
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
α-Mangostin is a major active compound of mangosteen (Garcinia mangostana L.) pericarp extract (MPE) that has potent antioxidant activity. Unfortunately, its poor aqueous solubility limits its therapeutic application. Purpose: This paper reports a promising approach to improve the clinical use of this substance through electrospinning technique.
Methods
Polyvinylpyrrolidone (PVP) was explored as a hydrophilic matrix to carry α-mangostin in MPE. Physicochemical properties of MPE:PVP nanofibers with various extract-to-polymer ratios were studied, including morphology, size, crystallinity, chemical interaction, and thermal behavior. Antioxidant activity and the release of α-mangostin, as the chemical marker of MPE, from the resulting fibers were investigated.
Results
It was obtained that the MPE:PVP nanofiber mats were flat, bead-free, and in a size range of 387–586 nm. Peak shifts in Fourier-transform infrared spectra of PVP in the presence of MPE suggested hydrogen bond formation between MPE and PVP. The differential scanning calorimetric study revealed a noticeable endothermic event at 119°C in MPE:PVP nanofibers, indicating vaporization of moisture residue. This confirmed hygroscopic property of PVP. The absence of crystalline peaks of MPE at 2θ of 5.99°, 11.62°, and 13.01° in the X-ray diffraction patterns of electrospun MPE:PVP nanofibers showed amorphization of MPE by PVP after being electrospun. The radical scavenging activity of MPE:PVP nanofibers exhibited lower IC50 value (55–67 µg/mL) in comparison with pure MPE (69 µg/mL). The PVP:MPE nanofibers tremendously increased the antioxidant activity of α-mangostin as well as its release rate. Applying high voltage in electrospinning process did not destroy the chemical structure of α-mangostin as indicated by retained in vitro antioxidant activity. The release rate of α-mangostin significantly increased from 35% to over 90% in 60 minutes. The release of α-mangostin from MPE:PVP nanofibers was dependent on α-mangostin concentration and particle size, as confirmed by the first-order kinetic model as well as the Hixson–Crowell kinetic model.
Conclusion
We successfully synthesized MPE:PVP nanofiber mats with enhanced antioxidant activity and release rate, which can potentially improve the therapeutic effects offered by MPE.
Introduction
Mangosteen (Garcinia mangostana L.) is a tropical plant that is cultivated mostly in Southeast Asian countries. Xanthone-rich pericarp of mangosteen has demonstrated numerous biological activities including anti-inflammatory, antiparasitic, antitumor, and antioxidant activities.Citation1–Citation5 Xanthones exist as either oxygenated or prenylated form. The latter, particularly α-mangostin, β-mangostin, and γ-mangostin as the most abundant xanthone-type compounds, has gained a special attention due to its beneficial effects for health.Citation6–Citation8 Various studies have reported α-mangostin to exhibit antioxidant,Citation9–Citation11 antimicrobial,Citation12–Citation14 anti-inflamatory,Citation15–Citation17 and anticancer activities.Citation18,Citation19 However, the limited aqueous solubilityCitation20,Citation21 property constricts bioavailability of α-mangostin via oral route. Hence, an appropriate delivery system is absolutely required to improve the efficacy of α-mangostin.
Nanotechnology seems to be a smart way to solve the clinical problem of α-mangostin. α-Mangostin can be incorporated into nano-sized biocompatible carriers such as bio-compatible nanofibers. Decreasing particle size to nanoscale can greatly increase the surface area for a given quantity of biodegradable polymer material, which in turn significantly enhances the release of drug via drug diffusion and matrix degradation/erosion mechanisms.Citation22–Citation26 Nanofibers have been recognized for their applications in drug delivery.Citation27–Citation32 Nanofibers as drug delivery systems with their very large surface area to volume ratios have the potential to improve drug release significantly. Furthermore, the small dimension of fibers combined with their microporous structure provided by the polymer mimics a protective shield, resulting in increased drug loading and drug stability.Citation33
Electrospinning is the most versatile technique for the synthesis of nanofibers.Citation34,Citation35 This technique involves Coulomb forces resulted from the applied electrical charge and elongation of polymer solution as a result of exposure to electrical charge. These events lead to the formation of fine fibers and accumulation of the fibers onto a grounded-collector.Citation36 The size and morphology of the resulting fibers are highly tunable by proper adjustment of polymer properties (structure, molecular weight, and tacticity), precursor solution (viscosity and conductivity), and electrostatic field.Citation37 Electrospinning offers great capability of producing fibers ranging from very small diameter to ≥10 nm and presents good mechanical characteristics with microporous structures and controlled surfaces,Citation29 which makes electrospun fibers potentially demonstrate promising results as a drug delivery system.
Recently, few studies have reported successful development of polymer/mangosteen pericarp extract (MPE) nanofibers intended for various purposes. For instance, MPE was loaded onto polyvinyl alcohol (PVA) nanofibers for dermal delivery purposeCitation38 and was spun in a mixture with chitosan (CS)/EDTA/PVA for wound healing.Citation39 In such cases, the choice of polymer must be considered carefully since polymer can affect aqueous solubility of active compound and its release profile. Moreover, the underlining mechanism on how the polymer influences the release profile of the active substance needs to be understood. Water-soluble polymers such as PVA are a suitable matrix for nanofibers with rapid release. However, a highly soluble polymer such as PVP might be required in some cases, particularly when a faster release is desired.Citation40 Therefore, the use of PVP for the production of MPE-loaded nanofibers intended for immediate release would be advantageous. Successful production of MPE:PVP nanofibers using electrospinning technique has been previously reported.Citation34 However, comprehensive studies that investigate contributing factors affecting in vitro performance of MPE-containing nanofiber mats, such as crystallinity, size, and drug-to-polymer ratio, are still scarce.
In this study, we synthesized PVP:MPE nanofiber mats using electrospinning technique, in which PVP is an US Food and Drug Administration–approved polymer matrix that is acceptable for food and pharmaceutical products with low toxicity and highly biocompatible.Citation34,Citation41 In addition, it has great water solubility and spinnability,Citation42 which are important for electrospinning process. It was expected that PVP would assist the release of α-mangostin more rapidly in comparison with other water-soluble polymers. The morphology, physicochemical characteristics, and in vitro release of α-mangostin, the marker compound for antioxidant activity of MPE, from the MPE:PVP nanofiber mats were examined. Moreover, in vitro antioxidant activity of α-mangostin was determined to study if a high voltage application during electrospinning impairs the biological activity of active compounds contained in MPE.
Materials and methods
Materials
Polyvinylpyrrolidone (PVP) with molecular weight of 1,300,000 kg mol−1, pure α-mangostin powder, and 1,1-diphenyl-2-picrylhydrazyl (DPPH) were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Mangosteen pericarp was collected from a local market in Bandung, Indonesia. Other chemical substances used for this study were of analytical grade.
α-Mangostin content assay
Pure MPE was prepared as follows. The fruit pericarps were rinsed with water thoroughly, cut into small pieces, and dried in oven at 50°C for 24 hours. The dried pericarps were milled into fine powder. The mangosteen pericarp powder was macerated with ethanol for 5 days. The as-macerated extract was filtered and concentrated using a rotary evaporator (RV 05-ST Janke & Kunkel IKA, Staufen, Germany) at 45°C and stored properly until used. α-Mangostin content in the MPE was analyzed using a high-performance liquid chromatography (HPLC) system as follows. MPE was accurately weighed and was dispersed in methanol with the final MPE content of 1,000 ppm. The mixture was homogenized for 20 minutes to allow for the extraction of α-mangostin. The mixture was then centrifuged at 10,000 rpm for 10 minutes, and the supernatant was collected. Dilution of the supernatant was performed accordingly.
The sample was injected into a C-18 column (250 × 4.6 mm, particle size of 5 µm; Phenomenex, CA, USA) using the HPLC system with a UV spectrophotometer detector (Agilent Technologies, Santa Clara, CA, USA). The mobile phase was water–methanol (95:5) with a flow rate of 1 mL/min. The pure MPE content was detected at a wavelength of 320 nm.
Preparation of nanofibers
PVP:MPE fibers were fabricated from a PVP:MPE precursor solution prepared with in situ process, in which PVP and MPE solutions were dissolved separately and then mixed together just before electrospinning process. The MPE solution (10 wt%) was prepared by dissolving MPE in ethanol at room temperature under constant stirring for 5 hours. Separately, the PVP solution was prepared in ethanol at 40°C and stirred for 2 hours. The PVP and MPE solutions were mixed and stirred for 1 hour at room temperature to form a homogeneous precursor solution.
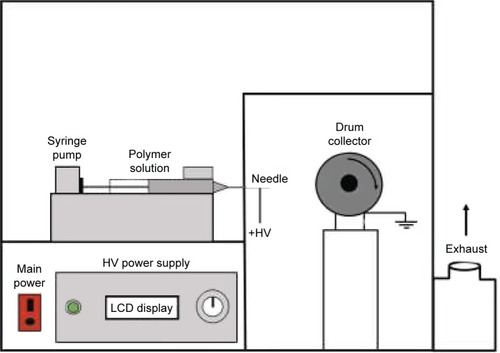
An electrospinning apparatus (Nachriebe 600, Nachriebe; Integrated Laboratory of Materials and Instrumentation, Department of Physics, ITB, Bandung, Indonesia), which is schematically described in , was used.Citation67 It consists of a syringe pump to discharge the precursor solution in a syringe with needle, a high-voltage power supply (HVPS) with the positive pole connected to the needle to induce fiber formation via a solution jet, and a drum collector connected to the negative pole of HVPS to collect fibers. This apparatus produced fibers with very large surface area to volume ratio. Each precursor solution to produce nanofibers as given in , which is labeled as MF0, MF1, MF2, or MF3, was loaded into a 10 mL syringe with 0.45 mm needle. During electrospinning process, the flow rate of syringe pump was maintained at 5 µL/min, the high voltage at 10 kV, and the distance between needle and collector at 12 cm. The resulting fiber mats were subjected to α-mangostin content assay by using the method described in the previous section.
Table 1 The mass ratio of MPE and PVP for electrospinning process
Figure 1 The schematic diagram of electrospinning apparatus used in the experiments.
Abbreviations: HV, high voltage; LCD, liquid crystal display.
Viscosity and conductivity
Viscosity and conductivity of the MF0, MF1, MF2, and MF3 precursor solutions were determined by using a Fenske-Ostwald viscometer (Thermo Fisher Scientific, Waltham, MA, USA) and a conductivity meter (Mettler Toledo) at 25°C, respectively.
Scanning electron microscopy
A scanning electron microscope (SEM, JSM-6510; JEOL, Tokyo, Japan) was used to examine the morphology of mats resulted from the MF0, MF1, MF2, and MF3 precursor solutions. Conductive coating of sample was applied prior to SEM imaging. SEM images of the mats were observed at the voltage of 10 kV and an optical magnification of 10,000 times. The size distribution of the fibers was determined using Origin ver.9 software (Origin Lab Corporation, MA, USA).
Fourier-transform infrared (FTIR) spectroscopy
FTIR analysis was performed to investigate the possible chemical interactions between PVP and MPE in the nanofibers. The FTIR spectra of nanofiber mats were acquired from an FTIR spectrophotometer (Alpha; Bruker, Germany) with spectral range of 500–4,000 cm−1.
X-ray diffraction (XRD)
An X-ray diffractometer (D8 Advance, Bruker) was used to obtain the XRD patterns of the pure MPE along with the MF0, MF1, MF2, and MF3 nanofiber mats. The sample was placed in a standard Cu tube and irradiated with Cu (Kα) with a wavelength of 1.5405 Å at an operating voltage of 40 kV and electric current of 35 mA. The angular position (2θ) of the diffraction pattern was recorded in the range of 5°–70°.
Differential scanning calorimetry
Thermal behaviors of the MF1, MF2, and MF3 nanofiber mats in comparison with the PVP (MF0) nanofiber mat were studied by a differential scanning calorimeter (DSC, STA PT1600; Linseis, NJ, USA). Accurately weighed samples were sealed in an aluminum crimp pan and scanned from 50°C to 270°C at a heating rate of 10°C/min.
Antioxidant activity
Scavenging activity of MPE/PVP nanofiber mats was performed by using a modified method reported by Blois.Citation43 Various concentrations of the pure MPE were prepared, ranging from 10 to 80 µg/mL. Each preparation was reacted with equal volume of DPPH solution at 50 µg/mL concentration. The mixture was incubated for 30 minutes. The MF1, MF2, and MF3 nanofiber mats were treated likewise. The absorbance was measured after 30 minutes of incubation by using a UV-Visible spectrophotometer (DU 7500i; Beckman Coulter, CA, USA) at the wavelength of 515 nm. Methanol was used for blank absorbance reading. An amount of 50 µg/mL of DPPH solution was used as a control solution, and ascorbic acid was used as a reference material for antioxidant activity. The analysis was conducted in triplicate for ascorbic acid, pure MPE, and the nanofiber mats. Antioxidant activities of the pure MPE, MF1, MF2, and MF3 nanofiber mats were determined based on the reduction of DPPH absorbance by calculating the percentage of antioxidant activity.Citation44
In vitro release study
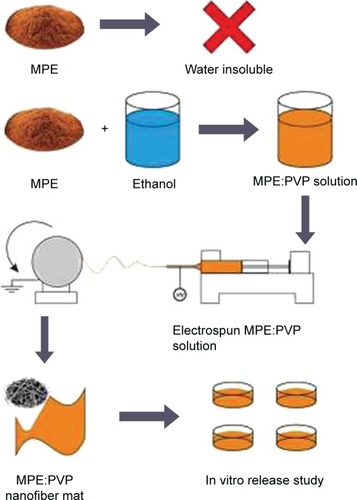
Release profile of α-mangostin from the MF1, MF2, and MF3 nanofiber mats were studied on a dissolution test apparatus (SR8 Plus; Hanson Research, Los Angeles, CA, USA). Dissolution apparatus 2 (paddle apparatus) was used with the following conditions: paddle rotation speed of 50 rpm, 400 mL of phosphate-buffered solution (pH 6.8) as the medium, and temperature controlled at 37°C±0.5°C. Each nanofiber mat containing 40 mg of MPE was carefully weighed and was dispersed in the phosphate-buffered solution. This procedure was run triplicate. About 5 mL of aliquot was taken at 5, 10, 15, 30, 45, 60, and 120 minutes and the medium was replaced with equal volume of fresh buffer. The amount of α-mangostin released over time was measured by using the HPLC system at 320 nm as described previously. The summary of our study is illustrated in .
Figure 2 Schematic illustration of the experiment.
Notes: α-Mangostin is poorly soluble in aqueous environment. Precursor solution was prepared by mixing MPE with PVP solution. The precursor solution was electrospun into fiber mat. Finally, in vitro evaluation and characterization were performed to predict the pharmacological effect in vivo.
Abbreviations: MPE, mangosteen pericarp extract; PVP, polyvinylpyrrolidone.
Release kinetics
The release kinetics of the pure MPE, PVP:MPE physical mixture, and nanofiber mats were investigated using zero-order model, first-order model,Citation45,Citation46 and Hixson–Crowell model,Citation47 as follows:
1. Zero-order model
In this model, the cumulative amount of drug released (Qt) is plotted against time. The pattern of drug release adopts the condition that the rate of drug release is independent of its concentration as shown by EquationEq (1)
(1) .
where Qt is the total drug released at time t (in percentage concentration) and k0 is the constant of zero-order model (in concentration/time).2. Fist-order model
In this model, the log of the cumulative percentage of drug remaining is plotted against time. In this model, the rate of drug release is assumed to be dependent on its concentration as presented in EquationEq (2)
where Q0 is the initial concentration of the drug, k is the constant of first-order model, and t is the time.3. Hixson–Crowell model
This model is based on Hixson and Crowell (1931) recognizing that the regular area of the particle is proportional to the cubic root of its volume. This model has been used to describe the release profile considering the diminishing surface of the drug particles during the dissolution as in EquationEq (3)
where Q0 is the initial concentration of the drug, Qt is the amount of drug released at time t, and k is the constant of Hixson–Crowell model. A straight line can be gained by plotting
Results and discussion
α-Mangostin content
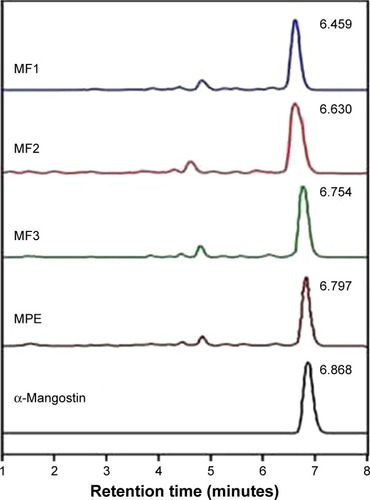
α-Mangostin as the major xanthone in mangosteen pericarp was identified as a marker compound for quantitative analysis and standardization of MPE.Citation12,Citation48 Standardization of the MPE is required to maintain the product quality. In this study, this was achieved by determining the amount of α-mangostin contained in each nanofiber mat and the amount of α-mangostin released over time. It was found that the amount of α-mangostin in the pure MPE, MF1, MF2, and MF3 nanofiber mats were 13%, 1.3%, 2.6%, and 3.4%, respectively. When incorporated into nanofibers, the release rate of α-mangostin increased threefold in comparison with that of pure MPE, with more than 90% of α-mangostin released in just an hour. shows HPLC chromatograms of standard α-mangostin, MPE, and MF1, MF2, MF3 nanofiber mats. α-Mangostin was detected from the nanofiber samples by using the HPLC system at retention time of 6.4–6.7 minutes. The chromatogram showed that α-mangostin peak was clearly distinguished from the baseline and the other peaks.
Figure 3 HPLC chromatograms of α-mangostin, MPE and MF1-, MF2-, MF3 nanofiber mats.
Abbreviations: MPE, mangosteen pericarp extract; MF0, mangosteen fiber 0; MF1, mangosteen fiber 1; MF2, mangosteen fiber 2; MF3, mangosteen fiber 3; HPLC, high-performance liquid chromatography.
Morphology and average diameter of nanofibers
The morphology of MF0, MF1, MF2, and MF3 fiber mats observed by SEM is shown in . The fibers were commonly ribbon-shaped without flaws such as bead formation or broken strands, with diameter in nanometer range. The presence of MPE with different mass concentrations in PVP fibers did not affect their morphologies. This finding is similar to our previous work in which the presence of curcumin in PVP nanofibers did not affect the shape of nanofibers.Citation22 Although there were some polar–polar interactions between curcumin and PVP, the nanofibers were still in a uniform shape. Therefore, the formation of PVP:MPE nanofibers were absolutely influenced by the concentration of PVP:MPE solution in electrospinning process.
Figure 4 SEM images of nanofiber mats and their fiber-size distributions.
Note: SEM image at 10,000× of MF0, MF1, MF2, and MF3 nanofiber mats.
Abbreviations: SEM, scanning electron microscope; MF0, mangosteen fiber 0; MF1, mangosteen fiber 1; MF2, mangosteen fiber 2; MF3, mangosteen fiber 3.
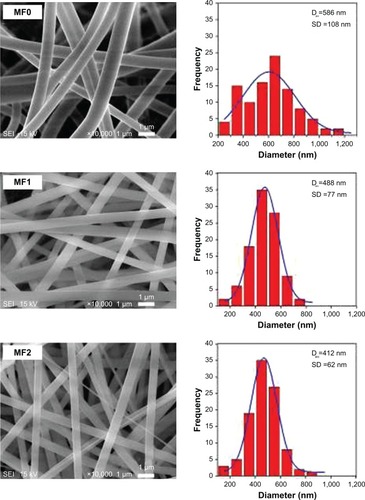
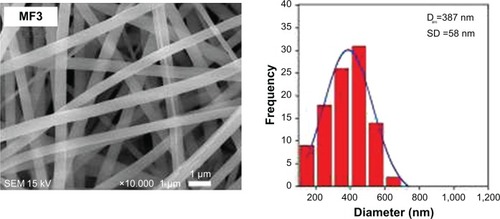
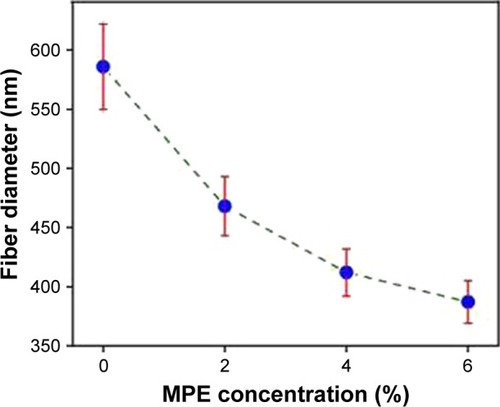
The diameters of MF0, MF1, MF2, and MF3 nanofibers of their mats varied in the range of 200 to 1,200 nm. The average diameters of these nanofibers were 586, 464, 412, and 387 nm, respectively. illustrates the average diameter of the nanofibers as a function of the MPE loaded to the nanofibers. As clearly depicted in the graph, the diameter of nanofibers decreased with the increase in MPE concentration. A possible reason is due to the influence of decreased viscosity and increased electric conductivity of the precursor solution. The viscosity of pure PVP precursor solution (MF0) was found to be 70.10 cP. In the presence of MPE, its viscosity decreased to 67.86, 61.76, and 55.87 cP for the MF1, MF2, and MF3 precursor solutions, respectively. The low-viscosity precursor solution resulted in less polymer unit per volume in the solution. Consequently, the polymer chains tend to interact with the solvent rather than to form a coiled structure and to entrap the solutes. Under this condition, the number of intermolecular interactions that can be formed between PVP and MPE such as van der Waals force, hydrogen bond, and dipole–dipole interaction is lower. These weak interactions caused PVP chains to straighten out in the solvent and be easily affected by the given electric field.Citation36,Citation49 The straightened PVP chains are more likely to achieve perfect elongation upon exposure to electrical charge during the electrospinning process, resulting in finer fibers. These presumptions can explain our findings where the diameter of the fibers became smaller when the viscosity decreased, with smallest diameter obtained in the MF3 nanofibers.
Figure 5 Correlation between the diameter of nanofibers and MPE concentration in the nanofibers.
Abbreviation: MPE, mangosteen pericarp extract.
It was found that the electrical conductivity of the PVP:MPE precursor solution increased as the concentration of MPE increased. The electrical conductivity of the MF3 precursor solution was the highest, that is, 258.3 µS/cm, while the MF1 had the lowest electrical conductivity at 118.2 µS/cm. Upon exposure to high voltage, the electrical conductivity of the precursor solution represents the number of ions at the surface of the solution. The MF3 precursor solution with a higher electrical conductivity underwent a rapid elongation due to higher ion formation on the surface of the solution.Citation36,Citation50 This phenomenon also explained a significant drop in the diameter of the electrospun nanofibers due to the increased electrical conductivity. These findings are in line with other studies that reported similar observation.Citation51
FTIR spectroscopy analysis
FTIR study was conducted to identify characteristic functional groups in MPE, PVP (MF0) nanofiber mat, and MPE:PVP (MF1, MF2, and MF3 nanofiber mats). The recorded spectra are shown in . Hydroxyl group in the MPE was identified from a broad peak at 3,326 cm−Citation1, which belongs to O-H stretching.Citation21 This peak confirmed the presence of polyphenolic xanthones in the MPE.Citation52 The peaks at 2,972 and 2,925 cm−Citation1 were assigned to asymmetric C-H stretching of the methyl group.Citation20,Citation21 The presence of ester group in the MPE was indicated by sharp peaks at 1,639 cm−Citation1 (C=O stretching of carbonyl group) and 1,279 cm−Citation1 (C-O-C stretching of methoxy group).Citation20,Citation52 The medium-intensity peak at 1,423 cm−Citation1 was assigned to asymmetric C=C stretching from aromatic ring.Citation20 Alkene groups in the MPE were confirmed by the presence of peaks at 838 and 583 cm−Citation1 for =C-H bending.Citation53 The characteristic peaks of PVP were noticed in the FTIR spectrum of MF0 nanofiber mat: O-H stretching at around 3,420 cm−Citation1, C=O stretching at 1,653 cm−Citation1, and C-N stretching at 1,288 cm−Citation1.Citation54,Citation55 Asymmetric stretching of CH2 in PVP chain was shown by the moderate peak at 2,952 cm−Citation1.Citation55
Figure 6 (A) FTIR spectra of MPE, MF0, MF1, MF2, and MF3 nanofiber mats; (B) hydrogen bonding between PVP and α-mangostin.
Abbreviations: FTIR, Fourier transform infrared spectroscopy; MPE, mangosteen pericarp extract; MF0, mangosteen fiber 0; MF1, mangosteen fiber 1; MF2, mangosteen fiber 2; MF3, mangosteen fiber 3; PVP, polyvinylpyrrolidone.
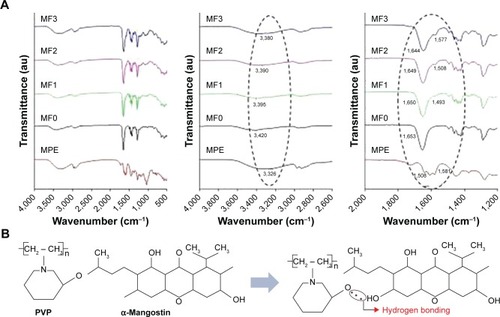
We observed two distinct features of infrared spectra of the MPE-containing nanofiber mats: 1) a higher MPE content led to sharper and more noticeable hydroxyl peaks occurring around 3,800–3,000 cm−Citation1 and 2) a higher MPE content caused hydroxyl and carbonyl peak of PVP to shift toward lower wavenumbers. The hydroxyl peak of PVP in the MF0 nanofiber mat at 3,420 cm−Citation1 shifted to 3,395 cm−Citation1 in the MF1 nanofiber mat, while it changed to 3,390 and 3,380 cm−Citation1 in the MF2 and MF3 nanofiber mats, respectively. The carbonyl stretching peak that belongs to pyrrolidone ring in PVP appeared at 1,653 cm−Citation1. This peak appeared at 1,650 cm−Citation1 in the MF1 nanofiber mat, while it existed at 1,649 and 1,644 cm−Citation1 in the MF2 and MF3 nanofiber mats, respectively. The peak shift of carbonyl stretching was thought to be a result of intermolecular interaction between MPE and PVP via hydrogen bond formation,Citation21 as shown in . The hydrogen bond is formed between hydroxyl group among the tricyclic rings in the MPE, as a strong hydrogen bond donor, and carbonyl group in the PVP, as a strong hydrogen bond acceptor. However, the presence of OH- stretching in the spectrum of pure PVP and its direction of peak shift indicated that the PVP chains were partially hydrated, owing to its hygroscopic nature.Citation22 The adsorbed moisture on the surface of PVP was also confirmed by thermal analysis, which is described later in our DSC results. As a result, the moisture contained in PVP chains also formed hydrogen bonds with carbonyl groups in MPE. This premise is supported by the peak shift of aromatic C=C stretching of MPE from 1,581 to 1,577 cm−Citation1 in MF3, to 1,508 cm−Citation1 in the MF2 nanofiber mat, and to 1,493 cm−Citation1 in the MF1 nanofiber mat. The peak shift of C=C stretching in the presence of PVP occurred as the result of decreased resonance of carbonyl-conjugated aromatic alkenesCitation56 as the free carbonyl groups of MPE became hydrogen-bonded with hydroxyl groups of PVP. Considering that the glass transition point (Tg) of PVP is around 178°C,Citation22 the PVP chains had sufficient rigidity to preserve this hydrogen bond-mediated intermolecular interaction under room temperature.Citation57
XRD analysis
XRD patterns of the MPE, MF0, MF1, MF2, and MF3 nanofiber mats are shown in . Using XPowder Ver.2004.04.70 PRO software, a typical diffraction pattern of MPE was found with intense peaks at 2θ of 5.99°, 11.62°, and 13.01°, indicating crystalline nature of MPE. The pattern and peaks exhibited by the MPE resemble those of mangosteen extract, despite slightly different 2θs (5.94°, 11.43°, and 12.69°).Citation58 Due to the high abundance of α-mangostin content in the MPE, it was then thought that the diffraction pattern demonstrated by the MPE was originated from α-mangostin.Citation45 Sharp and intense crystalline peaks in the MF1, MF2, and MF3 nanofiber mats were absent in the MF0 nanofiber mat. In contrast, two broad haloes were present at 2θs of 5° and 40°. The second halo at 2θ of 22.72° was a shouldered peak with relatively lower intensity. The XRD pattern of the MF0 nanofiber mat clearly indicated amorphous PVP, as reported in the previous study.Citation27
Figure 7 XRD patterns of MPE, MF0, MF1, MF2, and MF3 nanofiber mats.
Abbreviations: XRD, X-ray diffraction; MPE, mangosteen pericarp extract; MF0, mangosteen fiber 0; MF1, mangosteen fiber 1; MF2, mangosteen fiber 2; MF3, mangosteen fiber 3.
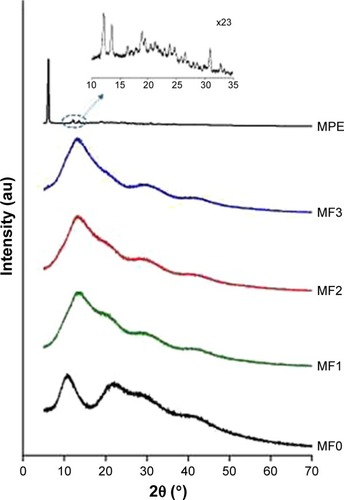
The XRD patterns of the MPE and PVP:MPE nanofiber mats suggested a crystalline-to-amorphous transformation of MPE, as indicated by the absence of crystalline peaks of the MPE in the MF1, MF2, and MF3 nanofiber mats. Note that PVP was reportedly capable of converting drug powder from crystalline state to amorphous state, particularly hydrophobic non-steroid anti-inflammatory drugs including indomethacin, ketoprofen, naproxen, and ibuproxam,Citation59–Citation62 in a binary system generated from co-milling, hot melt extrusion, or other mixing processes under a certain environment where PVP is in rubbery state (above the glass transition point, Tg).Citation60–Citation62 The degree of amorphization was dependent on the PVP content and the mechanism of amorphization involved loosening of crystal structure during exothermic mixing process, followed by dispersion in amorphous PVP chains and stabilization by intermolecular hydrogen bond between drug and PVP.Citation62 Amorphization power of PVP is noteworthy, owing to its spherical amorphous shape.Citation62 Below the Tg, PVP was able to induce amorphization of ibuprofen in a physical mixture over the storage time.Citation60 In our study, conversion into amorphous state most probably occurred during the electrospinning process although the environment temperature was below the Tg of PVP. The applied voltage caused the drug/polymer solution to rapidly migrate from the needle tip to the collector. At the same time, the polymer underwent elongation along with solvent evaporation, resulting in solidification into a fiber.Citation36 Within this extremely short time period, PVP and MPE molecules were incapable of rearranging their three-dimensional structure. As a result, the molecules were not highly ordered as in their crystalline state.
Our XRD study indicated molecular interactions between MPE and PVP, which strengthened the FTIR and DSC results. The XRD pattern of PVP was altered after being spun in a combination with MPE. The MPE nanofiber mats showed a broad halo instead of two haloes, peaking at 2θ of 13°–15°. Meanwhile, the characteristic halo of PVP at 2θ of 22.72° was not detected in the MF1, MF2, and MF3 nanofiber mats. This finding strengthened our presumption that MPE and MF0 (PVP) formed intermolecular interactions, particularly as hydrogen bonds, as explained in the FTIR studies.
Differential scanning calorimetry analysis
shows DSC thermograms of the MPE, MF0, MF1, MF2, and MF3 nanofiber mats. Three endothermic events were identified in MPE, peaking at 119°C, 177°C, and 218.1°C. The first endothermic peak was thought to originate from residual volatile compounds, mostly sesquiterpenes,Citation63 since MPE was not exposed to temperature of >50°C throughout our study. The endothermic peaks at 177°C and 218.1°C indicated the melting point and decomposition of MPE, respectively, in which MPE was represented by the α-mangostin content. The DSC thermogram of PVP showed a broad endothermic event from 35°C to 100°C due to the evaporation of adsorbed moisture. Our thermogravimetric analysis showed that the weight loss of the MF0 nanofiber mat at 100°C was 3.6%. The Tg of PVP was not detected in the MF0, MF1, MF2, and MF3 nanofiber mats, which was attributed to mechanically induced stress during electrospinning process.Citation22
Figure 8 DSC thermograms of MPE, MF0, MF1, MF2, and MF3 nanofiber mats.
Abbreviations: DSC, differential scanning calorimetry; MPE, mangosteen pericarp extract; MF0, mangosteen fiber 0; MF1, mangosteen fiber 1; MF2, mangosteen fiber 2; MF3, mangosteen fiber 3.
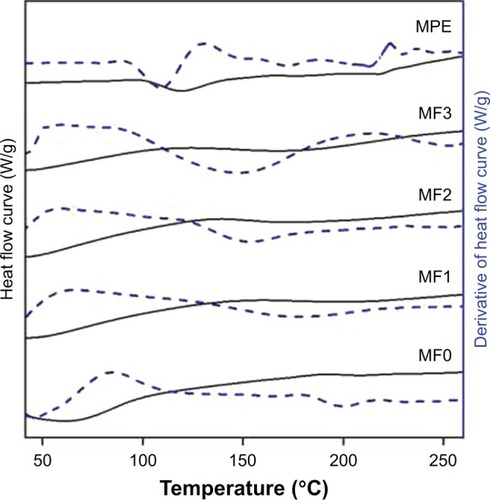
The DSC thermogram of the MF1, MF2, and MF3 nanofiber mats were similar except that the endothermic event taking place at 199.3°C in the MF1 nanofiber mat occurred at lower temperatures when the amount of MPE was increased, being 173.8°C in the MF2 nanofiber mat and 167.7°C in the MF3 nanofiber mat. These peaks were extremely broad, resembling a diffusion of melting points of MPE and PVP. Characteristic crystalline peaks of the MPE were unable to observe in the MF1, MF2, and MF3 nanofiber mats, most probably due to the condition where electrospinning process of the MPE:PVP solution inhibited the recrystallization of α-mangostin.Citation45 Moreover, the appearance of a single broad halo instead of fusion endotherms indicated complete amorphization.Citation62 The decrease in crystallinity of α-mangostin was also confirmed in the XRD analysis.
In vitro antioxidant activity
Antioxidant activity was detected in all MPE:PVP nanofiber mats (MF1, MF2, and MF3 nanofiber mats). The color of DPPH solution changed from purple to yellow after being reacted with MPE:PVP mats, indicating the quenching of DPPH free radicals by hydrogen donor groups in MPE:PVPV and, eventually, the formation of a stable compound.Citation64,Citation65 The hydrogen donor groups from α-mangostin, most commonly hydroxyl groups, play a fundamental role in scavenging the free radicals. The extent of antioxidant activity in this study was described by an IC50 value.Citation43,Citation66–Citation68 A very strong antioxidant will have an IC50 value between 1 and 50 µg/mL, while strong antioxidant and moderate antioxidant will demonstrate IC50 values in 50–100 and 101–150 µg/mL range, respectively. IC50 value of ≥150 µg/mL indicates weak antioxidant activity.Citation67,Citation68
The IC50 values of the MPE, MF1, MF2, and MF3 nanofiber mats were compared to ascorbic acid as the reference for antioxidant activity (). It is clear that the MF3 nanofiber mat had the strongest antioxidant activity among the nanofiber mats as shown by its lower IC50 value. Surprisingly, the antioxidant activity of MPE in the MF1, MF2, and MF3 nanofiber mats were stronger than the pure MPE. These findings suggest that the antioxidant activity of MPE was enhanced due to the nanostructure offered by the nanofiber mats. In nanofiber form, α-mangostin particles, in nanometer size, are distributed in a matrix with very high surface area.Citation69 As a result, the number of α-mangostin particles that can be released at a given time is significantly higher, and thus, α-mangostin can quench the free radicals more effectively. Our findings show that the stability of the biologically active compounds in MPE can still be maintained during the electrospinning process. Our previous study showed a similar report, in which the antioxidant activity of MPE was not weakened by the electrospinning process, despite the use of high voltage for a long period.Citation67
Table 2 Antioxidant activities of ascorbic acid, MPE and PVP:MPE composite nanofiber mats
In vitro release study
α-Mangostin possesses limited aqueous solubilityCitation20 yet good intestinal absorption,Citation70 which means that solubility of α-mangostin is the rate-limiting step. We attempted to increase the release rate of α-mangostin by incorporating MPE into nanosized fibers, presuming that there would be higher fraction of dissolved α-mangostin readily absorbed by the intestinal membrane at given time. The release study was adapted from standard protocol of dissolution test for immediate-release drug dosage form with modifications. Until recently, there is no single standardized method for release or dissolution study of drug from nano-sized dosage form.Citation71 However, this release study can be a predictive tool to investigate the release behavior, an important biopharmaceutical aspect of nanoparticulate delivery system.
Earlier, we intended to investigate pH-dependence of α-mangostin release by studying the release rate in two different media: acidic (pH 1.2) and basic (pH 6.8). However, α-mangostin was not detected when acidic medium (pH 1.2) was used. The phosphate-buffered solution (pH 6.8) was chosen since the pH mimics the small intestine environment, the region of gastrointestinal tract where most xanthones can be absorbed.Citation72 The amount of α-mangostin released from the three nanofiber preparations and pure MPE was carried out in phosphate buffer pH 6.8 at 37°C for 2 hours. The release profiles are shown in . Overall, the release of α-mangostin from the MF1, MF2, and MF3 nanofiber mats exhibited triphasic pattern. In such pattern, extremely high release rate or burst release effect takes place in the first phase as the result of accumulation of drug molecules on the surface of the polymeric-based system. The second phase, which is identified by a slower release rate, generally indicates drug release via mild polymer degradation or chain scission.Citation73,Citation74 Finally, as the polymer is continuously hydrated, water can eventually reach the fiber further and bulk erosion occurs. The time required to reach bulk erosion makes the release rate slower than those in the prior phases.Citation73,Citation75,Citation76 The multi-phase release pattern was typical of small-molecule release from a polymeric matrix, and this was not related to the overall release rate. In fact, release rate of α-mangostin was improved by incorporation into nanofiber, being three times higher than α-mangostin release from pure MPE.
Figure 9 The release profile of α-mangostin from MPE:PVP (MF1, MF2, and MF3) nanofiber mats and pure MPE at pH 6.8.
Abbreviations: MPE, mangosteen pericarp extract; PVP, polyvinylpyrrolidone; MF0, mangosteen fiber 0; MF1, mangosteen fiber 1; MF2, mangosteen fiber 2; MF3, mangosteen fiber 3.
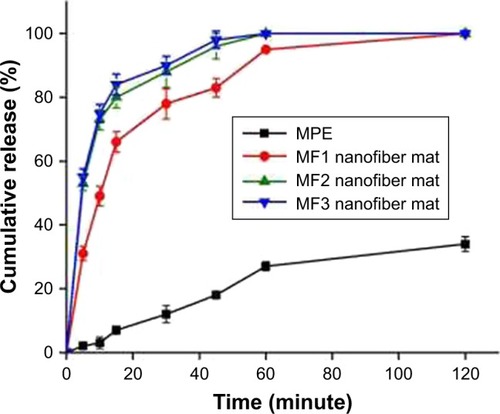
As seen in , it is clear that α-mangostin release from the pure MPE was lower in comparison with those of the MF1, MF2, and MF3 nanofiber mats. Less than 35% of α-mangostin was released from the pure MPE over 60 minutes. Meanwhile, α-mangostin in the nanofiber mats was immediately released with 90% average cumulative percentage in 60 minutes. It is clear that encapsulation of MPE into PVP-based nanofibers, regardless the ratio of MPE:PVP, has improved the rate of α-mangostin release dramatically. This indicated that the release enhancement was not only due to improvement of surface hydrophilicity by PVP.Citation77
The nature of nanofibers, where the surface area at a given volume is very high, played significant role for release rate enhancement of α-mangostin. This finding is in accordance with the Nernst–Brunner equation:
Our XRD and DSC studies indicated that conversion of MPE from crystalline state to amorphous state was due to electrospinning process. This can be another explanation for the rapid release of α-mangostin because an amorphous drug tends to have higher release rate. In amorphous state, the molecular interaction is relatively weaker and the molecular arrangement is rather irregular in comparison with crystalline state.Citation80,Citation81 Consequently, amorphous α-mangostin contained in the MF1, MF2, and MF3 nanofiber mats can be dissolved more rapidly, and thus, α-mangostin tends to diffuse out into the release medium on a higher rate.
The release rate of α-mangostin was found to be higher in nanofibers with smaller diameters. As shown in , the MF3 and MF2 nanofiber mats with respective diameters of 387 and 412 nm released 100% of α-mangostin within 60 minutes. Meanwhile, the MF1 nanofiber mat with a higher diameter of 468 nm only released 90% of α-mangostin over similar period. These results could be explained as follows. It was reported that polymer chains with smaller fibers have higher rate of absorbance of the molecules of solvent.Citation40,Citation82 Thus, it enlarges the volume of the polymer matrix which then allows the coil of polymer chains to be less tight, in which the PVP nanofibers reach a swollen state. As a result, the solvent can reach α-mangostin particles and dissolve them more easily. The dissolved α-mangostin can then immediately diffuse out of the matrix.
Release kinetics
A number of kinetic models represent release mechanisms of a certain drug from a certain matrix; zero-order, first-order, Higuchi, and Hixson–Crowell are examples of those kinetic models.Citation45–Citation47 To study the release mechanism of α-mangostin from the fibers, release patterns were fitted to those five models based on R-square value (). In vitro release of α-mangostin from the pure MPE followed the zero-order kinetic model, but it followed the first-order one when formulated into nanofibers. This means that the release of active compound in nanofibers was mainly controlled by drug concentration. In contrast, the release of α-mangostin from the pure MPE was concentration-independent. However, the release kinetic model of the MF1, MF2, and MF3 nanofiber mats also fitted well with the Hixson–Crowell model. This indicated that changes in surface area and diameter of matrix contributed for the release mechanism.Citation46,Citation83,Citation84
Table 3 Comparison of several release kinetic models for MPE:PVP (MF1, MF2, and MF3) nanofiber and MPE
The Hixson–Crowell kinetic model considers that 1) the mass of dissolved compound does not significantly change with time, 2) the dissolving particles are spherical and do not change with time, and 3) the particles do not disintegrate into several fragments during dissolution.Citation85 As illustrated in , the release rates of α-mangostin from the MF1, MF2, and MF3 nanofiber mats were significantly high in the first 20 minutes, but dropped quickly afterwards. Despite the presumed mechanism of triphasic release which was thought to be facilitated by drug accumulation on the matrix surface, polymer chain scission and bulk erosion,Citation77,Citation79 another possibility should be taken into consideration. This is because the first-order kinetic is based on the following assumptions: 1) the matrix is a non-swelling compound and 2) the matrix remains intact throughout the release process.Citation86
In the previous study, it was found that the solid dispersion of ketoconazole/PVP nanoparticles grew into micrometer-sized particles during dissolution study.Citation87 Therefore, it is also possible that the α-mangostin molecules, which have been released from the nanofiber mat during the first phase of release (ie, >60% in the first 20 minutes), undergo supersaturation and precipitate into nanoparticles. The nanoparticles were subjected to nucleation and thus particle growth occurred. This event might result in a decreased release rate in the subsequent phase, where the amount of released α-mangostin was <30% from 20 to 60 minutes.
Conclusion
Bead-free nanofibers were successfully synthesized from electrospinning of MPE:PVP solution. Size controlling was achieved by adjusting the viscosity and conductivity of precursor solution. Higher MPE content in the precursor solution lowered the viscosity while increased the conductivity. The SEM analysis revealed that the size of the nanofiber decreased as the MPE:PVP ratio was increased. However, the morphology of MPE:PVP nanofibers was identical in all preparations regardless the MPE content, confirming the excellent spinnability of PVP. Findings of the FTIR and DSC analyses suggested hydrogen-bond formation between MPE and PVP although PVP still retained its hygroscopic property. Crystalline-to-amorphous transformation was evident, as verified in the XRD study. α-Mangostin in the MPE:PVP nanofibers performed better radical scavenging and had significantly higher release rate in comparison with the pure MPE, as confirmed in antioxidant assay and in vitro release study. Enhanced antioxidant activity and release rate of α-mangostin from the MPE:PVP nanofibers was facilitated by high surface area of the nanofiber network. The mechanism of α-mangostin release from the MPE:PVP nanofibers was dependent on drug concentration and particle size since the release kinetic followed the first-order model as well as the Hixson–Crowell model. With the improvement of antioxidant activity and release rate, MPE:PVP nanofiber mats can potentially improve the clinical outcomes offered by MPE.
Acknowledgments
This research was financially supported by the Directorate of Research and Community Engagement of Ministry of Research, Technology and Higher Education, Republic of Indonesia under the University’s Excellence Research (PUPT) Grant in the fiscal year 2016–2017.
Disclosure
The authors report no conflicts of interest in this work.
References
- KosemNIchikawaKUtsumiHMoongkarndiPIn vivo toxicity and anti-tumor activity of mangosteen extractJ Nat Med20136725526322622784
- MoongkarndiPKosemNKaslungkaSLuanratanaOPongpanNNeungtonNAntiproliferation, antioxidation and induction of apoptosis by Garcinia mangostana (mangosteen) on SKBR3 human breast cancer cell lineJ Ethnopharmacol200490116116614698525
- WangJJSandersonBJZhangWCytotoxic effect of xanthones from pericarp of the tropical fruit mangosteen (Garcinia mangostana Linn.) on human melanoma cellsFood Chem Toxicol2011492385239121723363
- ChinY-WJungH-AChaiHKellerWJKinghornADXanthones with quinone reductase-inducing activity from the fruits of Garcinia mangostana (mangosteen)Phytochemistry20086975475817991497
- AkaoYNakagawaYLinumaMNozawaYAnti-cancer effects of xanthones from pericarps of mangosteenInt J Mol Sci2008935537019325754
- Pedraza-ChaverriJCárdenas-RodríguezNOrozco-IbarraMPérez-RojasJMMedicinal properties of mangosteen (Garcinia mangostana)Food and Chem Toxicol2008463227323918725264
- HemshekharMSunithaKSanthoshMSAn overview on genus Garcinia: phytochemical and therapeutical aspectsPhytochem Rev201110325351
- HanA-RKimJ-ALantvitDDCytotoxic xanthone constituents of the stem bark of Garcinia mangostana (mangosteen)J Nat Prod2009722028203119839614
- KondoMZhangLJiHKouYOuBBioavailability and antioxidant effects of a xanthone-rich mangosteen (Garcinia mangostana) product in humansJ Agric Food Chem2009578788879219807152
- YuLZhaoMYangBZhaoQJiangYPhenolics from hull of Garcinia mangostana fruit and their antioxidant activitiesFood Chem2007104176181
- IbrahimMYHashimNMMariodAAα-Mangostin from Garcinia mangostana Linn: an updated review of its pharmacological propertiesArab J Chem20149317329
- PalakawongCSophanodoraPPisuchpenSPhongpaichitSAntioxidant and antimicrobial activities of crude extracts from mangosteen (Garcinia mangostana L.) parts and some essential oilsInt Food Res J201017583589
- Al-MassaraniSMEl GamalAAAl-MusayeibNMPhytochemical, antimicrobial and antiprotozoal evaluation of Garcinia mangostana pericarp and α-mangostin, its major xanthone derivativeMolecules201318105991060824002136
- ArunrattiyakornPSuksamrarnSSuwannasaiNKanzakiHMicrobial metabolism of α-mangostin isolated from Garcinia mangostana LPhytochemistry20117273073421377704
- ChenL-GYangL-LWangC-CAnti-inflammatory activity of mangostins from Garcinia mangostanaFood Chem Toxicol20084668869318029076
- Gutierrez-OrozcoFChitchumroonchokchaiCLesinskiGBSuksamrarnSFaillaMLα-Mangostin: anti-inflammatory activity and metabolism by human cellsJ Agric Food Chem2013613891390023578285
- LiuS-HLeeL-THuN-YEffects of alpha-mangostin on the expression of anti-inflammatory genes in U937 cellsChin Med201271922920833
- ShibataM-AIinumaMMorimotoJα-Mangostin extracted from the pericarp of the mangosteen (Garcinia mangostana Linn) reduces tumor growth and lymph node metastasis in an immunocompetent xenograft model of metastatic mammary cancer carrying a p53 mutationBMC Med201196921639868
- JohnsonJJPetiwalaSMSyedDNα-Mangostin, a xanthone from mangosteen fruit, promotes cell cycle arrest in prostate cancer and decreases xenograft tumor growthCarcinogenesis20123341341922159229
- AhmadMYaminBMLazimAMA study on dispersion and characterisation of α-mangostin loaded pH sensitive microgel systemsChem Cent J201378523680098
- AishaAFIsmailZAbu-SalahKMMajidAMSASolid dispersions of α-mangostin improve its aqueous solubility through self-assembly of nanomicellesJ Pharm Sci201210181582522081501
- RahmaAMunirMMKhairurrijalKPrasetyoASuendoVRachmawatiHIntermolecular interactions and the release pattern of electrospun curcumin-polyvinyl(pyrrolidone) fiberBiol Pharm Bull20163916317326830478
- KumarMNano and microparticles as controlled drug delivery devicesJ Pharm Pharm Sci2000323425810994037
- SoppimathKSAminabhaviTMKulkarniARRudzinskiWEBiodegradable polymeric nanoparticles as drug delivery devicesJ Control Release20017012011166403
- AgnihotriSAMallikarjunaNNAminabhaviTMRecent advances on chitosan-based micro-and nanoparticles in drug deliveryJ Control Release200410052815491807
- KumariAYadavSKYadavSCBiodegradable polymeric nanoparticles based drug delivery systemsColloids Surf B Biointerfaces20107511819782542
- RasekhMKaravasiliCSoongYLElectrospun PVP-indomethacin constituents for transdermal dressings and drug delivery devicesInt J Pharm20144739510424997411
- RathinamoorthyRNanofiber for drug delivery systemPak Text J2012614548
- HuXLiuSZhouGHuangYXieZJingXElectrospinning of polymeric nanofibers for drug delivery applicationsJ Control Release2014185122124768792
- BhandariJMishraHMishraPKWimmerRAhmadFJTalegaonkarSCellulose nanofiber aerogel as a promising biomaterial for customized oral drug deliveryInt J Nanomedicine2017122021203128352172
- SonYJKimWJYooHSTherapeutic applications of electrospun nanofibers for drug delivery systemsArch Pharm Res201437697824234913
- YooHSKimTGParkTGSurface-functionalized electrospun nanofibers for tissue engineering and drug deliveryAdv Drug Deliv Rev2009611033104219643152
- ShenXYuDZhuLBranford-WhiteCWhiteKChattertonNPElectrospun diclofenac sodium loaded Eudragit® L 100-55 nanofibers for colon-targeted drug deliveryInt J Pharm201140820020721291969
- SriyantiIEdikresnhaDMunirMMRachmawatiHKhairurrijalKElectrospun polyvinylpyrrolidone (PVP) nanofiber mats loaded by Garcinia mangostana L. extractsMater Sci Forum20178801114
- MunirMMIskandarFKhairurrijalKOkuyamaKA constant-current electrospinning system for production of high quality nanofibersRev Sci Instrum20087947
- RamakrishnaSFujiharaKTeoWELimTCMaZAn Introduction to Electrospinning and NanofibersSingapuraWorld Scientific Publishing Company20051396
- HuangSZhouLZhouDPreparation and properties of electrospun poly(vinyl pyrrolidone)/cellulose nanocrystal/silver nanoparticle composite fibersMaterials20169523
- OpanasopitPRuktanonchaiUSuwantongOElectrospun poly(vinyl alcohol) fiber mats as carriers for extracts from the fruit hull of mangosteenJ Cosmet Sci20085923324218528591
- CharernsriwilaiwatNRojanarataTNgawhirunpatTSukmaMOpanasopitPElectrospun chitosan-based nanofiber mats loaded with Garcinia mangostana extractsInt J Pharm201345233334323680732
- YuD-GShenX-XBranford-WhiteCOral fast-dissolving drug delivery membranes prepared from electrospun polyvinylpyrrolidone ultrafine fibersNanotechnology20092005510419417335
- DaiXYNieWWangYCShenYLiYGanSJElectrospun emodin polyvinylpyrrolidone blended nanofibrous membrane: a novel medicated biomaterial for drug delivery and accelerated wound healingJ Mater Sci20122327092716
- BonanRFBonanPRBatistaAUIn vitro antimicrobial activity of solution blow spun poly (lactic acid)/polyvinylpyrrolidone nanofibers loaded with Copaiba (Copaifera sp.) oilMater Sci Eng C201548372377
- BloisMAntioxidant determinations by the use of a stable free radicalNature195818111991200
- DeoPHewawasamEKarakoulakisAIn vitro inhibitory activities of selected Australian medicinal plant extracts against protein glycation, angiotensin converting enzyme (ACE) and digestive enzymes linked to type II diabetesBMC Complement Altern Med20161643527809834
- SamprasitWRojanarataTAkkaramongkolpornPNgawhirunpatTKaomongkolgitROpanasopitPFabrication and in vitro/in vivo performance of mucoadhesive electrospun nanofiber mats containing alpha-mangostinAAPS PharmSciTech2015161140115225716329
- SinghviGSinghSReview: in-vitro drug release characterization modelsInt J Pharm Stud Res201127784
- CostaPLoboJMSModeling and comparison of dissolution profilesEur J Pharm Sci20011312313311297896
- ThongNMQuangDTBuiNHTDaoDQNamPCAntioxidant properties of xanthones extracted from the pericarp of Garcinia mangostana (mangosteen): a theoretical studyChem Phys Lett20156253035
- DoiMEdwardsSFThe Theory of Polymer DynamicsOxfordClarendon Press2009140143
- FongHChunIRenekerDBeaded nanofibers formed during electro-spinningPolymer19994045854592
- TanSInaiRKotakiMRamakrishnaSSystematic parameter study for ultra-fine fiber fabrication via electrospinning processPolymer20054661286134
- GhazaliSISLianGEAbd GhaniKDChemical constituent from roots of Garcinia mangostana (Linn.)Int J Chem20102134142
- AminahLNLeongSTWongYSOngSAKairulazamCKBiodiesel production of Garcinia mangostana Linn seeds by two-phase solvent extraction and alkali-catalyzed transesterificationInt J Chem Eng Appl2013436
- VijayaNSelvasekarapandianSHirankumarGStructural, vibrational, thermal, and conductivity studies on proton-conducting polymer electrolyte based on poly (N-vinylpyrrolidone)Ionics2012189199
- BorodkoYHabasSEKoebelMYangPFreiHSomorjaiGAProbing the interaction of poly (vinylpyrrolidone) with platinum nanocrystals by UV− Raman and FTIRJ Phys Chem B2006110230522305917107143
- RobinsonJWFrameESFrameGMIIUndergraduate Instrumental AnalysisFloridaCRC Press2014
- ImamuraKAsanoYMaruyamaYCharacteristics of hydrogen bond formation between sugar and polymer in freeze-dried mixtures under different rehumidification conditions and its impact on the glass transition temperatureJ Pharm Sci2008971301131217683061
- SamprasitWAkkaramongkolpornPNgawhirunpatTRojanarataTKaomongkolgitROpanasopitPFast releasing oral electrospun PVP/CD nanofiber mats of taste-masked meloxicamInt J Pharm201548721322225899284
- CirriMMuraPRabascoAGinesJMoyanoJGonzalez-RodriguezMCharacterization of ibuproxam binary and ternary dispersions with hydrophilic carriersDrug Dev Ind Pharm200430657415000431
- IvanovITTsokevaZEffect of chirality on PVP/drug interaction within binary physical mixtures of ibuprofen, ketoprofen, and naproxen: a DSC studyChirality20092171972718988257
- LimRTYNgWKTanRBDissolution enhancement of indomethacin via amorphization using co-milling and supercritical co-precipitation processingPowder Technol20132407987
- ZerroukNMenniniNMaestrelliFChemtobCMuraPComparison of the effect of chitosan and polyvinylpyrrolidone on dissolution properties and analgesic effect of naproxenEur J Pharm Biopharm200457939914729084
- MacLeodAJPierisNMVolatile flavour components of mangosteen (Garcinia mangostana)Phytochemistry198221117119
- SuttirakWManurakchinakornSIn vitro antioxidant properties of mangosteen peel extractJ Food Sci Technol20121235463558
- MoongkarndiPJaisupaNSamerJComparison of the biological activity of two different isolates from mangosteenJ Pharm Pharmacol2014661171117924641353
- NsimbaRYKikuzakiHKonishiYAntioxidant activity of various extracts and fractions of Chenopodium quinoa and Amaranthus spp. seedsFood Chem2008106760766
- SriyantiIEdikresnhaDRahmaAMunirMMRachmawatiHKhairurrijalKCorrelation between structures and antioxidant activities of polyvinylpyrrolidone/Garcinia mangostana L. extract composite nanofiber mats prepared using electrospinningJ Nanomater201720179687896
- FidriannyINataliaSInsanuMAntioxidant capacities of various fruit extracts from three varieties of tomato and correlation with total phenolic, flavonoid, carotenoid contentInt J Pharm Clin Res20157283289
- HuangZMZhangYZKotakiMRamakrishnaSAReview on polymer nanofibers by electrospinning and their applications in nanocompositesCompos Sci Technol20036322232253
- ChoiYHHanSYKimY-JKimY-MChinY-WD Absorption, tissue distribution, tissue metabolism and safety of α-mangostin in mangosteen extract using mouse modelsFood Chem Toxicol20146614014624472368
- ShenJBurgessDJIn vitro dissolution testing strategies for nanoparticulate drugDrug Deliv Transl Res2013340941524069580
- Gutierrez-OrozcoFFaillaMLBiological activities and bioavailability of mangosteen xanthones: a critical review of the current evidenceNutrients201353163318323945675
- FuYKaoWJDrug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systemsExpert Opin Drug Deliv2010742944420331353
- HolowkaEBhatiaSKDrug Delivery: Materials Design and Clinical PerspectiveNew YorkSpringer2014
- GrizziIGarreauHLiSVertMHydrolytic degradation of devices based on poly (DL-lactic acid) size-dependenceBiomaterials1995163053117772670
- Von BurkersrodaFSchedlLGöpferichAWhy degradable polymers undergo surface erosion or bulk erosionBiomaterials2002234221423112194525
- LiX-YWangXYuD-GElectrospun borneol-PVP nanocompositesJ Nanomater20122012731382
- BrunnerEReaktionsgeschwindigkeit in heterogenen Systemen. ZJ Phys Chem19044756102
- KaravasEGeorgarakisESigalasMPAvgoustakisKBikiarisDInvestigation of the release mechanism of a sparingly water-soluble drug from solid dispersions in hydrophilic carriers based on physical state of drug, particle size distribution and drug–polymer interactionsEur J Pharm Biopharm20076633434717267194
- MengFTrivinoAPrasadDChauhanHInvestigation and correlation of drug polymer miscibility and molecular interactions by various approaches for the preparation of amorphous solid dispersionsEur J Pharm Sci201571122425686597
- BaghelSCathcartHO’ReillyNJPolymeric amorphous solid dispersions: a review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugsJ Pharm Sci20161052527254426886314
- YuDGWangXLiXYChianWLiYLiaoYZElectrospun biphasic drug release polyvinylpyrrolidone/ethyl cellulose core/sheath nanofibersActa Biomater201395665567223099302
- RaoKSNagabhushanamMChowdaryKIn vitro dissolution studies on solid dispersions of mefenamic acidIndian J Pharm Sci20117324322303074
- RisdianCNasirMRahmaARachmawatiHThe influence of formula and process on physical properties and the release profile of PVA/BSA nanofibers formed by electrospinning techniqueJ Nano Res201531103116
- SiepmannJSiepmannFMathematical modeling of drug dissolutionInt J Pharm2013301224
- MulyeNTurcoSA simple model based on first order kinetics to explain release of highly water soluble drugs from porous dicalcium phosphate dihydrate matricesDrug Dev Ind Pharm199521943953
- KanaujiaPLauGNgWKNanoparticle formation and growth during in vitro dissolution of ketoconazole solid dispersionJ Pharm Sci20111002876288521290385