recurrence in hepatocellular carcinoma after curative resection. Hepatol Res. 2018;48(13):1140–1148. doi:10.1111/hepr.13220
- Huang Y, Chen S, Qin W, et al. A novel RNA binding protein-related prognostic signature for Hepatocellular Carcinoma. Front Oncol. 2020;10:580513. doi:10.3389/fonc.2020.580513
- Zhan L, Huang C, Meng XM, et al. Promising roles of mammalian E2Fs in hepatocellular carcinoma. Cell Signal. 2014;26(5):1075–1081. doi:10.1016/j.cellsig.2014.01.008
- Xanthoulis A, Tiniakos DG. E2F transcription factors and digestive system malignancies: how much do we know? World J Gastroenterol. 2013;19(21):3189–3198. doi:10.3748/wjg.v19.i21.3189
- Zimonjic DB, Popescu NC. Role of DLC1 tumor suppressor gene and MYC oncogene in pathogenesis of human hepatocellular carcinoma: potential prospects for combined targeted therapeutics (review). Int J Oncol. 2012;41(2):393–406. doi:10.3892/ijo.2012.1474
- Bhat M, Sonenberg N, Gores GJ. The mTOR pathway in hepatic malignancies. Hepatology. 2013;58(2):810–818. doi:10.1002/hep.26323
- Jiang P, Tang Y, He L, et al. Aberrant expression of nuclear KPNA2 is correlated with early recurrence and poor prognosis in patients with small hepatocellular carcinoma after hepatectomy. Med Oncol. 2014;31(8):131. doi:10.1007/s12032-014-0131-4
- Chen J, Rajasekaran M, Xia H, et al. The microtubule-associated protein PRC1 promotes early recurrence of hepatocellular carcinoma in association with the Wnt/beta-catenin signalling pathway. Gut. 2016;65(9):1522–1534. doi:10.1136/gutjnl-2015-310625
- Jeon T, Ko MJ, Seo YR, et al. Silencing CDCA8 suppresses Hepatocellular carcinoma growth and stemness via restoration of ATF3 tumor suppressor and inactivation of AKT/beta-catenin signaling. Cancers (Basel). 2021;13:5. doi:10.3390/cancers13051055
- Laurini E, Marson D, Fermeglia A, Aulic S, Fermeglia M, Pricl S. Role of Rad51 and DNA repair in cancer: a molecular perspective. Pharmacol Ther. 2020;208:107492. doi:10.1016/j.pharmthera.2020.107492