 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
The purpose of this study was to develop a transdermal ligustrazine patch containing a stable formulation and with good entrapment efficiency, release rate, and transdermal absorption.
Methods
Ligustrazine ethosomes were prepared by ethanol injection-sonication, with entrapment efficiency as an indicator. Using acrylic resin as the primary constituent, the ligustrazine ethosome patch was prepared by adding succinic acid as a crosslinking agent and triethyl citrate as a plasticizer. In vitro release and transdermal permeation studies were carried out. Finally, a pharmacokinetic study was carried out in rats to explore relative bioavailability. The formulations of ligustrazine ethosome were 1% (w/v) phospholipid, 0.4% (w/v) cholesterol, and 45% (v/v) ethanol.
Results
Ligustrazine ethosomes were obtained with an average particle size of 78.71 ± 1.23 nm and an average entrapment efficiency of 86.42% ± 1.50%. In vitro transdermal testing of the ligustrazine ethosome patches showed that the cumulative 24-hour amount of ligustrazine was up to 183 ± 18 μg/cm2. The pharmacokinetic results revealed that the relative bioavailability was 209.45%.
Conclusion
Compared with conventional ligustrazine administration, ligustrazine ethosome patches could promote better drug absorption and increase bioavailability. This study demonstrates that the transdermal action of the ligustrazine ethosome patch was comparatively good.
Keywords:
Introduction
Ligustrazine has the chemical name 4-methyl-pyrazine (tetramethylpyrazine), with a chemical formula of C8H12N2 and a molecular weight of 136.2. A traditional Chinese medicine for angina pectoris, ligustrazine plays a role in expanding blood vessels, increasing coronary and cerebral blood flow, preventing platelet aggregation, inhibiting thrombosis, and improving the microcirculation.Citation1 It has low toxicity, although 30 cases of adverse drug reactions have been reported, including anaphylactic shock, erythra, angioneurotic edema, angina pectoris, hypotension, acute transient ischemic attack, severe headache, prematurity, severe gastrointestinal reactions, and severe asthma.Citation2
Because angina pectoris is associated with high morbidity, is a chronic condition, and requires long-term medication, a transdermal patch for drug delivery would be a good choice for treatment. Compared with conventional oral administration, drug delivery by a dermal patch has the advantages of avoiding first-pass hepatic metabolism and destruction in the gastrointestinal tract, reducing side effects, improving compliance with medication, and dosage control.Citation3
Flexible liposomes are common vectors in transdermal drug-delivery systems, with relatively good liquidity and deformability. Currently there are three types of flexible liposomes, ie, transfersomes, ethosomes, and niosomes. In recent years, ethosomes have become new liposome carriers with high deformability, high entrapment efficiency, and a good transdermal permeation rate in the drug-delivery system, and are suitable for transdermal administration.Citation4,Citation5 Compared with other liposomes, the physical and chemical properties of ethosomes make these more effective for drug delivery through the stratum corneum into the blood circulation, which is very important in the design of a transdermal drug-delivery system.Citation6,Citation7 It has been reported that there were no significant changes in average particle size, distribution, and structure of ethosomes over two years.Citation8 One study has found that the size of liposomes significantly increased with time, while the average size of ethosomes basically remained constant over four weeks.Citation9 It has also been found that ethosomes are well distributed when cholesterol is included in the formulation, and that they are prone to aggregation in the absence of cholesterol. It is thought that cholesterol stabilizes into a bilayer when ethosomes are maintained in a gel state, and that the high concentration of ethanol in ethosomes can ensure mobility of the vesicles, and that a moderate amount of cholesterol could ensure stability.Citation10–Citation12 The purpose of this study was to develop a sustained-release dermal patch with a stable formulation and good entrapment efficiency, release rate, and transdermal absorption.
Materials and methods
Instruments and materials
The sonication instrument used in this study was purchased from Thermo Fisher Scientific Co (San Nicolas de los Garza, Mexico). The Zetasizer 1000 type laser particle size analyzer was obtained from Malvern Instruments (Malvern, UK). A MFP-3D type atomic force microscope was obtained from Asylum Research Co (Santa Barbara, CA). High-performance liquid chromatography (ZorbaxSB-C18 column, 150 mm × 4.6 mm, 5.0 μm) was from Dionex Co (Chelmsford, MA). The Sigma 3K30 type high-speed refrigerated centrifuge was from Sigma Co (Munich, Germany). Soybean lecithin was from Avanti Polar Lipids Inc (Alabaster, AL). The ligustrazine reference substance was obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). The ligustrazine drug substance was sourced from the Huapeng Natural Plant Development Co Ltd (Shanxi, China), and the poloxamer was from the Tianjin Chemical Reagent Research Institute (Tianjin, China). Acrylic resin IV was obtained from the Iodine Factory (Lianyungang, China), and succinate was from the Dongxing Reagent Factory (Shenyang, China). Triethyl citrate was obtained from the Qingpu Synthetic Reagent Factory (Qingpu, China). Azone was obtained from the Guangzhou Chemical Auxiliary Agent Factory (Guangzhou, China). Other reagents were of analytical pure grade. Twenty-eight male Sprague Dawley rats weighing 250–300 g were obtained from the Experimental Animal Center of Xiangya School of Medicine (Xiangya, China).
Preparation of echosome
Ethosome was prepared by the ethanol injection-sonication method,Citation13 and was composed of 1% (w/v) lecithin, 0.4% (w/v) cholesterol, 45% (v/v) ethanol, ligustrazine, and water. Lecithin was dissolved with ethanol in a glass bottle. Ligustrazine was dissolved in double-distilled water and mixed uniformly with a magnetic stirrer. A glass bottle was hermetically connected with a syringe, which allowed for addition of ethanol and avoidance of evaporation. After the ligustrazine was dissolved, an ethanol solution of lecithin was added at a flow rate of 200 μL/min, then homogenized with an ultrasonic probe for five minutes. Thereafter, ligustrazine ethosome suspension was filtered using 0.45 μm disposable filters. All the procedures were performed under nitrogen gas at room temperature. The final concentration of the drug was adjusted to 10 g/L by controlling the volume. The empty ethosome suspension was obtained by not adding any ligustrazine during the preparation process.
Determination of particle size
The particle size of the ethosomes was measured immediately after preparation using a laser dynamic scattering instrument. The same percentage of water–ethanol solution in the ethosome preparation (filtered with a 0.22 μm filter) was used for a sample diluent. Five different batches of ethosomes were investigated to calculate the average particle size.
Content and entrapment efficiency
Drug content was determined by high-performance liquid chromatography as described elsewhere.Citation14 The entrapment efficiency of ligustrazine ethosomes was determined as previously described.Citation15
Preparation of patch
The crosslinking agent has an impact on the performance of an acrylic pressure-sensitive adhesive to a certain extent. In order to improve performance, succinic acid was added along with an acrylic resin as a crosslinking agent, and triethyl citrate as a plasticizer. Appropriate amounts of acrylic resin, triethyl citrate, succinate, azone, and ligustrazine ethosomes were well stirred to form a gel, evenly coating the back layer of polyethylene film. After drying at room temperature, a protective layer of polystyrene film was coated on the gel, thus obtaining the ligustrazine ethosome transdermal patch.
Stability testing
Three batches of samples were sealed with aluminum foil pouch packaging, and kept at a constant temperature of 40°C and a relative humidity of 75%. The samples were collected at the first, second, and third month to determine their content and observe their stability.
In vitro release rate
Six ligustrazine ethosome patches were glued to the center of a round square stainless steel assembly plate with a diameter of 90 mm and a length of 78 mm. The silver square hard plastic film was carefully peeled off. A homogenated dissolution device was used, with 1000 mL of 0.9% sodium chloride solution as the solvent at 32 ± 0.5°C. The assembly plate was placed flat on the bottom of the container, keeping the release side upward and parallel to the homogenate edge as well as the surface of the dissolution medium. The bottom edge of the agitating vane was 25 ± 2 mm from the plate of the device. Air bubbles adsorbed on the release surface were removed. The device was then rotated at 50 rpm. A 10 mL sample of solution was taken, without filtering, as the test solution at hours 2, 4, 6, 12, and 24. At the same time, 10 mL of 0.9% sodium chloride solution at the same temperature was added to the container. The cumulative amount released was calculated by the drug concentration of the test solutions detected using high-performance liquid chromatography.
In vitro transdermal test of patch
Six healthy male Sprague Dawley rats (weight 250–300 g) were killed and the abdominal hair was removed. After washing with normal saline, the abdominal skin was peeled off with a scalpel, and the subcutaneous tissue and fat were removed. The intact rat skin was immersed into normal saline and stored in a refrigerator at 4°C until use within 24 hours. The self-modified Franz diffusion cell used in the experiment had a receiving chamber (central diameter 2 cm, cover area 3.14 cm2, and volume 11.0 mL). The isolated pretreated rat skin was removed from the refrigerator, fixed on the modified Franz diffusion cell, and the horny skin orientated toward the release chamber. The prepared transdermal ligustrazine ethosome patch was then fixed to the rat skin. The receiving chamber was filled with normal saline 11 mL as the diffusion medium because ligustrazine has better solubility in this medium. The surface of the isolated skin in the release chamber just touched the receiving solution in the receiving chamber. The entire diffusion chamber device was put on the constant-temperature magnetic stirrer and stirred continuously at 200 rpm and 37°C. The solution was removed from the receiving chamber at hours 1, 2, 3, 4, 5, 6, 8, 10, 12, 16, and 24, respectively. Fresh diffusion medium was then added at the same volume. The solution collected was filtered through a 0.45 μm microporous membrane. The primary filtrate was discarded, and 20 μL of the subsequent filtrate was collected as a sample for analysis by high-performance liquid chromatography. The cumulative transdermal amount of drug was calculated according to the formula: Qn = CnV/A, where Cn was the correction concentration of the drug at t time, V was the total volume of receptor solution, Qn was the cumulative transdermal amount, and A was the effective diffusion area. Linear regression analysis was conducted according to the Qn-t curve to obtain the linear slope (dQ/dt) and steady-state permeation rate of the drug, Js (μg·cm− 2·s−1).
Pharmacokinetic test of patch in rats
For the pharmacokinetic study, 81 male Sprague Dawley rats weighing 250–300 g were randomly divided into three groups, ie, Group A (intragastric ligustrazine, n = 27), Group B (transdermal ligustrazine ethosome patch, n = 27), and Group C (conventional transdermal ligustrazine patch, n = 27). The dose of ligustrazine administered was 100 mg/kg. Three rats in each group were killed at hours 0.1, 0.2, 0.5, 1, 2, 4, 8, 12, and 24 after administration. Thereafter, 3 mL blood (with heparin) was collected from the abdominal aorta, and centrifugated for 15 minutes at 4000 rpm. The supernatant sample of blood plasma was taken out and adjusted to a volume of 1 mL with normal saline. Five percent diethyldithiocarbamate solution was added and mixed thoroughly, and then bathed in water for 30 minutes at 37°C. The sample was taken out and cooled at room temperature, washed with 2 mL of chloroform, and vortexed for two minutes. The sample was centrifugated for 10 minutes at 2000 rpm and separated using chloroform, washed with chloroform again, and dried using dry air in a constant water bath at 37°C. The sample was dissolved in a 100 μL mobile phase and filtered for high-performance liquid chromatography detection. Finally, the plasma drug concentration in the three groups was analyzed using 3p97 software for pharmacokinetic parameters. Methodological tests showed that the plasma content was in line with linear regression at 1–250 μg/mL, with a regression equation of A = 226.8590C + 72.2584, r = 0.9994, which met the requirements of the experiment.
Statistical analysis
SPSS software (v. 13.0; SPSS Inc., Chicago, IL) was used for statistical analysis. Pharmacokinetic data were analyzed by 3p97 software (Chinese Pharmacological Society, Beijing, China).
Results
General characteristics
Diluted ligustrazine ethosomes were measured using a laser particle size analyzer, and had an average particle size of 78.71 ± 1.23 nm and a polydispersity index of 0.145 ± 0.042. The particle size of ligustrazine ethosomes had a narrow distribution, as shown in .
Figure 1 Particle size and size distribution of ligustrazine ethosomes.
The three prepared batches of ligustrazine ethosome samples were detected according to their compositions by high-performance liquid chromatography. The percentage content was 99.08%, 97.94%, and 98.62%, respectively, with an average entrapment efficiency of 86.42% ± 1.50%.
Stability testing showed that the appearance, content, and viscosity of the ligustrazine ethosome patches remained unchanged over three months, which indicated good stability of the patches.
In vitro release rate
The in vitro release results are shown in .
Table 1 The in vitro release result of ligustrazine ethosome patches
The correlation coefficients of these equations were compared, and the Higuchi equation was found to have the optimal fit. The drug release rate was 19.54 μg·cm−2·h−1/2 according to the linear slope of the Higuchi equation.
Transdermal performance
Cumulative transdermal and steady-state permeation rates are shown in .
Table 2 Cumulative transdermal and steady-state permeation rates
Pharmacokinetics
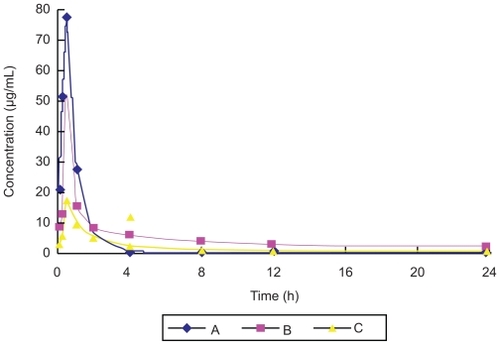
In the pharmacokinetic experiment, plasma drug concentrations decreased two hours after administration in Groups A, B, and C. The plasma concentration of Group B was high and stable after two hours, as shown in . The pharmacokinetic results for the three groups were consistent with a two-compartment model. The value of the area under the concentration-time curve (AUC) was 80.42 μg·ml−1·h in Group A, 168.38 μg·ml−1·h in Group B, and 79.31 μg·ml−1·h in Group C. The AUC value of Group B was 2.09 times greater than that of Group A, and 2.12 times greater than that of Group C. The relative bioavailability of ligustrazine ethosome patches in this study was 209.45%, but that of the conventional ligustrazine patches was only 98.63%. This demonstrated that ligustrazine ethosome patches could improve drug absorption and bioavailability compared with the other two preparations. In addition, the T1/2β value was 1.94 hours in Group A, 18.99 hours in Group B, and 29.89 hours in Group C, respectively. Compared with orally administered ligustrazine, ligustrazine ethosome patches had a significantly longer elimination half-life and more stable plasma concentrations at the high level. Although the elimination half-life of conventional ligustrazine patches was longer than that of ligustrazine ethosome patches, the plasma concentration was lower. Therefore, ligustrazine ethosome patches have an obvious advantage compared with the other two preparations.
Figure 2 Plasma concentration–time curve of three ligustrazine preparations (A: Intragastric administration group of ligustrazine; B: Transdermal drug delivery goup of ligustrazine ethosome patch; C: Transdermal drug delivery group of conventional ligustrazine patch).
Discussion
The combined methods of ethanol injection and sonication were used in the preparation of ligustrazine ethosomes in this study. We found that the particle size of the ligustrazine ethosomes decreased as ethanol concentration increased, but increased as the phospholipid concentration increased. Ligustrazine is a water-soluble drug. Ethanol could increase the solubility of ligustrazine, and the entrapment efficiency of the ethosomes containing a water-soluble drug was higher than that of conventional vesicle preparations.Citation7 Compared with liposomes, the most striking feature of ethosomes is the relatively high concentration of ethanol. On the one hand, the presence of ethanol increases the flexibility of the lipid bilayer in ethosomes; on the other hand, the interaction between ethanol and the lipid of the stratum corneum reduces the phase transition temperature of the latter, thus promoting mobility and drug penetration. It has been reported that a low ethanol concentration is not conducive to entrapment of drugs in ethosomes, thus affecting drug loading, and that a high alcohol concentration is not conducive to ethosome formation because phospholipids can easily dissolve in ethanol. Therefore, the ethanol concentration in the ethosome system should not be too high, and generally should be kept below 45%.Citation16
The molecular weight of the polymer is an important determinant of the properties of an acrylic pressure-sensitive adhesive. The crosslinking formed by the action between succinic acid and the quaternary ammonium group of acrylic resin IV may enhance the molecular weight. Generally, persistent adhesivity increases as the amount of crosslinking agent increases. Further increasing the amount of the crosslinking agent will increase both the vitrification temperature and the rigidity of the glue film, leading to a decrease in adhesivity of the patches. In this study, succinic acid and triethyl citrate were added to the polyacrylic acid resin to adjust the initial adhesivity, persistent adhesivity, and cohesive strength of the acrylic pressure-sensitive adhesive, so as to improve the biocompatibility of the acrylic pressure-sensitive adhesive patches.
Regardless of whether transdermal absorption has a systemic action or a local effect, drugs should pass through the stratum corneum. However, due to the barrier effect of the stratum corneum, many drugs cannot play a full role. Ethosomes have been used for transdermal drug delivery in recent years because they can enable a drug to pass through the skin and increase the accumulation of drug in the skin.Citation16 According to one report,Citation15 the steady-state transdermal rate of trihexyphenidyl hydrochloride ethosomes composed of phospholipid, ethanol, and water increased by 87-fold compared with normal liposomes.
In vitro transdermal release rates are important indicators of quality for a transdermal drug-delivery system. The transdermal drug absorption process can be divided into three stages, ie, release from the matrix, passage through the stratum corneum barrier, epidermis, and dermis, and absorption by blood vessels. Some active ingredients of Chinese traditional medicines cannot achieve a therapeutic dose by passive absorption alone. Therefore, a suitable method to promote penetration is needed. Ligustrazine was prepared in ethosome patches in this study. The cumulative amount of the transdermal drug was regarded as an evaluation index in our in vitro transdermal experiment. A self-prepared Franz diffusion cell was used. According to the test results, the 24-hour cumulative amount of transdermal drug release was up to 183 ± 18 μg/cm2, which greatly increased the transdermal effect of ligustrazine. In our release test, the in vitro release curve of ligustrazine ethosome patches was consistent with the Higuchi equation within 24 hours, which indicated a good sustained-release effect.
In order to improve the safety and bioavailability of ligustrazine and to avoid the first-pass hepatic effect of oral administration, the transdermal drug-delivery system has been used in medical treatment.Citation17 In that study, the AUC of the ligustrazine ethosome patch group was 2.09-fold greater than that of the oral ligustrazine group, and was 2.12-fold greater than that of the conventional ligustrazine patch group. Compared with oral ligustrazine, the relative bioavailability of ligustrazine ethosomes was up to 209.45%, while that of a conventional transdermal patch was only 98.63%. There was little difference in AUC values between the conventional ligustrazine patch group and the oral ligustrazine group, indicating a poor transdermal effect of the conventional ligustrazine patch. These results indicate that ligustrazine ethosome patches play a role in promoting drug absorption and increasing bioavailability compared with the other two preparations.
In a toxicity test of ethosomes, Paolino et al studied the difference between blank ethosomes, sodium chloride (0.9%, v/v), and ethanol–water solution. They found that an ethanol– water solution caused significant skin erythema, whereas blank ethosomes did not, even in a high concentration of ethanol. This indicates that ethosomes cause no significant irritation to human skin.Citation18 Histological testing of erythromycin ethosomes also revealed that these ethosomes caused no damage to skin.Citation12 For testosterone ethosomes (including 25% ethanol, w/w), skin irritation was not found during the treatment period.Citation19 Moreover, in a toxicity test of fibroblasts, ethosomes caused no significant toxicity to cultured cells in vitro.Citation20 For these reasons, we can conclude that transdermal drug delivery via ethosomes has potentially extensive applications in medicine as a result of its high efficiency, convenient administration, and limited toxicity.
Disclosure
The authors report no conflicts of interest in this work.
References
- DouYZTengHWangQSunYMMaSLPreparation, in vitro release and percutaneous penetration of ligustrazine hydrochloride transdermal delivery systemChinese Journal of Pharmaceutics20083910745749
- ZengCYMeiQXADRs induced by tetramethylpyrazine: Literature analysis of 30 casesJ China Pharm2008192419081910
- PrausnitzMRMitragotriSLangerRCurrent status and future potential of transdermal drug deliveryNat Rev Drug Discov20043211512415040576
- FangYPTsaiYHWuPCHuangYBComparison of 5-aminolevulinic acid- encapsulated liposome versus ethosome for skin delivery for photodynamic therapyInt J Pharm20083561214415218242901
- JainSTiwaryAKSapraBJainNKFormulation and evaluation of ethosomes for transdermal delivery of lamivudineAAPS Pharm Sci Tech200784E111
- BouwstraJAHoneywell-NguyenPLSkin structure and mode of action of vesiclesAdv Drug Deliv Rev200254Suppl 1S415512460715
- LiuXXRaoYfLiangWQStudy on transdermal penetration of ethinyl estradiol ethosome gelChin Pharmaceut J2006414284286
- ZhuWWZhaiGXZhaoJProgresson ethosomesFood and Drug20079014649
- Lopez-PintoJMGonzalez-RodriguezMLRabascoAMEffect of cholesterol and ethanol on dermal delivery from DPPC liposomesInt J Pharm2005298111215896932
- JainSJainPUmamaheshwariRBJainNKTransfersomes – a novel vesicular carrier for enhanced transdermal delivery: Development, characterization, and performance evaluationDrug Dev Ind Pharm20032991013102614606665
- CevcGLipid vesicles and other colloids as drug carriers on the skinAdv Drug Deliv Rev200456567571115019752
- GodinBTouitouERubinsteinEAthamnaAAthamnaMA new approach for treatment of deep skin infections by an ethosomal antibiotic preparation: An in vivo studyJ Antimicrob Chemother200555698999415857943
- ZengZWWangXLZhangYDLiNFPreparation of matrine ethosome, its percutaneous permeation in vitro and anti-inflammatory activity in vivo in ratsJ Liposome Res200919215516219241204
- LiaoCXLuoSMZhuYYDetermination of ligustrazine phosphate for injection by HPLCJ China Pharm2007183326002601
- DayanNTouitouECarriers for skin delivery of trihexyphenidyl HCl: Ethosomes vs liposomesBiomaterials200021181879188510919691
- TouitouEDayanNBergelsonLGodinBEliazMEthosomes – novel vesicular carriers for enhanced delivery: Characterization and skin penetration propertiesJ Control Release200065340341810699298
- QiuLWangQZhangJHuoNBStudies on inhibition of crystallization and in vitro percutaneous absorption of tetramethylpyrazine transdermal delivery systemChin Pharmaceut J20062116421646
- PaolinoDLucaniaGMardenteDAlhaiqueFFrestaMEthosomes for skin delivery of ammonium glycyrrhizinate: In vitro percutaneous permeation through human skin and in vivo anti-inflammatory activity on human volunteersJ Control Release2005106129911015970347
- AinbinderDTouitouETestosterone ethosomes for enhanced transdermal deliveryDrug Deliv200512529730316188729
- GodinBTouitouEErythromycin ethosomal systems: Physicochemical characterization and enhanced antibacterial activityCurr Drug Deliv20052326927516305429