
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
We report the development of an efficient antibody delivery system for the incorporation of trastuzumab (TZ) into poly(lactic-co-glycolic) acid nanoparticles (PLGA NPs). The aim of the work was to overcome the current limitations in the clinical use of therapeutic antibodies, including immunogenicity, poor pharmacokinetics, low tumor penetration and safety issues.
Materials and methods
Trastuzumab-loaded PLGA NPs (PLGA-TZ) were synthesized according to a double emulsion method. The same protocol was used to produce control batches of nonspecific IgG-loaded NPs and empty PLGA NPs. After release of TZ from PLGA NPs, the effects on the main biological activities of the antibody were evaluated on SKBR3 (human epidermal growth factor receptor 2 [HER2]-positive breast cancer cell line), including specific binding to HER2, phosphorylation of HER2 (Y1248), degradation of HER2 protein and antibody-dependent cell-mediated cytotoxicity (ADCC) mechanism. In addition, an MTT assay was performed for treating SKBR3 cells with PLGA NPs loaded with TZ and doxorubicin to evaluate the cytotoxic activity of the combined treatment.
Results and discussion
TZ was gradually released in a prolonged way over 30 days. The physical characterization performed with circular dichroism, Fourier transform infrared and fluorescence spectroscopy of TZ after release demonstrated that no structural alterations occurred compared to the native antibody. In vitro experiments using SKBR3 cells showed that TZ released from PLGA NPs maintained the same biological activity of native TZ. PLGA NPs allowed a good co-encapsulation efficiency of TZ and doxorubicin resulting in improved therapy.
Conclusion
With the TZ case study, we demonstrate that the distinctive features of therapeutic monoclonal antibodies, including molecular targeting efficiency, capability to inhibit or properly affect the regulatory signaling pathways of cancer cells and stimulation of the ADCC, are fully preserved after loading into and release from PLGA NPs. In addition, PLGA NPs are shown to allow for the simultaneous incorporation of TZ and conventional chemotherapeutics, resulting in a potent antitumor nanodrug well suited for in situ combination and neoadjuvant therapy.
Introduction
Therapeutic monoclonal antibodies (TMAs) have raised significant expectation in the past two decades due to obvious clinical efficacy combined with reduced systemic toxicity normally associated with conventional chemotherapy.Citation1–Citation3 Since the first Food and Drug Administration (FDA)-approved TMA in 1986, the market of TMAs has grown exponentially, with approximately 50 antibodies approved and over 70 expected in 2020.Citation4 Among them, TMAs targeting the tyrosine kinase receptors that are overexpressed in several tumor cell subsets (eg, trastuzumab [TZ; Trastuzumab-Herceptin®, Genetech, San Francisco, CA, USA] and cetuximab Erbitux®, Merck, Darmstadt, Germany, directed toward the human epidermal growth factor receptor 2, HER2, and the epidermal growth factor, respectively),Citation5,Citation6 or those addressing the tumor microenvironment, such as bevacizumab (Avastin®, Roche, Basel, Switzerland),Citation7 and the new anti check-point inhibitors,Citation8 have initiated a new paradigm for targeted therapy. TMAs operate through various mechanisms, exploiting both the targeted variable fragments Fab – ie, by interfering with transduction signaling cascades regulating cell growth and differentiation – and the constant fraction Fc, able to activate the antibody-dependent cell-mediated cytotoxicity (ADCC) and the complement-dependent cytotoxicity.Citation9–Citation11
However, despite the great benefit and improved success rate shown by antibody therapies in the treatment of lymphomas and solid tumors, the clinical practice has raised numerous limitations, mostly related to safety issues. Risks deriving from several adverse reactions, including infection, autoimmune disease and cardiotoxicity, have been associated to TMAs treatment.Citation12 In addition, the observed suboptimal efficacy in treating cancer might be attributed to a combination of different factors, including 1) limited tumor penetration of TMAs due to interstitial pressure gradient; 2) poor pharmacokinetics; and 3) high molecular weight that strongly hampers crossing of biological barriers, such as the blood brain barrier (BBB), resulting ineffective against metastatic tumors at the central nervous system.Citation13 More in general, the administration of peptide drugs, including TMAs, could be associated to short-term stability caused by immunogenic reactions and biodegradation by proteases.Citation14
Hence, in order to improve the efficacy, bioavailability and accumulation of TMAs in the tumor, new formulations are desired. One possible strategy to overcome these problems resides in the antibody encapsulation into biodegradable and biocompatible polymers creating a tailorable nanosystem.Citation15–Citation17 In this way, the loaded antibody is protected from degradation and from interaction with the immune system and can be released in a controlled manner. Poly(lactic-co-glycolic) acid (PLGA) is an excellent candidate, as it has been approved by FDA for human use because of its biodegradability and low systemic toxicity.Citation18–Citation20 Its in vivo degradation takes several days allowing a prolonged release of the entrapped material, which results in fewer administrations and improved patient compliance. PLGA nanoparticles (PLGA NPs) could exhibit several advantages over direct systemic injection of TMAs, including 1) prolonged residence in circulation; 2) reduced immunogenicity;Citation21 and 3) combined extracellular and intracellular effect. In addition, they can be exploited as permeation enhancer due to their fast uptake in cancer cells and tissues, and they can be tailored to cross biological barriers,Citation22 allowing their cargo to be delivered across the BBB and intestinal mucosa. Eventually, PLGA NPs are suitable to be orally administered.Citation23,Citation24
Major issues related to protein structural and functional preservation after incorporation and release from PLGA NPs were envisaged. Indeed, NPs assembly involves organic solvents and sonication steps, which could affect protein folding and biological functionality. Additionally, polymer degradation is correlated with the release of free lactic acid monomers, which is likely to modify the pH of microenvironment with possible effects on protein denaturation.Citation25 To this end, preliminary evidence on structural conservation of bevacizumab has been already provided through investigation of protein stability and functionality.Citation26 In the present study, we loaded for the first time TZ into PLGA NPs. After confirming the structural conservation of TZ after loading into and release from PLGA NPs, we studied the maintenance of antitumor efficiency of released TZ against breast cancer cells through a multiparametric in vitro investigation. We demonstrated that either the released antibodies or the nano-complexes are able to exert an effective antitumor activity in HER2-positive breast cancer cells both by direct action on cell signaling transduction and by cell-mediated cytotoxicity.
Materials and methods
Materials
PLGA (Resomer® RG 504 H, 50:50 lactide:glycolide, acid terminal, MW 40.000 Da) and poloxamer (Pluronic F-127) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Doxorubicin hydrochloride (DOXO) (Adriblastina®) was supplied by Pfizer, NY, USA, and used after HCl removal by incubation with three molar excess of trimethylamine in CHCl3. TZ (Herceptin®) was supplied by Genentech (South San Francisco, CA, USA). TZ was used after dialysis in phosphate-buffered saline (PBS) at 4°C on a Slyde-A-Lyzer dialysis cassette (Waltham, MA, USA) with a 7 kDa cutoff. IgG from rabbit serum and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich Co. Amicon® Ultra 2 mL 100 K MWCO were purchased from Merck Millipore (Billerica, MA, USA). Zeba™ spin desalting columns 2 mL 7 K MWCO were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All solvents and common reagents were purchased from Sigma-Aldrich Co.
Synthesis of TZ-loaded polymeric NPs
TZ-loaded PLGA NPs (PLGA-TZ) were synthesized according to the double emulsion method.Citation27 In total, 500 µL of PLGA (35 mg) solution in ethyl acetate was emulsified with 400 µL of an aqueous solution of TZ (1 mg) and poloxamer (5 mg), as protecting agent, as widely demonstrated in literature.Citation27 The first w/o emulsion was obtained with sonication tip (Sonifier Sound, Branson Ultrasonics, Shanghai, People’s Republic of China), 30 s +30 s, in ice bath, 38% intensity. Then, the first emulsion was poured into 2 mL of 2% w/v PVA aqueous solution and sonicated under the same conditions as already described, thus obtaining a w/o/w double emulsion. Then, 4 mL of 2% w/v PVA was added to harden the NP suspension for 4 h at room temperature (RT), under magnetic stirring (750 rpm). After curing, NPs were collected by centrifugation at 14,400 rpm, 20 min, 4°C (Heraeus Fresco 21; Thermo Fisher Scientific), washed with double distilled water and freeze-dried through Alpha 1-2 LD freeze drier (Christ, Memmingen, Germany) at 0.340 mbar, −51°C, 12 h, without the addition of any cryoprotectant. The process yield was calculated after freeze-drying process as the ratio between collected NPs and starting raw materials (TZ and polymers).
The same protocol was used to produce control batches of nonspecific IgG-loaded NPs (PLGA-IgG). PLGA NPs, without antibody, were generated using only poloxamer solution as the aqueous phase for the first emulsification step. Additional batches were obtained by co-encapsulating TZ and DOXO inside PLGA NPs (PLGA-TZ-DOXO), intended as a combined immunochemotherapeutic drug model. For the preparation, DOXO (350 µg) was dissolved in the polymeric organic solution, while TZ was dissolved in the poloxamer aqueous solution. The other process parameters were kept as already described. A schematic description of the synthetic process is represented in . TZ-FITC was properly synthesized incubating TZ (5 mg) with FITC (1 mg) in NaHCO3 0.1 M (pH 8–9) for 12 h at 4°C, and then purified from unreacted FITC with Amicon® and Zeba™. The amount of recovered TZ-FITC was quantified using NanoDrop (Thermo Fisher Scientific). TZ-FITC was then loaded into PLGA NPs according to the above described procedure.
All NPs were characterized in terms of size, size distribution and zeta potential through dynamic light scattering (Zetasizer Nano ZS; Malvern Instruments). NP suspension in ultrapure water at 1 mg mL−1 concentration was analyzed using a disposable 1 cm optical length cuvette at 25°C. The scattered light from the NPs in suspension was used to calculate NPs’ hydrodynamic diameter considering medium viscosity. The results are reported as zeta average diameter. NP size distribution is described by polydispersity index (PDI) as PDI <0.2 corresponds to monodisperse NP population. The same NP sample was used to measure zeta potential using the same instrument with an appropriate cuvette. Transmission electron microscope (TEM) images of PLGA, PLGA-TZ and PLGA-TZ-DOXO were acquired. TEM samples were prepared by deposition of an NP suspension drop (20 µL) on the formvar coated copper grid. After drying on air, the grids were stained by uranyl acetate solution (1% in PBS) for 5 min and visualized by a TEM (Tecnai G2 Spirit BioTWIN operating on 120 kV; FEI, Hillsboro, OR, USA).
Release test and drug loading
PLGA-TZ were suspended in 1 mL of PBS 1×, pH 7.4, in a low-protein binding centrifuge tube and incubated at 37°C. At predetermined time points, NP suspension was centrifuged and 500 µL of the supernatant was withdrawn and replaced with the same volume of fresh release medium in order to keep TZ sink conditions. The amount of released TZ was quantified through MicroBCA assay kit (Cyanagen, Bologna, Italy), following the manufacturer’s instructions. The release test was performed over 30 days. At the end, the release medium was completely removed and the NPs collected were incubated with 1 mL of 14 mM NaOH for 7 days in order to induce polymer degradation and to completely extract the not-released amount of TZ.Citation28 The total loaded amount of TZ was calculated as the sum of TZ released and TZ extracted after the release test. The TZ encapsulation efficiency (EE%) and relative drug content (DC%) were calculated as follows:
DOXO release from PLGA-TZ-DOXO NPs was performed at the same conditions as described for TZ. The amount of DOXO loaded into NPs was calculated by dissolving 2 mg of freeze-dried PLGA-TZ-DOXO in dimethyl sulfoxide (DMSO). DOXO concentration in DMSO was evaluated through FluoroMax-4 fluorometer (Horiba Scientific, Italy) at λem=590 nm and quantified with a standard calibration curve in DMSO (0.2–1 µg mL−1). EE% and DC% of DOXO were calculated with Equationeq 1(1) and Equation2
(2) . Released DOXO was quantified measuring DOXO emission with fluorometer, as already described, and calculated with a proper calibration curve prepared with DOXO samples in PBS 1× (0.2–1 µg mL−1).
Evaluation of TZ structural integrity after release
Samples of TZ released from NPs were subjected to a deep investigation in order to check the antibody structural integrity compared to the native antibody used as reference.
The primary structure of the antibody was evaluated through sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE with 12% of acrylamide). Released TZ (over the first 48 h) was loaded into the gel and compared with references: control TZ, TZ discarded during washing steps and TZ extracted after incubation with NaOH. All samples were concentrated with Amicon® in order to load the same amount of TZ for all the samples (2–3 µg/sample). The proteins in the gel were stained with Coomassie dye R-250 and scanned to obtain the relative image.
The secondary structure of the released TZ (3.3 µM) was evaluated with circular dichroism (CD) (Jasco J-815 spectropolarimeter; Jasco Corp., Tokyo, Japan) at 25°C and in a 0.1 cm path-length quartz cell under the following conditions: 260–195 nm spectral range, 2 nm of bandwidth and 20 nm min−1 of scan speed. Spectra were averaged over four scans and presented after buffer subtraction. Mean residue ellipticity [θ] was calculated as:Citation29
A quantitative estimation of the protein secondary structures was obtained by using the BeStSel method.Citation30 For Fourier transform infrared (FTIR) measurements in attenuated total reflection (ATR), 2 µL of 3.3 µM TZ was deposited on the diamond plate of the single reflection ATR device (Quest; Specac, Orpington, UK). Spectra were collected after solvent evaporation to allow the formation of a hydrated protein film.Citation31 The Varian 670-IR spectrometer (Varian Australia Pty Ltd, Mulgrave VIC, AU, Australia), equipped with a nitrogen-cooled Mercury Cadmium Telluride detector and an air dryer purging system, was employed under the following conditions: 2 cm−1 spectral resolution, 25 kHz scan speed, 1,000 scan co-additions and triangular apodization. The second derivative analysis of the protein absorption spectra was performed by the Savitzky–Golay procedure (). The Resolutions Pro software (Varian Australia Pty Ltd) was employed for FTIR measurements and analyses.
The tertiary structure stability was assessed with fluorescence emission exploiting the intrinsic fluorescence of tyrosine and tryptophan residues.Citation32 Released TZ samples in PBS were excited at 280 nm and the emission was recorded in the range of 300–500 nm. Alterations in the peak intensity and/or wavelength were checked and compared to native TZ at the same concentrations, as indicator of structural modifications.
Cell culture
SKBR3 cell line (American Type Cell Culture) was chosen as HER2-positive breast cancer model and cultivated in Dulbecco’s Modified Eagle’s Medium high glucose – Ham’s F12 nutrient factors (1:1) supplemented with 10% of fetal bovine serum (FBS), 1% of penicillin-streptomycin and 1% L-glutamine at 37°C in a humidified incubator with 5% CO2.
Binding of TZ-FITC
In total, 5×105 SKBR3 were collected in flow cytometry tubes and incubated with 50 µg mL−1 of TZ, released TZ and released IgG labeled with FITC in PBS/1% bovine serum albumin (BSA) for 30 min at 4°C. After incubation, cells were washed with PBS, and FITC fluorescence of all samples was analyzed by Gallios™ Flow Cytometer (Beckman Coulter Inc., Brea, CA, USA).
To test the selectivity of TZ binding to HER2 after release from PLGA NPs, SKBR3 were plated at a density of 3×105 in 12 wells and after 24 h at 37°C were incubated with 2 µg mL−1 and 20 µg mL−1 of TZ, released TZ and released IgG for 24 and 48 h at 37°C. After incubations, cells were washed with PBS and incubated for 30 min at 4°C with TZ-FITC (50 µg mL−1) in PBS/1% BSA. Cells were washed and analyzed by flow cytometry. The mean fluorescence intensity of FITC signals was analyzed acquiring 10,000 events per sample and data were expressed as mean ± standard deviation (SD) of three independent replicates.
Confocal microscopy
SKBR3 were plated at a density of 2×105 in 12 wells on coverslips for confocal microscopy. After 48 h at 37°C, cells were incubated with 2 µg mL−1 of TZ and released TZ for 24 h at 37°C. After incubation, SKBR3 were washed twice with PBS and incubated with TZ-FITC (50 µg mL−1) for 30 min at 4°C. Cells were then washed again twice with PBS and fixed with 4% paraformaldehyde for 10 min at RT, washed twice with PBS and labeled with DAPI (1:5,000; for nuclei staining – Thermo Fisher Scientific) and WGA-555 (1:1,000; for cell membrane staining – Thermo Fisher Scientific). SKBR3 were mounted on microscope slides using ProLong Antifade reagent (Thermo Fisher Scientific) and analyzed by Nikon A1 Confocal Microscope (Nikon Instruments Europe BV, Amsterdam, the Netherlands).
To study the different endocytosis mechanisms of TZ and PLGA-TZ, SKBR3 were plated at a density of 2×105 in 12 wells on coverslips. Cells were incubated for 48 h at 37°C to reach confluence. After that, cells were incubated with 10 μg mL−1 of TZ and PLGA-TZ for 30 min and 24 h at 37°C. Cells were than washed twice with PBS, fixed with 4% paraformaldehyde for 10 min at RT and washed twice with PBS. To label early endosomes and lysosomes, cells were incubated for 2 h at RT with anti-EEA1 (1:1,000 – BD Biosciences, San Jose, CA, USA) and anti-CatD (1:50 – Calbiochem, Merck, Darmstadt, Germany), respectively, in PBS +5% Goat serum +0.3% Triton X-100. Then, cells were washed twice with PBS and incubated with anti-mouse secondary antibody labeled with Alexa Fluor-555 (1:300 – Thermo Fisher Scientific) for 1 h at RT. After washing with PBS, nuclei were labeled with DAPI (1:5,000 – Thermo Fisher Scientific) and cells were mounted on microscope slides using ProLong Antifade reagent (Thermo Fisher Scientific) and analyzed by Nikon A1 Confocal Microscope.
Western blot
SKBR3 were plated at 106 in a 6-well plate for 24 h at 37°C, and then incubated with 2 µg mL−1 TZ, released TZ, PLGA-TZ, IgG, released IgG and PLGA-IgG for 4 and 24 h. Cells were washed with PBS, lysed with lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 10% glycerol, 1% NP-40, protease inhibitor cocktail [Thermo Fisher Scientific] and phosphatase inhibitor [Roche Life Science, Indianapolis, IN, USA]), sonicated with 3 cycles of 10 s at 10% of amplitude, gently rotated for 20 min at 4°C and centrifuged for 15 min at 13,200 rpm at 4°C. Supernatants were quantified by BCA assay (Cyanagen), separated by 12% SDS-PAGE and blotted onto a PVDF membrane. The membranes were blocked with TBS – Tween 0.1% solution +5% milk and then immunodecorated alternatively with monoclonal anti-HER2/ErbB2 antibody (Cell Signaling Technology, Leiden, the Netherlands) or polyclonal anti-phosphorylated HER2 antibody (Tyr1248; Cell Signaling Technology) or monoclonal antibody anti-β-actin (Santa Cruz Biotechnology Inc., Dallas, TX, USA) in TBS – Tween 0.1% solution +5% BSA overnight at 4°C. After washing with TBS – Tween 0.1% solution, PVDF membranes were incubated with a goat anti-rabbit or a goat anti-mouse IgG HRP-linked secondary antibody (Cell Signaling Technology) in TBS – Tween 0.1% solution +5% milk for 1 h at RT. Protein bands were detected with an enhanced chemiluminescent system (Euroclone SpA, Milan, Italy). Densitometric analyses were done by ImageJ software.
PBMC isolation
Peripheral blood from healthy donor was layered on Ficoll-Paque in 50 mL conical tube and centrifuged for 30 min at 1,500 rpm without brake. At the end, the peripheral blood mononuclear cell (PBMC) layer was carefully transferred in a new 50 mL conical tube and diluted with PBS, centrifuged for 6 min at 1,400 rpm and the supernatant was eliminated to remove platelets. This step was repeated for four times decreasing the rpm of centrifugation up to 1,000 rpm. Washed PBMCs were resuspended in RPMI-1640 medium with 10% of decomplemented FBS and 1,000 U mL−1 IL-2 (BioLegend, San Diego, CA, USA) for 24 h at 37°C. The described procedure was approved by Ethical Committee of the University of Milano-Bicocca (prot.#351, 13th November 2017) after submission of the project together with informed consent by the healthy volunteer.
ADCC assay
ADCC was evaluated using CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega Corporation, Madison, WI, USA). Assays were done adding effector (PBMCs) onto target cells (SKBR3) at an effector:target (E:T) ratio of 20:1 and 40:1, plating SKBR3 at a density of 5×103 in 96 wells. Firstly, target cells were coated with 0.02, 0.2 and 2 µg mL−1 of TZ, released TZ and released IgG for 30 min at 4°C in RPMI-1640 medium. After coating, PBMCs at exact concentrations were added and after 4 h at 37°C, LDH release from target cells was measured by EnSight™ multimode plate reader (Perkin Elmer, Waltham, MA, USA) setting absorbance wavelength at 490 nm. Percentage of ADCC was calculated using the formula following protocol instruction:
Data were represented as percentage ± SD of three independent biological replicates.
Viability assay
To test PLGA, PLGA-DOXO, PLGA-TZ, PLGA-TZ-DOXO, DOXO and TZ toxicity, an MTT assay (CellTiter 96 Non-Radioactive Cell Proliferation Assay, Promega) was performed seeding 104 SKBR3 in a 96-well plate making four replicates per each concentration. First set of experiments was done comparing the cytotoxic effect of DOXO, PLGA-DOXO and PLGA-DOXO-TZ. Tested NPs’ concentration was established on the basis of their DOXO content in order to compare the concentration of encapsulated drug inside PLGA NPs with free molecule (0.01, 0.1, 1 and 5 µM) (). The other set of experiments compared the antitumor effect of TZ and PLGA-TZ keeping constant the concentration of TZ, both free and loaded (0.0236, 0.236, 2.36 and 11.8 µg mL−1) (). In the case of empty PLGA NPs, we used the same amount of PLGA used for PLGA-TZ: 0.78, 7.8, 78 and 390 µg mL−1 ().
Table 1 Specific concentrations of each sample used for viability experiments
According to the manufacturer’s instructions, at the end of the exposure time (24, 48 and 72 h), MTT was added and formazan product was detected after 4 h at 37°C reading the absorbance at 570 nm with EnSight™ Multimode Plate Reader (Perkin Elmer) subtracting the absorbance of background at 620 nm. Results were obtained by normalizing the untreated cells set at 100% and expressed as percentage ± SD of three independent biological replicates.
Results and discussion
TZ-loaded PLGA NP characterization
In our study, TZ, a monoclonal antibody used for the treatment of HER2-positive breast cancer, was selected as a model antibody to assess the potential of TMA loading into PLGA NPs. TZ-loaded PLGA NPs were produced according to a conventional w/o/w double emulsion method,Citation27,Citation33,Citation34 with some adjustments (). Hydrophilic poloxamer was added to the synthetic protocol to maintain TZ integrity during NP preparation, as widely demonstrated in the literature.Citation27 Dynamic light scattering data, summarized in , confirmed that the optimized protocol was suitable to generate both unloaded and TZ-loaded PLGA NPs having size below the 200 nm threshold and homogeneous size distribution ().
Table 2 Dimensional characterization and process yield of PLGA, PLGA-TZ and PLGA-TZ-DOXO NPs
The presence of loaded TZ did not change significantly the morphological and dimensional features of NPs (). A detectable decrease in size was recorded upon incorporation of TZ, suggesting stabilizing effect in the close packing of the NPs attributable to the favorable interaction of polyanionic PLGA NPs with the positively charged protein. By observing TEM pictures, it is clear that the presence of loaded TZ did not change the surface morphology of NPs significantly. As expected, the surface charge of TZ-loaded NPs measured by zeta potential was appreciably less negative compared to unloaded NPs, which is an indirect confirmation of the antibody incorporation (IPTZ=8.45). The method was also adaptable to the co-encapsulation of cytotoxic drugs useful for combination therapy, ie, DOXO, obtaining NPs with dimensional properties on the same order ( and ).
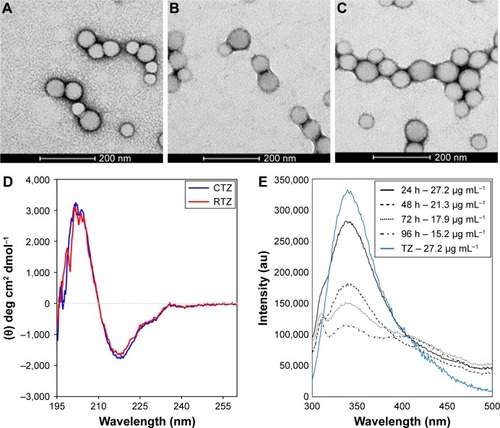
Figure 1 Characterization of nanoencapsulated and released TZ.
Notes: TEM images of (A) PLGA, (B) PLGA-TZ and (C) PLGA-TZ-DOXO NPs. (D) Far-UV CD spectra of CTZ and RTZ. (E) Fluorescence emission spectra of different released TZ (24, 48, 72 and 96 h) compared to a standard solution of native TZ (27.2 µg mL−1).
Abbreviations: TZ, trastuzumab; TEM, transmission electron microscope; PLGA, poly(lactic-co-glycolic) acid; DOXO, doxorubicin; NP, nanoparticle; CD, circular dichroism; CTZ, control TZ; RTZ, released TZ.
TZ encapsulation efficiency in PLGA NPs, with or without DOXO, was 37.5%±1.4% with a corresponding drug content of 2.9%±0.1%. TZ was released from PLGA NPs in a controlled manner, reaching 78% in 30 days (). A prolonged release was obtained also for DOXO with 35% released after 30 days (), confirming the ability of PLGA NPs to deliver their payload in a sustained manner (DOXO %EE: 21.8%±8.6%; %DC: 0.54%±0.25%).
Structural characterization of TZ released from PLGA NPs
First, we assessed the structural integrity of TZ released from PLGA NPs. The conservation of the primary structure was confirmed by SDS-PAGE, in which TZ released at different time points (30 min to 48 h) showed the typical bands corresponding to the molecular weight of the heavy and light chains (). No differences in the band profiles were observed along the release process, confirming that no primary structure degradation occurred. The maintenance of the secondary structure was investigated by CD and FTIR spectroscopy comparing the structural features of TZ released by PLGA NPs (RTZ) with those of native TZ used as control (CTZ). shows the far-UV CD spectrum of RTZ compared with CTZ. It is clear that RTZ spectrum was superimpos able to that of CTZ. In particular, it showed a maximum at ~202 nm and a minimum at ~218 nm corresponding with a β-structure-rich proteins. For both native and released TZ, a β-sheet content of ~43% was determined from the CD spectra ().Citation30 FTIR second derivative spectra of control and released TZ () are dominated by two main components at ~1,692 cm−1 and at ~1,641 cm−1 due to the native β-sheet protein structure.Citation35
Finally, the tertiary structure of RTZ was confirmed through tryptophan fluorescence measurements at 330 nm. Fluorescence intensity was concentration dependent as we can clearly observe in . We do not observe a shift in emission wavelength among each RTZ samples but we can appreciate a reduction in RTZ emission intensity compared to a standard solution of CTZ at the same concentration (27.2 µg/mL). Despite the signal reduction observed, we decided to deeply evaluate RTZ activity with in vitro experiments.
HER2 binding of released TZ
A first set of experiments to achieve a functional characterization of RTZ was focused on the direct activity of the antibody. We assessed whether released TZ maintained the binding selectivity for its receptor (HER2). For this purpose, we selected SKBR3 cell line as HER2-positive breast cancer cells.Citation36 SKBR3 cells were pre-treated with 2 and 20 µg mL−1 of RTZ for 24 and 48 h, respectively, at 37°C and then incubated with dye-labeled TZ (TZ-FITC) at 4°C. At this temperature, endocytic processes are globally inhibited and the contribution of ligand-receptor binding could be better appreciated through competition experiments by flow cytometry. As positive and negative controls, we used CTZ and released IgG (RIgG), respectively, at the same concentrations of RTZ. and show a significant reduction of TZ-FITC binding on SKBR3 surface after pre-treatment with RTZ and CTZ, confirming a competition between RTZ and TZ-FITC for HER2 binding. By contrast, RIgG pre-treatment did not alter the TZ-FITC binding. After 24 and 48 h of treatment with RTZ, we observed a significant reduction of TZ-FITC binding on SKBR3 surface of 88.9% and 92.2%, respectively, comparable to CTZ. Once again, no binding inhibition of TZ-FITC was detected using RIgG (). Altogether, these results demonstrated that the specific binding ability of TZ was not altered after PLGA NP loading and release.
Figure 2 HER2 binding of released TZ from PLGA NPs.
Notes: (A) TZ-FITC binding on SKBR3 cells after treatment with RIgG, CTZ and RTZ at two different concentrations (2 and 20 µg mL−1) for 24 and 48 h. Data are represented as the mean of fluorescence intensity of three independent replicates ± SD acquiring 10,000 events for each sample by flow cytometry. (B–E) Confocal microscopy images of TZ-FITC binding on SKBR3. (B) Basal binding of TZ-FITC; (C, D and E) binding of TZ-FITC after pre-treatment of RIgG, CTZ and RTZ, respectively, for 24 h at 37°C. Cells were labeled with DAPI (nuclei) and WGA-555 (membranes). Scale bar =50 µm. ****P<0.01 vs UNTR; **P<0.05 vs UNTR.
Abbreviations: HER2, human epidermal growth factor receptor 2; TZ, trastuzumab; PLGA NPs, poly(lactic-co-glycolic) acid nanoparticles; FITC, fluorescein isothiocyanate; RIgG, released IgG; CTZ, control TZ; RTZ, released TZ; SD, standard deviation; CNTR, cells without any labeling; UNTR, cells coated with TZ-FITC.
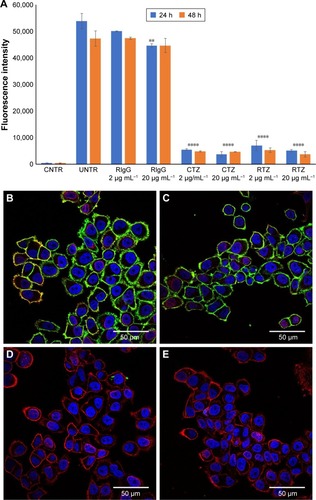
Data obtained by flow cytometry were confirmed by confocal microscopy analysis (), where pretreatment with either CTZ or RTZ caused complete decrease of TZ-FITC binding (green signal – , respectively) compared to untreated cells (), whereas RIgG treatment proved to be unreactive ().
Nanoformulation of TZ enhances HER2 degradation
A known mechanism by which TZ exerts its antitumor efficacy is reducing downstream signaling triggering endocytosis and degradation of HER2, altering HER2-mediated mitogenic and pro-survival signaling, inhibiting homo and heterodimerization, and preventing HER2 extracellular domain shedding.Citation5,Citation37 Whether TZ actually induces HER2 downregulation or not remains unclearCitation38 and the direct effect on HER2 intracellular partners is still under debate.Citation39 However, previous evidence suggested that major impact of TZ on HER2 expression upon endocytosis should be expected as mediated by NP delivery.Citation40,Citation41 Hence, TZ nanoencapsulation might benefit the antitumor activity harnessing extracellular release of TMA and intracellular accumulation favored by NP-assisted endocytosis.
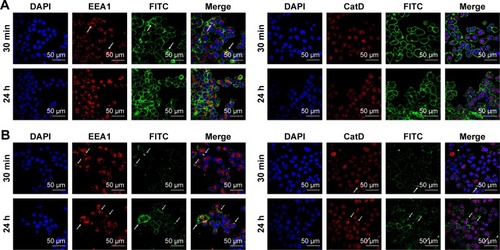
Western blot analysis of HER2 expression from SKBR3 extracts after treatment with 2 µg mL−1 RTZ, CTZ () and TZ-loaded NPs (PLGA-TZ) () exhibited higher HER2 downregulation associated to PLGA-TZ compared to native and released TZ. The same experiment was performed with unspecific IgG as control (). To assess if the observed HER2 downregulation caused by PLGA-TZ was actually due to a pathway degradation enhancement, we performed a confocal microscopy experiment by labeling early endosomes (recycling pathway) and lysosomes (degradation pathway). Confocal images of cancer cells incubated with TZ-FITC () or PLGA-TZ () showed important differences in the mechanism of internalization of TZ-HER2 complex. Indeed, while CTZ caused antibody-receptor complex internalization arresting at an early endosome stage, PLGA-TZ could evolve to the lysosomal pathway, resulting in improved degradation efficiency of HER2, corroborating western blot results. shows untreated cells as control.
Figure 3 HER2 phosphorylation.
Notes: (A) Analysis of pHER2 (Y1248) and HER2 expression on SKBR3 cells after treatment with control CTZ and RTZ at 2 µg mL−1 for 4 and 24 h; (B) Analysis of HER2 expression on SKBR3 cells after treatment with PLGA-TZ at 2 µg mL−1 for 4 and 24 h. Values were calculated as ratio between pHER2/HER2 and HER2/b-actin and normalized with untreated cells (UNTR).
Abbreviations: HER2, human epidermal growth factor receptor 2; CTZ, control trastuzumab; RTZ, released trastuzumab; PLGA-TZ, trastuzumab-loaded poly(lactic-co-glycolic) acid nanoparticles; UNTR, cells without treatment.
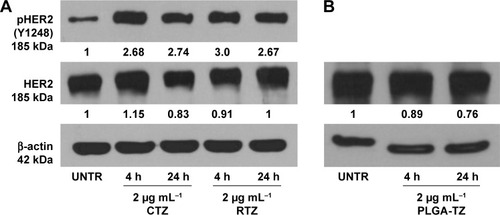
Figure 4 Interaction of TZ with intracellular processes.
Notes: Confocal microscopy images of endocytosis mechanism of (A) TZ-FITC and (B) PLGA (TZ-FITC) in SKBR3. Cells were labeled with DAPI (nuclei), mouse AbII-Alexa555 against EEA1 or CatD. Arrows indicate the co-localization of FITC with Alexa555 of AbII. Scale bar = 50 µm.
Abbreviations: TZ, trastuzumab; FITC, fluorescein isothiocyanate; PLGA, poly(lactic-co-glycolic) acid.
Effect of TZ on the phosphorylation of HER2 (Y1248)
The interaction of TZ with HER2 signal transduction cascade in sensitive cells (eg, BT474 and SKBR3 breast cancer cell lines) was associated with its ability to induce HER2 phosphorylation at Y1248, which in turn correlated to the inhibition of cell proliferation.Citation42 This event affects signaling through the PI3K/Akt pathway initiating processes that lead to cell survival, suppression of apoptosis and cell cycle control.Citation43 Thus, we tested the capability of released TZ to interfere with the first step of the tyrosine kinase signal transduction in SKBR3 cells. After treating cancer cells with 2 µg mL−1 of RTZ or CTZ, western blot analysis revealed that the extent of Y1248 phosphorylation in cells treated with RTZ was on the same level of those treated with CTZ, maintaining the effect at least for 24 h (). PLGA-TZ NPs showed a similar ability in inducing HER2 phosphorylation ().
PLGA NPs to combine immunotherapy and chemotherapy
Over the past years, adjuvant therapy of HER2-positive breast cancer using TZ, as a single agent following multimodal anthracycline treatment or as a part of combination therapy including doxorubicin, has significantly decreased the risk of tumor relapse.Citation44 The administration of TZ combined with chemotherapy allowed by co-encapsulation in PLGA NPs might improve the beneficial effect of this advantageous strategy. After demonstrating that TZ maintained the same structure and activity upon release by PLGA NPs, we studied the potential of PLGA-DOXO-TZ NPs to induce cytotoxicity in SKBR3 cells. We firstly compared the effect of PLGA-DOXO-TZ NPs with that of DOXO and PLGA-DOXO NPs (). Notably, in the first 24 h cytotoxicity of PLGA-DOXO-TZ NPs was much higher compared to PLGA-DOXO NPs and DOXO alone. This result could be explained by the combination of DOXO activity together with TZ, which induces an antitumor activity through the degradation of HER2 ().
Figure 5 Combination therapy with PLGA nanocarriers.
Notes: (A) Viability assay of SKBR3 treated with different concentrations of DOXO, PLGA-DOXO and PLGA-DOXO-TZ keeping constant the concentration of DOXO (0.01, 0.1, 1 and 5 µM) for 24, 48 and 72 h. (B) Viability assay of SKBR3 treated with PLGA, PLGA-TZ and TZ (for concentrations see ) for 24, 48 and 72 h. Data are represented as a mean ± SD of three independent replicates. **P<0.05 vs UNTR (100%); ***P<0.01 vs UNTR (100%); #P<0.01 vs PLGA-DOXO-TZ; @P<0.01 vs PLGA-TZ.
Abbreviations: PLGA, poly(lactic-co-glycolic) acid; DOXO, doxorubicin hydrochloride; TZ, trastuzumab; SD, standard deviation; UNTR, cells without treatment.
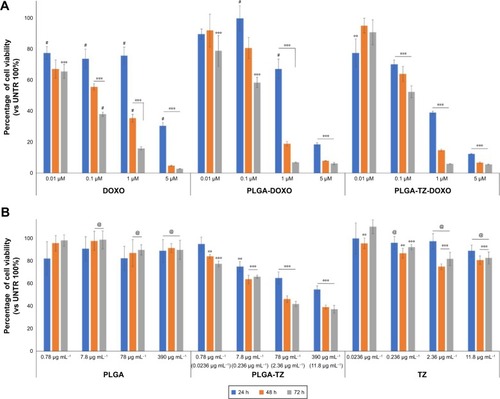
Most interesting was the comparison between the effect of PLGA-TZ and native TZ. In , PLGA-TZ were remarkably more cytotoxic compared to unloaded TZ at the same antibody concentration, supporting the assumption that the intracellular action of TZ allowed by PLGA nanodelivery exhibited more pronounced antitumor effect, as already suggested by western blot () and confocal microscopy analysis ().
Released TZ induces ADCC mechanism
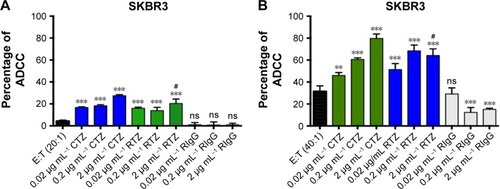
Among the multiple biological activities exerted by TZ, the greatest clinical impact is probably attributable to its capability to promote ADCC.Citation45 The recognition of TMAs Fc already bound to cancer cells (target [T] cells) by natural killer (NK) cells (effector [E] cells) through CD16 receptor induces activation of innate immune cells to release lytic granules, including perforins and granzyme B, leading to cancer cell death.Citation46 To evaluate if RTZ was still able to activate ADCC mechanism, SKBR3 cells were treated with CTZ, RTZ and RIgG at different concentrations (0.02, 0.2 and 2 µg mL−1) upon addition of PBMC population, which contained approximately 5% NK cells. The experiment was conducted at two different E:T ratios (20:1 and 40:1). After 4 h of treatment, lysis of tumor cells induced by NK cells was quantified using a lactate dehydrogenase (LDH) assay, which correlates the release of LDH enzymes by target cells to cytolytic activity of NK cells. shows that RTZ induced the same extent of ADCC activity compared to CTZ, with a more pronounced effect using the 40:1 E:T ratio. As expected, cytolysis was not observed using RIgG at both the tested conditions. These data confirmed the ability of TZ in inducing immune response against cancer cells after binding HER2 receptor. This effect, combined with HER2 degradation and phosphorylation, as demonstrated previously, contributed to the overall activity of TZ in tumor treatment.
Figure 6 RTZ induced the same level of ADCC mechanism as that of control TZ.
Notes: ADCC mechanism induced by TZ, RTZ and RIgG on SKBR3 cells with E:T ratio of 20:1 (A) and 40:1 (B). Data are represented as percentage of mean ± SD of three independent replicates. **P<0.05 vs E:T; ***P<0.01 vs E:T; #P<0.01 vs TZ at the same concentrations ns vs E:T.
Abbreviations: RTZ, released trastuzumab; ADCC, antibody-dependent cell-mediated cytotoxicity; RIgG, released IgG; E:T, effector:target; SD, standard deviation; ns, not significant.
Conclusion
In this study, we reported the feasibility of loading TZ inside PLGA NPs with a reliable and efficient method and in association with recent results from a study Sousa et al.Citation26 We demonstrated the wide application of this encapsulation method to several TMAs maintaining their structural and functional features.
We synthesized PLGA NPs loaded with TZ achieving a prolonged release of the antibody preserving its structural integrity. In addition, we demonstrated that the released antibody was fully effective in targeting HER2-positive breast cancer cell, interfering with receptor-mediated signaling transduction and also maintaining its ability to induce ADCC mechanism by innate immune cells. PLGA NPs added up further advance allowing the simultaneous incorporation of TMAs and cytotoxic drugs, such as doxorubicin, opening new perspective in combination therapy of cancer disease.
In summary, with the help of our work it is possible to appreciate the great potentiality of TZ-loaded PLGA NPs since both encapsulated and released antibody is useful in cancer therapy. NPs can be exploited as drug carriers to increase antibody intracellular concentration, resulting in an improved cytotoxic activity compared to native antibody. At the same time, if properly functionalized, they can overcome biological barriers (ie, BBB), releasing their payload in the extracellular matrix, without affecting the antibody activity.
Future in vivo validation will be necessary to appreciate the pharmacological parameters, including pharmacokinetics, systemic toxicity and biodistribution. In the light of the great potential of FDA-approved PLGA NPs as safe nanocarriers for use in humans, TMA-PLGA NPs could be realistically proposed for clinical translation.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Acknowledgments
This work was partly supported by Fondazione Regionale per la Ricerca Biomedica, Fondazione Cariplo-Regione Lombardia (Grant 2016-0886) and Academic Funding Unimib 2016. CB is thankful to Fondazione Umano Progresso for fellowship.
Supplementary materials
Figure S1 Synthesis of TZ-loaded polymeric NPs.
Note: Schematic description of the synthetic procedure to obtain double-loaded PLGA-NPs.
Abbreviations: TZ, trastuzumab; NPs, nanoparticles; PLGA, poly(lactic-co-glycolic) acid.
Figure S2 Evaluation of TZ secondary structure integrity after release.
Notes: (A) Protein secondary structure content determined from CD spectra by using the BeStSel method. Error bars represent the standard deviations of the results obtained from the analysis of six independent CD spectra for each sample. (B) Second-derivatives of the FTIR absorption spectra of CTZ and RTZ in the Amide I region. Second derivative minima allow for identification of the main absorption components. The main peaks at ~1,692 cm−1 and at ~1,641 cm−1 are assigned to the native β-sheet structures of the proteins in agreement with previous FTIR characterizations.
Abbreviations: TZ, trastuzumab; CD, circular dichroism; FTIR, Fourier transform infrared; CTZ, control TZ; RTZ, released TZ.
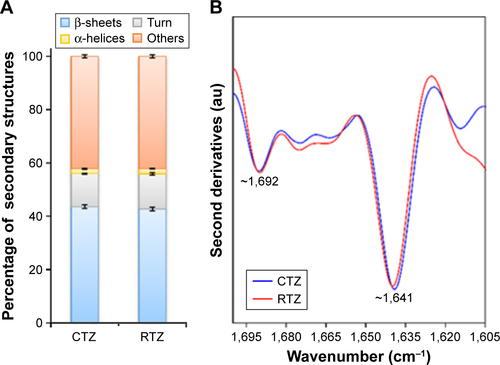
Figure S3 Dynamic light scattering measurement of NPs.
Notes: Size distribution of PLGA, PLGA-TZ and PLGA-TZ-DOXO. Results are expressed as intensity distribution. Data are representative of the average size distribution of at least three batches of nanoparticles.
Abbreviations: NPs, nanoparticles; PLGA, poly(lactic-co-glycolic) acid; TZ, trastuzumab; DOXO, doxorubicin.
Figure S4 In vitro release test.
Note: (A) TZ and (B) DOXO cumulative release in vitro.
Abbreviations: TZ, trastuzumab; DOXO, doxorubicin.
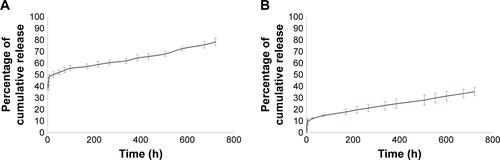
Figure S5 Evaluation of TZ primary structure integrity after release.
Note: SDS-PAGE performed on RTZ, TZ extracted with NaOH (TZ_NaOH) and unloaded TZ (TZ_wash) compared to CTZ as reference.
Abbreviations: SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; TZ, trastuzumab; RTZ, released TZ; CTZ, control TZ.
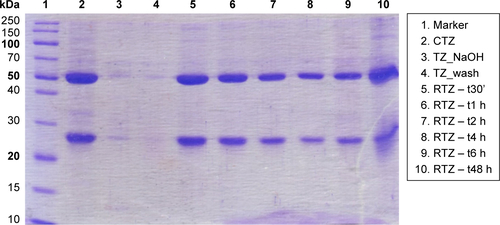
Figure S6 Binding of TZ-FITC.
Notes: (A) Basal binding of CTZ, RTZ and RIgG labeled with FITC to SKBR3 cell line. (B) Percentage of TZ-FITC binding on SKBR3 after 24 and 48 h of treatment with 2 µg mL−1 and 20 µg mL−1 of CTZ, RTZ and RIgG. ****P<0.01 vs UNTR.
Abbreviations: TZ, trastuzumab; FITC, fluorescein isothiocyanate; CTZ, control TZ; RTZ, released TZ; RIgG, released IgG; UNTR, cells coated with TZ-FITC.
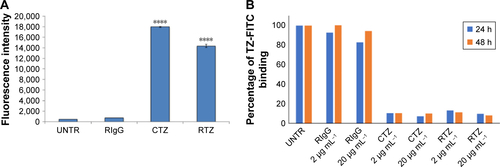
Figure S7 Western blot of unspecific IgG.
Notes: Analysis of HER2 expression on SKBR3 cells after treatment with CIgG, RIgG and IgG@PLGA at 2 µg mL−1 for 24 h. Values were calculated as ratio between HER2/β-actin and normalized with untreated cells (UNTR).
Abbreviations: HER2, human epidermal growth factor receptor 2; CIgG, control IgG; RIgG, released IgG; IgG@PLGA, IgG-loaded PLGA NPs; UNTR, cells without treatment.
Figure S8 Confocal microscopy.
Notes: Confocal microscopy untreated cells labeled with DAPI (nuclei), EEA1 (early endosomes) and CatD (lysosomes). Scale bar =50 µm.
Abbreviations: DAPI, 4′,6-diamidin-2-fenilindolo; EEA1, Early Endosome Antigen 1; CatD, Cathepsin D.
Figure S9 Western blot of unspecific IgG.
Notes: Analysis of pHER2 expression on SKBR3 cells after treatment with TZ@PLGA at 2 µg mL−1 for 4 h and 24 h. Values were calculated as ratio between pHER2/HER2 and normalized with untreated cells (UNTR).
Abbreviations: HER2, human epidermal growth factor receptor 2; TZ@PLGA, trastuzumab-loaded poly(lactic-co-glycolic) acid nanoparticles; UNTR, cells without treatment.
Disclosure
The authors report no conflicts of interest in this work.
References
- ScottAMWolchokJDOldLJAntibody therapy of cancerNat Rev Cancer201212427828722437872
- GotwalsPCameronSCipollettaDProspects for combining targeted and conventional cancer therapy with immunotherapyNat Rev Cancer201717528630128338065
- ZumlaARaoMWallisRSHost-Directed Therapies Network consortiumHost-directed therapies for infectious diseases: current status, recent progress, and future prospectsLancet Infect Dis2016164e47e6327036359
- EckerDMJonesSDLevineHLThe therapeutic monoclonal antibody marketMAbs20157191425529996
- HudisCATrastuzumab – mechanism of action and use in clinical practiceN Engl J Med20073571395117611206
- BriaECupponeFDi MaioMCetuximab for metastatic colorectal cancerN Engl J Med200936119596
- SawyersCTargeted cancer therapyNature2004432701529429715549090
- HuangACPostowMAOrlowskiRJT-cell invigoration to tumour burden ratio associated with anti-PD-1 responseNature20175457652606528397821
- WeinerLMSuranaRWangSMonoclonal antibodies: versatile platforms for cancer immunotherapyNat Rev Immunol201010531732720414205
- ChildsRWCarlstenMTherapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakensNat Rev Drug Discov201514748749826000725
- Gras NavarroABjörklundATChekenyaMTherapeutic potential and challenges of natural killer cells in treatment of solid tumorsFront Immunol2015620225972872
- HanselTTKropshoferHSingerTMitchellJAGeorgeAJThe safety and side effects of monoclonal antibodiesNat Rev Drug Discov20109432533820305665
- BöttgerRHoffmannRKnappeDDifferential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serumPLoS One2017126e017894328575099
- MittapalliRKAdkinsCEBohnKAMohammadASLockmanJALockmanPRQuantitative fluorescence microscopy measures vascular pore size in primary and metastatic brain tumorsCancer Res201777223824627815391
- SousaFCastroPFontePKennedyPJNeves-PetersenMTSarmentoBNanoparticles for the delivery of therapeutic antibodies: dogma or promising strategy?Expert Opin Drug Deliv201629114
- VarshochianRRiazi-EsfahaniMJeddi-TehraniMAlbuminated PLGA nanoparticles containing bevacizumab intended for ocular neovascularization treatmentJ Biomed Mater Res A2015103103148315625773970
- LiFHurleyBLiuYLeonardBGriffithMControlled release of bevacizumab through nanospheres for extended treatment of age-related macular degenerationOpen Ophthalmol J20126545822798970
- DanhierFAnsorenaESilvaJMCocoRLe BretonAPréatVPLGA-based nanoparticles: an overview of biomedical applicationsJ Control Release2012161250552222353619
- KamalyNYameenBWuJFarokhzadOCDegradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug releaseChem Rev201611642602266326854975
- KhangGRheeJMJeongJKLocal drug delivery system using biodegradable polymersMacromol Res2003114207223
- ManishMRahiAKaurMBhatnagarRSinghSA single-dose PLGA encapsulated protective antigen domain 4 nanoformulation protects mice against Bacillus anthracis spore challengePLoS One201384e6188523637922
- FornagueraCDols-PerezACalderóGGarcía-CelmaMJCamarasaJSolansCPLGA nanoparticles prepared by nano-emulsion templating using low-energy methods as efficient nanocarriers for drug delivery across the blood-brain barrierJ Control Release201521113414326057857
- JainAKSwarnakarNKDasMAugmented anticancer efficacy of doxorubicin-loaded polymeric nanoparticles after oral administration in a breast cancer induced animal modelMol Pharm2011841140115121557558
- JainAKSwarnakarNKGoduguCSinghRPJainSThe effect of the oral administration of polymeric nanoparticles on the efficacy and toxicity of tamoxifenBiomaterials201132250351520934747
- LiuHSlamovichEBWebsterTJLess harmful acidic degradation of poly(lactic-co-glycolic acid) bone tissue engineering scaffolds through titania nanoparticle additionInt J Nanomedicine20061454154517722285
- SousaFCruzAFontePPintoIMNeves-PetersenMTSarmentoBA new paradigm for antiangiogenic therapy through controlled release of bevacizumab from PLGA nanoparticlesSci Rep201771373628623267
- SánchezATobíoMGonzálezLFabraAAlonsoMJBiodegradable micro- and nanoparticles as long-term delivery vehicles for interferon-alphaEur J Pharm Sci2003183–422122912659933
- Carrillo-CondeBRDarlingRJSeilerSJRamer-TaitAEWannemuehlerMJNarasimhanBSustained release and stabilization of therapeutic antibodies using amphiphilic polyanhydride nanoparticlesChem Eng Sci201512598107
- GreenfieldNJUsing circular dichroism spectra to estimate protein secondary structureNat Protoc2006162876289017406547
- MicsonaiAWienFKernyaLAccurate secondary structure prediction and fold recognition for circular dichroism spectroscopyProc Natl Acad Sci U S A201511224E3095E310326038575
- GoormaghtighERaussensVRuysschaertJMAttenuated total reflection infrared spectroscopy of proteins and lipids in biological membranesBiochim Biophys Acta19991422210518510393271
- DetermanASWilsonJHKipperMJWannemuehlerMJNarasimhanBProtein stability in the presence of polymer degradation products: consequences for controlled release formulationsBiomaterials200627173312332016504288
- FontePSoaresSSousaFStability study perspective of the effect of freeze-drying, using cryoprotectants on the structure of insulin Loaded into PLGA nanoparticlesBiomacromolecules201415103753376525180545
- SainzVPeresCCimanTOptimization of protein loaded PLGA nanoparticle manufacturing parameters following a quality-by-design approachRSC Adv20166106104502104512
- OcchipintiEVerderioPNatalelloAInvestigating the structural biofunctionality of antibodies conjugated to magnetic nanoparticlesNanoscale20113238739020877896
- GinestierCAdélaïdeJGonçalvèsAERBB2 phosphorylation and trastuzumab sensitivity of breast cancer cell linesOncogene200726507163716917525746
- ColomboMCorsiFFoschiDHER2 targeting as a two-sided strategy for breast cancer diagnosis and treatment: outlook and recent implications in nanomedical approachesPharmacol Res201062215016520117211
- AustinCDDe MazièreAMPisacanePIEndocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycinMol Biol Cell200415125268528215385631
- NagataYLanKHZhouXPTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patientsCancer Cell20046211712715324695
- ColomboMFiandraLAlessioGTumour homing and therapeutic effect of colloidal nanoparticles depend on the number of attached antibodiesNat Commun201671381827991503
- CorsiFFiandraLDe PalmaCHER2 expression in breast cancer cells is downregulated upon active targeting by antibody-engineered multifunctional nanoparticles in miceACS Nano2011586383639321790185
- DokmanovicMWuYShenYChenJHirschDSWuWJTrastuzumab-induced recruitment of Csk-homologous kinase (CHK) to ErbB2 receptor is associated with ErbB2-Y1248 phosphorylation and ErbB2 degradation to mediate cell growth inhibitionCancer Biol Ther20141581029104124835103
- DokmanovicMWuWJTrastuzumab-induced HER2 phosphorylation: exploring the mechanisms and implicationsReceptor Clin Invest201414e340
- EspelinCWLeonardSCGerettiEWickhamTJHendriksBSDual HER2 targeting with trastuzumab and liposomal-encapsulated doxorubicin (MM-302) demonstrates synergistic antitumor activity in breast and gastric cancerCancer Res20167661517152726759238
- CollinsDMO’DonovanNMcGowanPMO’SullivanFDuffyMJCrownJTrastuzumab induces antibody-dependent cell-mediated cytotoxicity (ADCC) in HER-2-non-amplified breast cancer cell linesAnn Oncol20122371788179522056974
- GuillereyCHuntingtonNDSmythMJTargeting natural killer cells in cancer immunotherapyNat Immunol20161791025103627540992