 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Purpose
The purpose of the study was to develop and evaluate different lipid-based formulations for parenteral administration, as potential novel carrier systems for lipophilic drugs, and to turn an unstable drug such as chlorambucil into a useful one.
Methods
A two-stage, high-pressure homogenizer was used to yield a very fine monodispersed lipid nanosphere. The strategy of combining egg yolk phospholipid and nonionic emulsifier (Lutrol F 68 and Tween 80) as an emulsifier mixture was adopted to increase safety and tolerance. The final lipid nanospheres, in a lipophilic mixture consisting of three components, monostearin, medium-chain triglycerides and soya oil, were evaluated for physicochemical properties, such as particle size, surface morphology, drug-entrapment efficiency, drug-loading capacity, lyophilization and in vivo drug-release behavior.
Results
A monodispersed lipid nanosphere with a mean particle size ranging from 90 to 150 nm was achieved. The optimized injectable cryoprotectants for lipid nanosphere were sucrose (7.5%) and mannitol (7.5%), which can stabilize the particle size (LD50) at approximately 129 nm after reconstitution. The results show that the formulation can effectively administer anticancer drugs and thus improve patient quality of life.
Conclusions
The novel lipid nanosphere complex developed is a useful anticancer drug delivery vehicle for parenteral administration. The formulation strategy has the potential for the development of further methods of drug delivery for a wide variety of anticancer drugs.
Introduction
Inhibition of tumor-associated angiogenesis has become one of the most promising developments in cancer treatment, which has resulted in the evaluation of a large number of angiogenesis inhibitors in clinical trials.Citation1 Chlorambucil (CHL), a nitrogen mustard, is the primary treatment for patients with chronic lymphocytic leukemia (CLL) and is used in the pharmacotherapy of lymphomas, and advanced ovarian and breast carcinomas.Citation2 It contains a nitrogen mustard group that transfers alkyl groups to amino, carboxyl, sulfhydryl and phosphate moieties ().Citation3 In the early development programs, it became apparent that nitrogen mustards were the most effective antitumor agents, probably through their ability to form interstrand cross-links in DNA, and also to form adducts with RNA and protein. It was also shown that alkylating cytostatic drugs influence the regulation of angiogenesis, already in nontoxic concentrations,Citation4 and lead to a reduction in endothelial precursors in blood after chemotherapy.Citation5 In this study, CHL was chosen as a model drug, because of the need to find new formulations to administer the drug more effectively and thus provide a useful lipid-based formulation for parenteral administration.
Figure 1 Chemical structure of chlorambucil.
Over the last two decades, emulsions,Citation6 liposomes,Citation7 solid lipid nanoparticles (SLN), nanostructured lipid carriers,Citation8–Citation11 and lipid nanospheres (LNS)Citation12 have been developed and used as parenteral drug delivery carriers. These various lipid carriers can enhance the efficacy and reduce the toxicity of antitumor drugs,Citation13 and lipid carriers are receiving an increasing amount of attention as drug delivery systems for cancer chemotherapy. However, the development of intravenous lipid carriers is a very challenging task, and problems such as drug entrapment, crystallization, solubility and chemical degradation are still difficult to overcome, especially degradation of unstable antitumor drugs caused by high-temperature sterilization or hydrolysis. In this study, a novel injectable LNS formulation system was developed based on three main production principles, to solve many of the production issues involved in turning an unstable anticancer agent into an effective drug. First, the particle diameter and cumulative distribution were maintained at less than 200 nm to meet the filtration sterilization requirements. This diameter is essential for anticancer drugs because most anticancer drugs are susceptible to heating-induced decomposition. Second, a biocompatible surfactant mixture (Lutrol F 68 and Tween 80) was used for intravenous injection. The use of this combination may have the beneficial effect of reducing drug formulation toxicity by decreasing the content of each surfactant. Third, high-quality, freeze-dried characteristics of the anticancer drug-loaded LNS were obtained by replacing some of the liquid oils with the safest solid lipid, monostearin, in the formulation. We determined whether the use of LNS-containing complex oils and a mixture of emulsifiers could efficiently deliver anticancer drugs for intravenous administration. This study aimed to evaluate different lipid-based nanospheres that were formulated by combining medium-chain triglycerides/long-chain triglycerides (MCT/LCT) and monostearin as a lipophilic mixture, and using egg yolk phospholipid and nonionic emulsifiers (Lutrol F 68 and Tween 80) as an emulsifier mixture. It was expected that this novel delivery carrier would give a smaller particle size and a much safer formulation strategy for parenteral administration than currently available delivery systems.
Materials and methods
Materials
Egg lecithin 80 was purchased from Lipoid Corp. (Lipoid K G, Germany). Poloxamer 188 (Lutrol F 68) was supplied by BASF (Ludwigshafen, Germany). Tween 80 for parenteral use was purchased from Shenyu Medicine and Chemical Industry Corp. (Shanghai, China). Soybean oil (LCT) for parenteral use was a gift from Beiya Corp. (Tieling, China). Miglyol® 812 (caprylic/capric triglycerides, MCT) was obtained from Huls (Witten/Ruhr, Germany). Monostearin was purchased from Shanghai Chemical Reagent Corp. (Shanghai, China). PEG-40 was purchased from Sigma (St. Louis, MO). Chlorambucil (purity 98.8%) was purchased from Beijing Lunarsun Pharmaceutical Corp. (Beijing, China).
Solubility study of chlorambucil
The solubility of drugs in the lipophilic mixture (soybean oil and MCT) was determined. Samples were prepared by adding an excess amount of drugs to 1 mL of lipophilic mixture and shaking reciprocally on a shaking water bath at 37 ± 0.5°C for 24 hours. After centrifugation at 12,000 rpm for 20 minutes, the supernatant was diluted with ethanol and assayed by reversed phase high-performance liquid chromatography (RP-HPLC).
Drugs were analyzed according to the method of Ganta et alCitation14 with modification. The HPLC system consisted of a 200 mm × 4.6 mm RP 18 BonChrom column packed with 5-μm particles (Agela Tech, Newark, DE), a Shimadzu LC-10AT pump and a Shimadzu SPD-10A ultraviolet detector (Shimadzu, Kyoto, Japan). The mobile phase consisted of methanol and a 0.2% (v/v) acetic acid solution (80:20, v/v), the injection volume was 20 μL, the detection was set at 258 nm at a flow rate of 1.0 mL·min−1, and the column was set at 30°C.
Formulation and preparation of lipid nanospheres
High-pressure homogenization, which is a reliable and powerful technique for industrial scale-up, was used to prepare fine LNS. The lipophilic mixture differed in composition, with monostearin and various amounts of soybean oil or MCT or binary liquid–oil mixtures () while maintaining a constant final concentration of lipid. The mixture was heated at 55°C. Then, drugs (0.7 mg) were added to the lipophilic mixtures and dispersed by magnetic stirring. The aqueous phase was prepared by dissolving a mixture of emulsifiers with different ratios of Lutrol F 68, Tween 80 and E80 () in distilled water under magnetic stirring. Aqueous phases were heated separately to 60°C, and then the hot water and surfactants were poured into the hot lipid phase and mixed using a magnetic stirrer. Next, 30 mL of the coarse dispersion was passed through a high-pressure homogenizer with a two-stage homogenizing valve assembly (ATS, AH100D) at a pressure of 700 bars for 10 cycles, and, finally, the LNS were obtained (pH ~7.0).
Table 1 Formulation components of the lipophilic mixture of the final lipid nanospheres
Table 2 Formulation components of the surfactant mixture of the final lipid nanospheres
The following formulations were optimized as a whole, with different ratios of lipophilic mixtures and surfactant mixtures. The formulation and production process was optimized according to Bock et al.Citation15
Drug entrapment efficiency determination and loading capacity
The total LNS drug content (Winitial drug) and free-drug content (Wfree drug) in the final suspension were determined by HPLC for encapsulation.
Free drugs were separated using an ultrafiltration method. The LNS were diluted with 0.2% (v/v) acetic acid approximately 10 times, instead of the mobile phase, to avoid dissolving LNS. Based on a previous report,Citation16 the diluted sample was placed in the outer chamber and then ultrafiltrated at 4000 rpm for 10 min using an Amicon Ultra 10K device (MW 10000 Amicon Ultra centrifugal filter devices with low-binding Ultracel@ membranes); the ultrafiltrate was assayed by HPLC after diluting with the mobile phase, and then the free drug content was determined.
A total of 1 mL of LNS was dissolved in 25 mL of methanol under a water bath at 50°C for 2 minutes. Then, the sample was diluted 20 times with the mobile phase, 20 μL were subsequently injected into HPLC for analysis, and, finally, the total drug content in the LNS suspension was calculated. All sample analyses were performed within 12 hours. Entrapment efficiency (EE%) was determined by the following equation:
Addition acetic acid was added to lipophilic mixtures, adjusting the pH to 3.0 against drug hydrolysis during the preparation process of the optimized LNS formulation. After adding acetic acid to the oil phase, a high CHL entrapment efficiency (>99.0%) was achieved.
LNS with a moderate loading capacity of 0.7 mg·mL−1 were produced and could be dissolved in liquid–lipophilic mixtures (MCT and LCT) according to solubility studies. The other materials played a significant role in the drug solubilization in LNS, including the presence of phospholipids and surfactants. It was possible to incorporate higher concentrations of drugs into the LNS than the original drug load according to solubility studies, and LNS with CHL concentrations of 1.0 and 1.5 mg·mL−1 were produced.
Particle size distribution
Laser diffraction (LD) was used to determine the volume average diameter (Nicomp PSS ZW 380, Santa Barbara, CA) for the fresh and reconstituted lipid nanospheres. Prior to measurement, lyophilized LNS were reconstituted with filtered water. In this work, D (v,10), D (v,50) and D (v,90) were used, representing diameters at 10%, 50%, and 90% cumulative volume, respectively, and D (v,50) is the median diameter.
Transmission electron microscopy (TEM)
A TEM instrument (TEM-1200EX; JEOL, Tokyo Japan) was used to examine the morphology of the optimized LNS. A drop of drug-loaded LNS was placed on the copper grids to form a film, followed by negative staining with a drop of 2% (w/v) phosphotungstic acid. The air-dried sample was then viewed by TEM.
Differential scanning calorimetry (DSC)
DSC was performed to determine the phase transition of optimized LNS complex. About 25 mg of sample were weighed into standard aluminium pans using an empty pan as reference. The samples were heated from 0 to 90°C at a rate of 10°C·min−1 (DSC-60; Shimadzu Corp.).
Freeze-drying of lipid nanospheres
As acetic acid was used to prevent drug hydrolysis during formulation preparation, the optimized LNS were readjusted to neutral pH with 0.1 M sodium hydroxide before lyophilization. To optimize the cryoprotectants for the LNS dispersion during the freeze-drying process, mannite, lactose, glucose, sorbitol, trehalose and mannose were chosen for intravenous application. Various amounts of cryoprotectants were added to the LNS dispersions, each sample was pre-cooled at −70°C in a deep-freeze for 4 hours, and then freeze-drying was carried out in a refrigerator (Eyela FDU-110; Tokyo, Rikakikai Co, Ltd). Primary drying was performed at a shelf temperature of −40°C for 14 hours, followed by a secondary drying step at a shelf temperature of 10°C for 2 hours; finally, the powdered LNS were obtained. The lyophilized products were reconstituted in distilled water by manual shaking and then sonication for 1 minute using an ultrasonic water-bath sonicator (KQ-100DB; Kunshan Co. Ltd., China).
In vivo studies
The experiments were carried out using Kunming female mice (28–32 g). Two groups of 36 mice were used to evaluate the pharmacokinetics of CHL-LNS, and, for comparison, CHL injections were prepared based on a reported method.Citation17 The mice were purchased from Shenyang Pharmaceutical University’s animal center. Three mice were used at each time point. After the intravenous administration of 10 mg·kg−1 of drug, blood samples were obtained from the retro-orbital sinus at 5, 15, 30, 45, 60 and 90 minutes. The blood was instantly centrifuged at 6000 rpm for 5 minutes. Blood sample treatment was performed by adding 0.3 mL (2 μg·mL−1 internal standard) of methanol-acetonitrile (50:50, v/v) to 0.1 mL of plasma. A tissue distribution study was carried out at the same time for 15, 45, 60, and 90 minutes, respectively. Tissues (heart, liver, spleen, lung, and kidney) were cut and rinsed with cold saline with 0.2% (v/v) acetic acid. Then, they were homogenized with 4 volumes of methanol-acetonitrile (50:50, v/v) with a 2 μg·mL−1 internal standard. Protein precipitation of all plasma and tissue samples was carried out after vortexing for 5 minutes. The samples were kept on ice for 30 minutes and then centrifuged at 6000 rpm for 5 minutes to precipitate the proteins; all samples were maintained at −20°C until 20 μL of the supernatant was injected into the HPLC for analysis.
The separation was carried out with the mobile phase consisting of 0.03 M KH2PO4:MEOH = 30:70 (v/v, pH 3.0, adjusted with 0.5 M H3PO4) at a flow rate of 1.0 mL·min−1. Chromatograms were monitored at 258 nm, and the temperature of the column was kept at 30°C. The selected internal standard is (2-chloro-N-[[(2-chlorophenyl)amino]carbonyl]-2-chloro-N-(2-chlorophenylcarbamoyl)acetamide).
The study was approved by the University Animal Ethics Committee and complied with the Animal Ethics Committee Guidelines.
Results and discussion
Solubility study
The solubilities of CHL in soybean oil and MCT were 36.4 and 78.2 mg·mL−1 at 37°C, respectively. LCT, MCT or a combination of both was used as the lipophilic mixture for the preparation of the LNS, as it is important to incorporate the drug into the inner phase of the LNS.
Because of the solid state of monostearin, it was very difficult to determine the real solubility of CHL under normal temperatures; therefore, the whole-drug loading capacities in LNS with increasing drug concentrations of 0.7, 1.0, and 1.5 mg·mL−1 were determined as described in .
Table 5 Drug loading amount and entrapment efficiency (EE%) of the optimized lipid nanospheres (formulation composed of lipophilic mixture 2: MCT: 10 mg·mL−1, LCT: 10 mg·mL−1, monostearin 40: mg·mL−1, and surfactant mixture 1: E80: 20 mg·mL−1, Tween 80: 30 mg·mL−1, Lutrol F 68:10 mg·mL−1)
Development of formulations and drug-loading capacity
The aim of this formulation study was to develop a parenteral lipid LNS system with a low toxicity and a high anticancer-drug loading capacity and to offer an appealing alternative to the intravenous administration of CHL and other drugs with poor stability. The LNS system is based on the rational selection of individual components and composition ratios by using a complex formulation composition that presents a less toxic and more biocompatible formulation strategy and leads to small droplet sizes that are suitable for sterilization by filtration.
Since 1984, MCT/LCT emulsions have been in clinical use, and they are more rapidly available to tissues than LCT emulsions and show beneficial effects on the immune system,Citation18 such as allowing the drug to interact with water-soluble proteins (eg, lipases) for more efficient in vivo hydrolysis and reducing the toxicity associated with pure LCT-based lipid emulsions.Citation19 So, the oil composition of the lipid nanospheres, was investigated for the production of different LNS formulations. shows three different types of LNS formulations with different particle sizes, which could promote the incorporation of the lipophilic drug and subsequently decrease the injected volume. The formulations with MCT or LCT/MCT had smaller mean particle sizes, and specifically, the MCT/LCT formulations resulted in the smallest mean particle sizes. These results indicate that different oil components might lead to formulations with different structures and might further influence the interfacial tension and viscosity of the LNS system.
Table 3 Effect of different lipophilic mixtures on lipid nanospheres droplet size and entrapment efficiency (EE%); aqueous phase composed of surfactants 1 (E80: 20 mg·mL−1, Tween 80: 30 mg·mL−1, Lutrol F 68: 10 mg·mL−1), and 0.7 mg drugs added per mL of lipid nanospheres
Additionally, 2% (w/v) E80 was selected to avoid LNS gelatinization. In order to formulate a nano-sized lipid drug carrier that would be acceptable for intravenous administration and would achieve a maximum drug-stabilization effect by forming a high-energy barrier at the oil–water interface,Citation20 additional stabilizing surfactant combinations (Lutrol F 68 and Tween 80) were used. The percentage of the total surfactant combination amount was up to 6% (w/v), which was necessary to cover the total interfacial oil–water surface during the LNS preparations. The three surfactants were used to reduce the toxicity of each surfactant. shows the average particle size with the change of surfactant combination. Therefore, different LNS were obtained and compared by selecting system surfactant combinations of Tween 80, Lutrol F 68 and phospholipids with different hydrophile–lipophile balance (HLB) values, which were about 15, 29 and 8 (10), respectively. Furthermore, the use of MCT and Tween 80 in this complex LNS formulation facilitates particle size reduction and filter-sterilization through a 0.22-μm membrane. The optimized intravenous LNS formulation (MCT: 10 mg·mL−1, LCT: 10 mg·mL−1, monostearin 40: mg·mL−1, E80: 20 mg·mL−1, Tween 80:30 mg·mL−1, Lutrol F 68:10 mg·mL−1) showed a very narrow distribution range from 90–150 nm in diameter with a polydispersity index of 0.017.
Table 4 Effect of composition of surfactants on lipid nanospheres droplet size and entrapment efficiency (EE%); lipid mixtures composed of lipophilic mixture 2 (MCT: 10 mg·mL−1, LCT: 10 mg·mL−1, monostearin 40: mg·mL−1), 0.7 mg drugs added for per mL of lipid nanospheres
The results of three different batches of the formulation are presented individually in , in which the drug incorporation and loading capacity can be compared for the amount of added drug.
From the results, we can conclude that acetic acid was effective in minimizing drug hydrolysis and was essential for a high drug-encapsulation efficiency. CHL was also easily dissolved in the oil phase by adding acetic acid to the oil phase. However, when drug loading increased from 0.7 to 1.5 mg·mL−1, a decreased drug-entrapment efficiency trend was observed; thus, 0.7 mg·mL−1 was deemed to be an appropriate drug loading content.
Freeze-drying
The reconstitution of lyophilized LNS is one of the most important factors in the whole process of the preparation and practical application of nanospheres. For long-term storage considerations, aqueous suspensions of LNS are essential for lyophilization and must be reconstituted into physiological suspensions as the original aqueous suspensions before use.
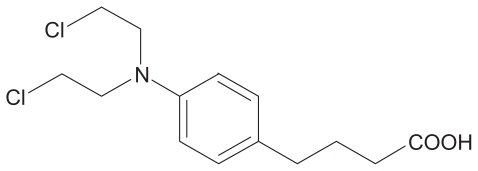
The protective effect of cryoprotectants (15%, w/v) on the physical properties and redispersion of lyophilized LNS was investigated. shows that these formulations had relatively good redispersion rates. The optimized combination of cryoprotectants was 7.5% sucrose and 7.5% mannitol. Sucrose can inhibit mannitol crystallization of the LNS to achieve elegant lyophilized products.Citation21 Such excipients can protect the lipid matrix more efficiently during the freeze-drying process. The particle size and range of distribution of the lyophilized LNS after reconstitution are shown in . Three distribution peaks are shown, indicating that a new distinct population of particles had formed. Freeze-drying is a complex drying process during the conversion of LNS solutions into solid materials, and it leads to particle growth and aggregation. The heterogeneous distribution of freeze-dried LNS mainly depended on the composition of this distinct formulation, such as the effects of monostearin, Tween 80 and phospholipid. The presence of Tween 80 led to a large size-distribution during the freeze-drying process. This finding was also reported by Lim and Kim,Citation22 where lyophilized SLN with 54:46, 100:0 or 0:100 weight ratios of egg phosphatidylcholine: Tween 80 for the controlled release of all-trans-retinoic acid resulted in mean particle sizes of SLN of 329.5, 440.3 and 1459.7 nm, respectively. Zimmermann et alCitation23 reported that liposomes form after reconstitution, which may explain why LNS size aggregation occurred in this present study.
Figure 2 Mean particle size diameters of formulations after reconstitution with an optimized combination of cryoprotectants.
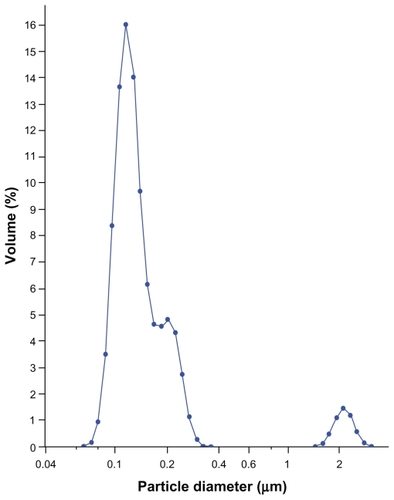
Table 6 Effects of cryoprotectants on morphological characteristics of reconstituted lipid nanospheres from freeze-dried suspension (cryoprotectant concentration %)
Because the formulation effect by which excipients preserve the native structure of LNS during freeze-drying is incompletely understood, the optimization of intravenous LNS remains a challenge. Therefore, to better understand the effect of LNS components on particle size after reconstitution, two types of LNS formulations were prepared and lyophilized, with a MCT:LCT:monostearin mixture used as the oil. One was a PEG-40 formulation in which we replaced Tween 80 in optimized LNS with PEG-40; after reconstitution, the particle size distribution values of the PEG-40 formulation were 87.7 nm (D10), 114 nm (D50) and 1422 nm (D90), while the particle size distribution values of the Tween 80 formulation were 98.4 nm (D10), 129 nm (D50) and 231 nm (D90), respectively. The D50 of the Tween 80 formulation was slightly increased, but it still had a relatively homogeneous particle size distribution. The final results showed that the Tween 80 component could improve the particle size distribution.
As all the LNS particles were smaller than 3 μm, its parenteral use by intravenous injection is acceptable.Citation24 No droplets should be larger than 5 μm to avoid possible embolism in the lung capillaries. We would expect that in clinical use the LNS would be administered through a 0.45-μm needle-filtrate for safety considerations.
Transmission electron microscopy
TEM was used to investigate the morphology of this transparent LNS system to characterize the LNS after high-pressure homogenization. The final LNS had spherical or ellipsoidal particle shapes , with most particle diameters <100 nm; however, the image diameters determined by TEM were smaller than those determined by LD. Drug crystals were invisible, and separated oil droplets were also not visible in the electron micrographs, indicating that the LNS components are not completely separated in the LNS dispersion. The different types of morphologies of lipid nanospheres could be explained by the formation of extra solid lipid nanoparticles and liposomes, hence the term LNS. For special formulations, however, this formulation also could be called a lipid nanocomplex (LNC).
Figure 3 A transmission electron microscopy image of 2% phosphotungstic acid-stained lipid nanospheres (pH adjusted to 7.4 with 0.1 mol/L−1 NaOH after preparation) (×40,000, bar = 200 nm).
DSC investigations
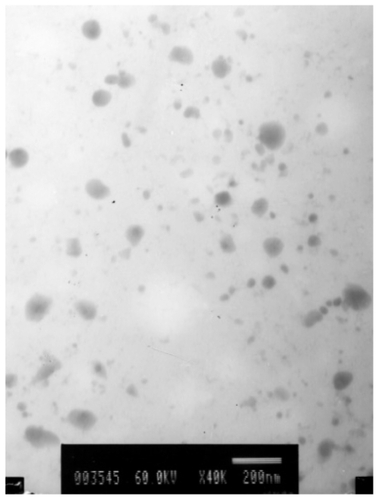
The polymorphic modification of the lipid particle matrix is one of the most important factors affecting drug incorporation. DSC gives information on the melting and recrystallization behavior of the lipophilic mixture. After heating, the crystal of the LNS lattice breaks down. Typical DSC tests of the nanospheres as well as those of blank nanospheres and raw materials are displayed in . The corresponding DSC data show the melting processes of the bulk matrix, lyophilized drug-free LNS and lyophilized drug-loaded LNS powder over a wide melting range. Furthermore, the impurities or less-ordered crystals of LNS that were influenced by the oil mixture of LCT and MCT are also reflected by the heating curve.
Figure 4 Differential scanning calorimetry scan of lyophilized lipid nanospheres (LNS) powder heating from 30°C to 120°C at a rate of 10°C·min−1. 1) The physical mixture material; 2) Placebo LNS; 3) Drug-loaded LNS (containing 0.7 mg·mL−1 of the model drug).
To compare the maximum melting temperatures of the physical mixture material and LNS, the melting peaks of drug (60°C), monostearin (55°C) and Lutrol F 68 (52°C) were all established. The physical mixture’s peak was 49.17°C, and those of drug-free and drug-loaded LNS were 47.07°C and 47.10°C, respectively. The decrease in the temperature peaks of drug-loaded and drug-free LNS according to the physical mixture of their components, could be explained by the fact that a monostearin polymorph might be formed during the previous heating to obtaining the LNS.Citation25 Another possible reason for the decrease in the maximum temperature could be the small-size effect,Citation26 as explained by the Thomson equation.
In vivo studies
A rapid and sensitive RP-HPLC method was developed to determine the CHL levels in rat plasma. A new internal standard substance was selected as an alternative to praziquantel, which was reported by another research group,Citation14 according to the chosen absorption spectrum of CHL at a wavelength of 258 nm to achieve a high selectivity against endogenous compounds in the chromatograms. Compared with a previous study,Citation14 the present method is more suitable, had a relatively simple sample preparation process and eliminated the potential degradation of CHL. A value of 0.2 μg·mL−1 was identified as the lower limit of quantification. The calibration curve was linear in the range of 0.2–20 μg·mL−1. The plasma concentration–time curves of CHL in rats after intravenous administration of 10 mg·kg−1 CHL-LNS or injection are shown in The main pharmacokinetic parameters for each formulation were calculated using Drug and Statistics (DAS 2.1.1; Mathematical Pharmacology Professional Committee of China, Shanghai, China), and the data on both curves fit a two-compartment model, with the parameters shown in . Statistical analysis of concentration–time curve (AUC), clearance, and steady-state apparent volume of distribution of CHL after infusion of the LNS indicated slight differences from those values obtained after infusion of the solution. This could be caused by rapid drug release and degradation, when the concentration of the drug in vivo compared with that in plasma was reduced rapidly. We believe that drugs that degrade rapidly are not appropriate to be used as model drugs in the study of drug release characteristic of this LNS. Furthermore, to further understand this LNS structure and its drug release mechanism, the study of other stable lipophilic model drugs is suggested. According to Zhigaltsev,Citation7 rapid release of a drug will eliminate the benefits of formulation encapsulation, resulting in efficacy similar to that of an equivalent dose of free drug. The balance between retention and release must be established to maximize therapeutic activity.
Figure 5 Mean plasma log concentration–time profiles after iv administration of lipid nanospheres of chlorambucil (CHL-LNS) and free chlorambucil (CHL) at a dose of 10 mg·kg−1 in mice.
Note: Results are shown as mean ± SD.
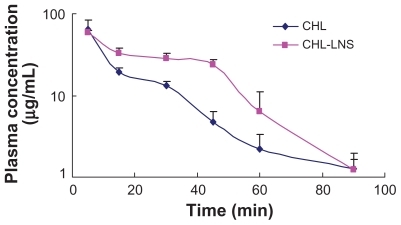
Table 7 Pharmacokinetic parameters after iv administration of chlorambucil (CHL) injection and lipid nanospheres (CHL-LNS) to mice
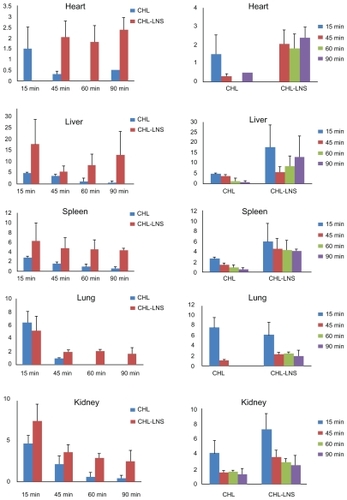
Tissue levels of CHL are shown in . When tissue drug concentrations of both CHL and CHL-LNS are taken into account, these five organs were exposed to higher CHL-LNS drug concentrations than after infusion of the solution. This result was due to the redistribution of the LNS. It should be noted that drugs within the lipid of the LNS contributed to the increased uptake by tissues. In accord with the protection against hydrolysis afforded by the LNS, we speculate that the special structure of LNS attenuates tissue penetration relative to the solution formulation.
Figure 6 Distribution profiles in various organs after iv administration of lipid nanospheres of chlorambucil (CHL-LNS) and free chlorambucil (CHL) at a dose of 10 mg·kg−1 in mice.
Note: Bars represent standard error of 3 determinations.
In summary, we successfully evaluated a new type of lipid nanosphere with a mixture of emulsifiers for intravenous use in a pharmacokinetics study after the intravenous administration of 10 mg·kg−1 of CHL to healthy rats via tail vein injection. In vivo data showed that CHL in LNS was rapidly eliminated following intravenous injection. The following three factors should be considered in complex LNS formulation development: small particle size, a certain type of lipid matrix and an optimized drug solubility (drug/lipid ratio). Small particle sizes lead to large surface areas on the nanospheres, and some drugs are adsorbed on to the surface of the lipid matrix; structurally different complex lipid matrices are also incorporated into each other. These in vitro formulation factors allow the interaction of LNS with lipases for more efficient hydrolysis and lead to rapid in vivo drug release. The drug/lipid ratio is the main formulation parameter in LNS formulation development responsible for enhancing the drug concentration distribution in the in vivo tissues.
Conclusions
The LNS were designed for parenteral use by incorporating the anticancer drug CHL in to the lipophilic mixtures. This process may provide a means for optimizing the composition and production parameters of LNS. The inclusion of monostearin in the LNS formation led to different drug-loading properties. The characteristics of this formulation were evaluated, and the results indicated that the new LNS are a good intravenous drug carrier after reconstitution. The LNS have advantages for intravenous use, such as a small particle size, a low toxicity and good reconstituting dispersion qualities after reconstitution. Although this formulation strategy led to rapid drug release, it offers the potential for the development of further methods of drug delivery for a wide variety of anticancer drugs.
Acknowledgment
The authors would like to thank Dr. Shufang Nie and the reviewers for valuable suggestions to improve the paper.
Disclosure
The authors disclose no conflicts of interest.
References
- KerbelRSClinical trials of antiangiogenic drugs: opportunities, problems, and assessment of initial resultsJ Clin Oncol20011945S51S11560971
- ArmitageJOLeukeran (chlorambucil)Current Clinical Guide2nd edNew York, NYWellcome Oncology NCM19933745
- FarmerPRMetabolism and reactions of alkylating agentsPharmacol Ther1987353013324117
- AlbertssonPLennernasBNorrbyKChemotherapy and antiangiogenesis: drugspecific effects on microvessel sproutingAPMIS2003111995100314629265
- ShakedYEmmeneggerUManSOptimal biologic dose of metronomic chemotherapy regimens is associated with maximum antiangiogenic activityBlood20051063058306115998832
- TonMNChangCCarpentierYADeckelbaumRJIn vivo and in vitro properties of an intravenous lipid emulsion containing only medium chain and fish oil triglyceridesClin Nutr20052449250116054521
- ZhigaltsevIVMaurerNLiposome-encapsulated vincristine, vinblastine and vinorelbine: A comparativie study of drug loading and retentionJ Control Release2005410311115866338
- PugliaCBlasiPRizzaLLipid nanoparticles for prolonged topical delivery: an in vitro and in vivo investigationInt J Pharm200835729530418343059
- SaupeAGordonKCRadesTStructural investigations on nanoemulsions, solid lipid nanoparticles and nanostructured lipid carriers by cryo-field emission scanning electron microscopy and Raman spectroscopyInt J Pharm200614566216574354
- MullerRHRadtkeMWissingSANanostructured lipid matrices for improved microencapsulation of drugsInt J Pharm200224212112812176234
- JenningVThunemannAFGohlaSHCharacterisation of a novel solid lipid nanoparticle carrier system based on binary mixtures of liquid and solid lipidsInt J Pharm200019916717710802410
- SekiJSasakiHDoiMLipid nano-sphere (LNS), a protein-free analog of lipoproteins, as a novel drug carrier for parenteral administration. IVJ Control Release199428352353
- WissingSAKayserOMullerRHSolid lipid nanoparticles for parenteral drug deliveryAdv Drug Deliv Rev2004561257127215109768
- GantaSPaxtonJWBaguleyBCGargSPharmacokinetics and pharmacodynamics of chlorambucil delivered in parenteral emulsionInt J Pharm200836011512118508212
- BockTKLucksJSKleinebuddePMullerRHMullerBWHigh-pressure homogenization of parenteral fat emulsions - influence of process parameters on emulsion qualityEur J Pharm Biopharm199440157160
- DipaliSRKulkarniSBBetageriGVComparative study of separation of non-encapsulated drug from unilamellar liposomes by various methodsJ Pharm Pharmacol199648111211158961156
- LeeFYCoePWorkmanPPharmacokinetic basis for the comparative antitumour activity and toxicity of chlorambucil, phenylacetic acid mustard and beta, beta-difluorochlorambucil (CB 7103) in miceCancer Chemother Pharmacol19861721293698173
- WirtitschMWessnerBSpittlerAEffect of different lipid emulsions on the immunological function in humans: a systematic review with meta-analysisClin Nutr20072630231317449147
- TamilvananSOil-in-water lipid emulsions: implications for parenteral and ocular delivering systemsProg Lipid Res20044348953315522762
- BenitaSFriedmanDWeinstockMPhysostigmine emulsion - a new injectable controlled release delivery systemInt J Pharm1986304755
- HaweAFriessWImpact of freezing procedure and annealing on the physico-chemical properties and the formation of mannitol hydrate in mannitol-sucrose-NaCl formulationsEur J Pharm Biopharm20066431632516875806
- LimSJKimCKFormulation parameters determining the physicochemical characteristics of solid lipid nanoparticles loaded with all-trans retinoic acidInt J Pharm200224313514612176302
- ZimmermannEMullerRHMaderKInfluence of different parameters on reconstitution of lyophilized SLNInt J Pharm200019621121310699720
- BenitaSLevyMYSubmicron emulsions as colloidal drug carriers for intravenous administration - comprehensive physicochemical characterizationJ Pharm Sci199382106910798289116
- DongzhiHouChangshengXieThe production and characteristics of solid lipid nanoparticles (SLNs)Biomaterials2003241781178512593960
- SiekmannBWestesenKThermoanalysis of the recrystallization process of melt-homogenized glyceride nanoparticlesColloids Surf B Biointerfaces19943159175