
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
Red blood cell membrane-coated nanoparticle (RBCm-NP) platform, which consist of natural RBCm and synthetic polymeric core, can extend circulation time in vivo with an improved biocompatibility and stability of this biomimetic nanocarrier. To achieve better bioavailability of antitumor drugs that were loaded in RBCm-NPs, the functionalization of coated RBCm with specific targeting ability is essential. Bispecific recombinant protein anti-EGFR-iRGD, containing both tumor penetrating peptide (internalizing RGD peptide) and EGFR single-domain antibody (sdAb), seems to be an optimal targeting ligand for RBCm-NPs in the treatment of multiple tumors, especially colorectal cancer with high EGFR expression.
Materials and methods
We modified the anti-EGFR-iRGD recombinant protein on the surface of RBCm-NPs by lipid insertion method to construct iE–RBCm–PLGA NPs and confirmed the presentation of active tumor-targeting ability in colorectal cancer models with high EGFR expression when compared with RBCm–PLGA NPs. In addition, potential anti-tumor drug gambogic acid (GA) was loaded into the NPs to endow the antitumor efficiency of iE–RBCm–GA/PLGA NPs. It was simultaneously evaluated whether GA can reach better biocompatibility benefiting from the improved antitumor efficiency of iE–RBCm–GA/PLGA NPs in colorectal cancer models.
Results
We successfully modified anti-EGFR-iRGD proteins on the surface of biomimetic NPs with integrated and stable “shell–core” structure. iE–RBCm–PLGA NPs showed its improved targeting ability in vitro (multicellular spheroids [MCS]) and in vivo (nude mice bearing tumors). Besides, no matter on short-term cell apoptosis at tumor site (terminal deoxyribonucleotidyl transferase-mediated dUTP nick end labeling [TUNEL]) and long-term tumor inhibition, iE–RBCm–GA/PLGA NPs achieved better antitumor efficacy than free GA in spite of the similar effects of cytotoxicity and apoptosis to GA in vitro.
Conclusion
We expect that the bispecific biomimetic nanocarrier can extend the clinical application of many other potential antitumor drugs similar to GA and become a novel drug carrier in the colorectal cancer treatment.
Introduction
Nanoparticle (NP)-based drug delivery platforms are often used for various drugs to achieve good bioavailability in complex biological environments.Citation1–Citation3 With the development of multi-type prepared materials for NPs, it is not limited to the biocompatible and biodegradable synthetic materials,Citation4,Citation5 such as polylactic acid (PLA), poly(lactic-co-glycolic acid) (PLGA),Citation6 polycaprolactone (PCL), and polyethylene glycol (PEG). Natural liposomeCitation7 cell membrane with inimitable structure and function attracts more attention for another new material of cell membrane-camouflaged NPs.Citation8–Citation11 Different from conventional NPs, the biomimetic nanocarriers served as the new class of NPs integrated two distinct materials – synthetic copolymer with customizability and flexibility and natural biomembrane with functionality and complexity. Biomembrane-cloaked NPs can indicate immune evasion to prolong circulation time in vivo, and NPs are able to accumulate at tumor sites subsequently via enhanced permeability and retention (EPR) effect. Hence, the application of biomimetic nanocarriers will be of great importance in antitumor therapy.Citation12–Citation15
Many kinds of cell membranes have been used to build the biomimetic NPs, and red blood cell membrane (RBCm) was the earliest and most widely explored,Citation16–Citation24 benefiting from the enormous quantity in vivo as well as simple structure and properties. However, compared with passive target through EPR effect, active target can optimize the bioavailability of antitumor drugs and minimize off-target side effects in vivo. The surface of RBCmCitation25 lacks specific proteins or molecules that can induce active targeting ability. Tumor-targeting ligands can make it possible by modified on the surface of RBCm-coated polymeric nanoparticles (RBCm-NPs). To retain as much biological components with immune evasion capabilities as possible on the RBCm, physical lipid insertion approach instead of chemical conjugation technique was selected to modify ligands onto the bilayered RBC lipid membranesCitation26,Citation27 instead of chemical conjugation techniqueCitation28,Citation29 was selected to modify ligands onto the bilayered RBC lipid membranes. Except modification of targeting ligands, other specific cell membranesCitation30–Citation32 such as cancer cell membranes have been used to achieve active targeting ability for NPs in recent research. Due to adhesion proteins on the surface, cancer cell membrane-coated polymeric NPs (CCNPs) seem to have self-targeting ability to homotypic tumor site in vivo.Citation33–Citation36 Nevertheless, process to capture cancer cells from blood circulation is complicated and the tumor heterogeneity between cancer cells is non-negligible as well. Therefore, available RBCm with active targeting ligands is likely to be a better option for biomimetic NPs in antitumor therapy.
Due to special microenvironment of solid tumor, which shows abnormal homeostatic regulation, hypoxic cancer cells, increased interstitial fluid pressure, and existence of extracellular matrix (ECM), antitumor drugs can accumulate preferentially at tumor site via targeting nanocarriers, but the poor penetration of drugs into solid tumor tissues may further prevent the antitumor efficiency. For this reason, the selection of targeting ligands is not only to ensure active targeting ability but also to promote drugs to penetrate into solid tumor tissues efficiently. As tumor penetrating and cell internalizing peptide, iRGD peptideCitation37–Citation39 with the sequence of CRGDK/RGPD/EC can target αvβ3/αvβ5 integrin receptor and neuropilin-1 (NRP-1)Citation40,Citation41 that highly expressed in tumor tissues. It has been reported that iRGD peptide promoted co-administered drugs to penetrate into extravascular tumor tissue by increasing vascular and tissue permeability.Citation38,Citation39,Citation42–Citation46 In addition, epidermal growth factor receptor (EGFR) is overexpressed in multiple human solid tumors including colorectal cancer and showed 27.3% high expression of colorectal cancer in the previous report.Citation47 Thus, EGFR targeting-based therapy has always been of important significance for colorectal cancer treatment. Therefore, the combination of iRGD peptide and EGFR antibody seems to be desired for targeting ligands in colorectal cancer. In our previous research, we have constructed a new recombinant protein named anti-EGFR–iRGD,Citation48,Citation49 containing both iRGD peptide and EGFR single-domain antibody (sdAb).Citation50–Citation53 Besides, we also confirmed the dual targeting ability and improved synergistic antitumor effect with antitumor drugs for anti-EGFR-iRGD recombinant protein.Citation48,Citation49,Citation54,Citation55 Thus, it can be seen that anti-EGFR-iRGD recombinant protein is expected to an optimal targeting ligand for RBCm NPs to achieve better tumor-targeting ability and antitumor efficiency.
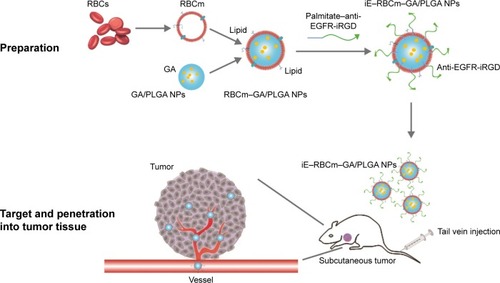
Gambogic acid (GA) is served as the major active ingredient of gamboge and can be potentially developed as a new antitumor drug.Citation56–Citation58 Recently, the incredible anti-tumor effect of GA on colorectal cancer has been reported in various researches.Citation59,Citation60 Therefore, GA is seemed to be an appropriate drug loaded in the bispecific targeting biomimetic NPs. Here, we lipidated anti-EGFR-iRGD proteins with palmitate chemicallyCitation61,Citation62 by detergent dialysis methodCitation63–Citation68 and, then, the lipid chain of palmitate–anti-EGFR-iRGD proteins could insert into bilayered RBC lipid membranes to obtain RBCm NPs modified with anti-EGFR-iRGD proteins (iE–RBCm–PLGA NPs). We evaluated the targeting ability of iE–RBCm–PLGA NPs and antitumor effect of iE–RBCm–GA/PLGA NPs loaded with potential antitumor drug GA in colorectal cancer ().
Figure 1 Schematic representation of preparations of iE–RBCm–GA/PLGA nanoparticles and re-transfusion into mice to evaluate the targeting ability and anti-tumor efficacy.
Abbreviations: GA, gambogic acid; PLGA, poly(lactic-co-glycolic acid); RBCm, red blood cell membrane.
Materials and methods
Materials and reagents
GA (purity ≥99%), PLGA (molecular weight =40,000), N-N-succinimidyl palmitate, dichloromethane (DCM), and polyvinyl alcohol (PVA) were purchased from Aladdin Industrial Corporation (Shanghai, People’s Republic of China).
Anti-EGFR-iRGD recombinant proteins
Escherichia coli BL21 (DE3), which express recombinant proteins anti-EGFR-iRGD, was preserved by our laboratory.Citation48,Citation49 The E. coli massively proliferated when cultured in Lysogeny broth (LB) after resuscitation. Proteins anti-EGFR-iRGD were then expressed in E. coli BL21 (DE3) after induction by isopropyl β-d-1-thiogalactopyranoside (IPTG) at 37°C for 4 h. The cells were collected and suspended in 5 mM imidazole buffer solution and subsequently disrupted using probe sonicator (Sonics, Newtown, CT, USA). The supernatant of cell lysate was purified using the protein liquid chromatography system (ÄKTA™ Pure; GE Healthcare UK Ltd, Little Chalfont, UK), and the purified proteins were obtained after dialysis to remove midazole.
Cells and cell culture
Human colon cancer cell lines Caco-2 (ATCC® HTB 37™), HT-29 (ATCC® HTB-38™), and SW480 (ATCC® CCL 228™) were preserved in our laboratory and purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA). The cells were cultured in Roswell Park Memorial Institute (RPMI-1640) medium (MultiCell, Calgary, AB, Canada) with 10% calf serum (Every Green, Hangzhou, People’s Republic of China) at 37°C in a humidified atmosphere with 5% CO2.
Animals
Experimental Animal Center of Nanjing Drum Tower Hospital supplied male BALB/c mice (nu/nu, weight: 18-25 g, age: 5–8 weeks). All animal-handling procedures were performed in compliance with guidelines set by the Animal Care Committee, Nanjing Drum Tower Hospital. All animal experiments were approved by the Animal Care Committee, Drum Tower Hospital, Nanjing, People’s Republic of China.
Preparation of iE–RBCm–GA/PLGA NPs
Preparation of RBCm–GA/PLGA NPs
The membranes of RBCs were collected in low osmotic pressure from broken RBCs, which separated from whole blood of BALB/c mice. Then, RBCm vesicles were prepared using sonication approach. Blank PLGA and GA-loaded PLGA NPs’ core were prepared using O/W emulsion method with the optimal ratio of GA and PLGA in weight. The fusion of RBCm-derived vesicles with GA/PLGA NPs’ core was conducted via mini-extruder (Avanti Polar Lipids, Alabaster, AL, USA). All the preparation of RBCm–GA/PLGA NPs was described in detail in our previous report.Citation60
Modification of proteins anti-EGFR-iRGD on the surface of RBCm–GA/PLGA NPs
Recombinant proteins anti-EGFR-iRGD were dissolved at 1.5 mg/mL in phosphate-buffered saline (1× PBS), containing 0.3% sodium deoxycholate (DOC), 0.1% sodium azide, and 0.1% sodium bicarbonate. N-succinimidyl palmitate was added to the protein solution, which was prewarmed to 37°C to yield a final concentration of 0.1 mg/mL. This mixture was then incubated at room temperature for 12–18 hours. The lipid-derivatized proteins (palmitate–anti-EGFR-iRGD proteins) were purified on a 10 mL sephadex G-25 column equilibrated with PBS containing 0.1% DOC.
The resulted palmitate–anti-EGFR-iRGD proteins were incubated with RBCm–GA/PLGA NPs at 37°C for >30 min and centrifuged to remove excess proteins. Finally, the targeting moieties (anti-EGFR-iRGD proteins) were incorporated into RBCm through the aid of lipid tethers to build iE–RBCm–GA/PLGA NPs.
Characterization of iE–RBCm–GA/PLGA NPs
To confirm that the recombinant proteins anti-EGFR-iRGD were modified on the surface of RBCm–GA/PLGA NPs successfully, the chemical states of N of iE–RBCm–GA/PLGA NPs and RBCm–GA/PLGA NPs were characterized by X-ray photoelectron spectroscopy (XPS; Kratos XSAM 800; Kratos Analytical, Manchester, UK). The samples were frozen dried before examination.
The composite structure of the iE–RBCm–GA/PLGA NPs was examined using a transmission electron microscopy (TEM; JEM-100S, Tokyo, Japan). A drop of the NPs suspension was deposited onto the cupper grid and, then, air-dried at room temperature. The sample of iE–RBCm–GA/PLGA NPs was stained with 1% uranyl acetate before observation.
To measure the diameter and polydispersity of iE–RBCm–GA/PLGA NPs, the dynamic light scattering (DLS) method was conducted by using the Brookhaven BI-900AT instrument/zeta potential analyzer (Brookhaven Instruments Corporation, Holtsville, NY, USA). To evaluate the stability of the NPs, the iE–RBCm–GA/PLGA NPs in 1× PBS buffer (pH =7.4) were kept at 25°C and the diameter was determined by DLS everyday for 7 days.
To study drug release, 1 mL of iE–RBCm–GA/PLGA NPs in dialysis bag with a molecular weight cutoff of 10 kDa was immersed in 10 mL of 1× PBS buffer (pH =7.4). And the dialyzing sample was kept in thermostated shaking water bath (Taichang Medical Apparatus Co., Jiangsu, People’s Republic of China) at 37°C for 14 days. Every day, the release buffer was removed for analysis, and 10 mL of fresh PBS buffer was added for continuing incubation. Then, the GA content in the release buffer was detected using a HPLC system (Agilent Technologies, Santa Clara, CA, USA).
Targeting ability in colorectal cancer
EGFR expression levels of colorectal cancer cell lines
Western blot was used to confirm EGFR expression levels of colorectal cancer cell lines (Caco-2, HT-29, and SW480). The proteins of three cell lines were collected from each supernatant fraction after lysed on ice for 10 min. Samples containing equivalent amounts of proteins were then subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto nitrocellulose membranes (EMD Millipore, Billerica, MA, USA). Membranes were then blocked with 5% skim milk and subsequently incubated with the EGFR-specific polyclonal antibody (Cell Signaling Technology, Danvers, MA, USA) in Tris-Buffered Saline and Tween 20 (Beyotime, Haimen, People’s Republic of China) overnight at 4°C. After several washes with TBST buffer, the membranes were incubated for 1 h with secondary antibody diluted 1:5,000. The membranes were then processed with enhanced chemiluminescence (ECL) western blot detection reagents. Signals were digitally captured using a MicroChemi chemiluminescent image analysis system (DNR Bio-imaging Systems, Jerusalem, Israel).
Formation of HT-29 MCS, uptake, and penetration in MCS
To form multicellular spheroids (MCS), colorectal cancer cells HT-29 were seeded in 96-well ultra-low attachment plates (Corning Incorporated, Corning, NY, USA) at a density of 2,000 cells/well and incubated at 37°C with 5% CO2 for 3–4 days (MCS iŝ200 µm in diameter).
PLGA NPs were fluorescence labeled with green 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO; Beyotime) dye or red 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI; Beyotime). The fluorescent PLGA NPs were coated with RBCm and modified with proteins anti-EGFR-iRGD. NPs were added into the cell culture medium for 4 h of incubation. Then, the HT-29 MCS were collected, washed with 1× PBS buffer, and placed in confocal dishes (In Vitro Scientific, Gerasdorf bei Wien Austria). The cellular uptake of NPs and penetration in MCS were observed using inverted fluorescence microscope (Olympus Corporation, Tokyo, Japan) and confocal laser scanning microscopy (CLSM; Olympus Corporation).
Target and penetration into tumor tissues
A total of 1×107 Caco-2 cells were implanted subcutaneously into the right flank of BALB/c mice. The PLGA NPs were fluorescence labeled with green DiO dye and divided into four equal groups. The four groups acted as PLGA NPs (P group), RBCm–PLGA NPs (RP group), RBCm–PLGA NPs with free anti-EGFR-iRGD proteins (iE + RP group), and RBCm–PLGA NPs modified with anti-EGFR-iRGD proteins (iE–RP group). The samples were injected to Caco-2 tumor-bearing nude mice via the tail vein. Tumor-bearing nude mice were sacrificed after 24 h, and subcutaneous tumors were collected. Immunofluorescence analysis was carried out with frozen tumor tissues. For immunostaining, slides were first blocked with 3% bull serum albumin (BSA) for 1 h at room temperature and then exposed to the primary antibody against CD31 (1:300 dilution; GeneTech, Shanghai, People’s Republic of China) at 4°C overnight and the second antibody (1:300 dilution) at 37°C for 30 min. Finally, nuclei were stained with 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI; Beyotime) and slides were visualized using CLSM.
Fluorescence images of real-time tumor targeting in vivo
To investigate the tumor targeting and accumulation of NPs in vivo, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotric-arbocyanine iodide (DiR; Bridgen, Beijing, People’s Republic of China) dye was used to label the different NPs as abovementioned. The four different groups (P group, RP group, iE + RP group, and iE–RP group) were injected to Caco-2 tumor-bearing nude mice via the tail vein. At different time points (24, 72, and 168 h), the mice were anesthetized and imaged using the Maestro EX in vivo fluorescence imaging system (CRi, Inc., Hopkinton, MA, USA). At the end of test, the tumor-bearing mice were sacrificed to collect tumors and main organs including heart, liver, spleen, lungs, and kidneys for images of biodistribution in vivo.
Antitumor efficiency of iE–RBCm–GA/PLGA NPs in colorectal cancer
Cytotoxicity
MTT assay was carried out to evaluate the cytotoxity of iE-RBCm–GA/PLGA NPs against colorectal cancer cells HT-29 in vitro. Cells were seeded in 96-well plates at the density of 5,000 cells/well and then incubated at 37°C with 5% CO2 for attachment. Each well was added with 20 µL of 5 mg/mL MTT solution after cells were exposed to various concentrations of NPs and free GA for 12, 24, and 48 h and then incubated for another 4 h. The supernatant was discarded, and 150 µL of dimethylsulphoxide (DMSO) was added to each well to dissolve the formazan crystals. The absorption at 490 nm was measured by Multiskan Spectrum Microplate Reader (BioTek, Winooski, VT, USA), and the cell viabilities were calculated by the following formula:
Apoptosis assay in vitro and in vivo
Apoptosis in vitro was detected using Annexin-V-FLUOS Staining Kit (Hoffman-La Roche Ltd., Basel, Switzerland) according to the instruction. A total of 1×106 HT-29 cells were exposed to certain various concentrations of iE-RBCm–GA/PLGA NPs and free GA for 12, 24, 48, and 72 h, and then, cells in each wall were collected and washed with 1× PBS buffer. Washed cells were resuspended in 100 µL of Annexin-V-FLUOS-labeling solution containing 100 µL of 4-(2-hydroxyethyl)-1-piperazineethanesulf onic acid; PI: propidium iodide (HEPES) buffer, 2 µL of propidium iodide (PI), and 2 µL of Annexin-V-Fluorescein, and then, samples were incubated in the dark for 15 min at room temperature. The apoptosis of HT-29 was analyzed on flow cytometer (EMD Millipore).
For in vivo experiment, iE–RBCm–GA/PLGA NPs (iE–RGP group), RBCm–GA/PLGA NPs (RGP group), GA, iE–RBCm–PLGA NPs (iE–RP group), and normal saline (NS) were injected to Caco-2 tumor-bearing nude mice via tail vein. After 48 h, mice were sacrificed. Subcutaneous tumors were collected and fixed with paraformaldehyde. Terminal deoxyribonucleotidyl transferase-mediated dUTP nick end labeling (TUNEL; Hoffman-La Roche Ltd.) method was used to detect the apoptotic cells of tumor tissues in vivo. Then, nuclei were stained with DAPI, and slides were visualized using CLSM.
In vivo antitumor effect and safety
Caco-2 tumor-bearing nude mice were randomly divided into five groups (n=6 per group) and were injected with iE–RBCm–GA/PLGA NPs (6 mg/kg, iE–RGP group), RBCm–GA/PLGA NPs (6 mg/kg, RGP group), GA solution (6 mg/kg, GA group), iE–RBCm–PLGA NPs (iE–RP group), and normal saline (NS group) via the tail vein once a week, a total of four times. The tumor size and weight of mice were measured on day 7 postimplantation and every other day after injection. Tumor volume was calculated as V=D×d2/2, where D and d are the longest and the shortest diameters of the tumor in millimeters, respectively. Survival was predefined as tumor size <2,000 mm3 prior to the initiation of the study. In order to reduce the impact of initial weight differences among different groups, relative weight was calculated by the following formula:
When mice were sacrificed, the tumor tissues and main organs including hearts, lungs, livers, spleens, and kidneys of mice in each group were collected for histology observation. The Ki67 immunohistochemistry was used to evaluate the proliferation of the cells in the tumors. And systemic toxicity was assessed via organ slices that stained by hematoxylin and eosin (H&E).
Statistical analysis
Statistical analysis was performed by the SPSS V.23.0 software (IBM Corporation, Armonk, NY, USA). One-way analysis of variance (ANOVA) or Independent Student’s t-test was used to determine the differences between various treatments. Data are presented as the mean ± SD. Statistically significant results were subjected to Tukey’s post hoc test (GraphPad Prism 6.0; GraphPad Software, Inc., La Jolla, CA, USA). All quantitative data reported represent the mean of at least three independent experiments. The resultant P-value was expressed as ***P≤0.001, **P≤0.01, and *P<0.05. Differences were considered to be statistically significant if P-value was <0.05.
Results
Preparation and physicochemical characterization of iE–RBCm–GA/PLGA NPs
The lipid terminus of palmitate–anti-EGFR-iRGD proteins can be inserted into the coating RBCm of GA/PLGA NPs to modify the bispecific targeting. To confirm that the proteins were modified on the surface of NPs, the surface chemistry of the iE–RBCm–GA/PLGA NPs and RBCm–GA/PLGA NPs was characterized using XPS and the spectra of nitrogen (N) were depicted in . Compared with RBCm–GA/PLGA NPs, the peak of elemental N of iE–RBCm–GA/PLGA NPs moved upward obviously,Citation69 demonstrating that the proteins were modified on the surface of NPs successfully.
Figure 2 Preparation and characterization of iE–RBCm–GA/PLGA NPs.
Notes: (A) XPS spectrum of N in iE–RBCm–GA/PLGA NPs and RBCm–GA/PLGA NPs. (B) TEM visualization of iE–RBCm–GA/PLGA NPs with uranyl acetate negative staining. (C) DLS measurements of the size and PDI of iE–RBCm–GA/PLGA NPs over 7 days. (D) GA release behavior from iE–RBCm–GA/PLGA NPs for 14 days.
Abbreviations: DLS, dynamic light scattering; GA, gambogic acid; NPs, nanoparticles; PDI, polydispersity index; PLGA, poly(lactic-co-glycolic acid); RBCm, red blood cell membrane; TEM, transmission electron microscopy; XPS, X-ray photoelectron spectroscopy.
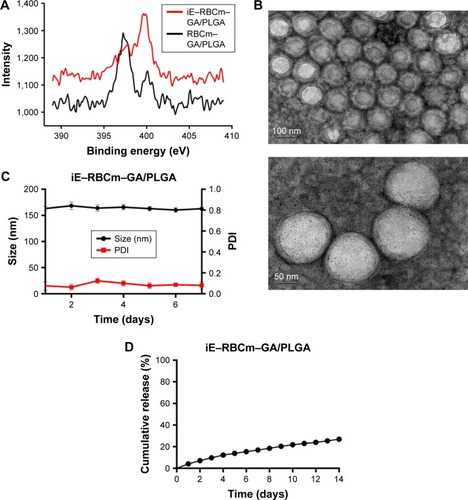
The structure of the iE–RBCm–GA/PLGA NPs with uranyl acetate negative staining was visualized by TEM and the shell–core structure was revealed in the image (). The hydrodynamic diameter of iE–RBCm–GA/PLGA NPs was about 153±3.83 nm, and little change in the particle size and polydispersity index (PDI) of iE–RBCm–GA/PLGA NPs could be observed in a week (). The NPs suspended in 1× PBS buffer (pH =7.4) showed the long-term stability in vitro. Besides, the iE–RBCm–GA/PLGA NPs also displayed a slow and sustained release in 14 days (). Approximately 30% of GA was released from iE–RBCm–GA/PLGA NPs in neutral PBS buffer (pH =7.4) over 14 days.
Targeting ability in colorectal cancer
To select suitable cell line, the EGFR expression of three different colorectal cancer cells (Caco-2, HT-29, and SW480) was measured. The result showed that EGFR was highly expressed in Caco-2 and HT-29 (). And compared with Caco-2 and HT-29, the EGFR levels in SW480 cells could not be detected. Besides, due to that cells HT-29 could form MCS with symmetrical and spherical shapes and cells Caco-2 could be implanted to form subcutaneous tumors, HT-29 was used in following experiments in vitro, while Caco-2 was used in in vivo.
Figure 3 Investigation of the total EGFR expression levels in three different colorectal cancer cell lines as well as cellular uptake of iE–RBCm–PLGA NPs and penetration in MCS.
Notes: (A) Western blot analysis of the whole cell line lysates for EGFR and GAPDH. (B) Images of HT-29 MCS incubated with DiO-labeled iE–RBCm–PLGA NPs (iE–RP), RBCm–PLGA NPs with free anti-EGFR-iRGD protein (iE + RP), and RBCm–PLGA NPs (RP) for 4 h under inverted fluorescence microscope; scale bar =100 µm. (C) CLSM images of HT-29 MCS incubated with DiI-labeled iE–RBCm–PLGA NPs (iE–RP), RBCm–PLGA NPs with free anti-EGFR-iRGD protein (iE + RP), and RBCm–PLGA NPs (RP) for 4 h; scale bar =100 µm.
Abbreviations: CLSM, confocal laser scanning microscopy; DiO, 3,3′-dioctadecyloxacarbocyanine perchlorate; EGFR, epidermal growth factor receptor; MCS, multicellular spheroids; NPs, nanoparticles; PLGA, poly(lactic-co-glycolic acid); RBCm, red blood cell membrane.
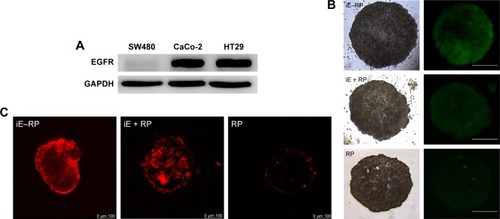
The penetration ability of the three groups of NPs (iE–RP, iE + RP, and RP) were analyzed with MCS. The result showed that green fluorescence intensity of iE–RP group was higher than the other two groups under the fluorescence microscope (), which reflected that more iE–RBCm–PLGA NPs were taken up by MCS. We also further evaluated the penetration of the three groups of NPs in MCS under CLSM. As shown in , the fluorescence signal of iE–RP group was localized in both interior and periphery of MCS, while RP group in the vicinity of periphery. The image of iE + RP group also showed a little increased penetration when compared with RP group. It may benefit from tumor-penetrating peptide iRGD, allowing co-administered NPs to penetrate into MCS. However, the penetration of iE–RBCm–PLGA NPs was deeper than RBCm–PLGA NPs without anti-EGFR-iRGD proteins modified on the surface. Therefore, RBCm–PLGA NPs modified with anti-EGFR-iRGD proteins on the surface could obtain positive penetration ability, while the increased penetration ability from free anti-EGFR-iRGD proteins was limited.
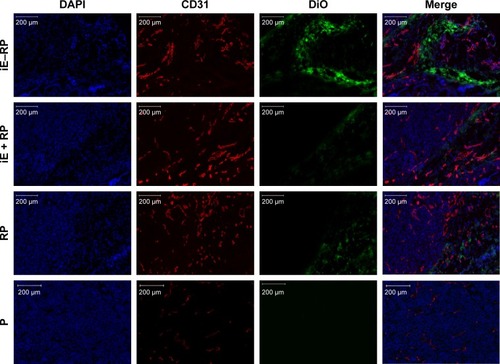
Based on the result that iE–RBCm–PLGA NPs could get deep penetration into MCS in vitro, target and penetration into tumor tissues in vivo were also observed and evaluated by both immunostaining tumor tissue slices and real-time near-infrared (NIR) fluorescence imaging. In the image of tumor tissue slice (), green fluorescence signal represents NPs visualized with DiO dye, red fluorescence signal represents blood vessels of tumor issue, and blue fluorescence signal represents tumor cells. Except P group, green fluorescent NPs could be seen in the images of other three groups (iE–RP, iE + RP, and RP). Besides, considerable amounts of green fluorescence NPs were around blood vessels and tumor cells of tumor issue in the iE–RP group, which was much more than that in the iE + RP group and the RP group. It meant that iE–RBCm–PLGA NPs could accumulate efficiently at tumor site with active targeting ability. In addition, a certain number of RBCm–PLGA NPs accumulated at tumor site benefiting from acceptable biocompatibility and EPR effect. Only free anti-EGFR-iRGD proteins instead of being modified on the surface could not obtain obvious active targeting delivery function for RBCm–PLGA NPs.
Figure 4 Immunofluorescence images of tumor tissue slices in four groups (iE–RP, iE + RP, RP, and P) at 24 h postinjection.
Notes: DiO-labeled nanoparticles (green), CD31-labeled blood vessels (red), and DAPI-labeled cancer cells are shown in the images of tumor sections. Scale bar =200 µm.
Abbreviations: DiO, 3,3′-dioctadecyloxacarbocyanine perchlorate; iE–RP, iE-RBCm-PLGA NPs; iE+RP, RBCm-PLGA NPs with free anti-EGFR-iRGD protein; RP, RBCm-PLGA NPs; P, PLGA NPs.
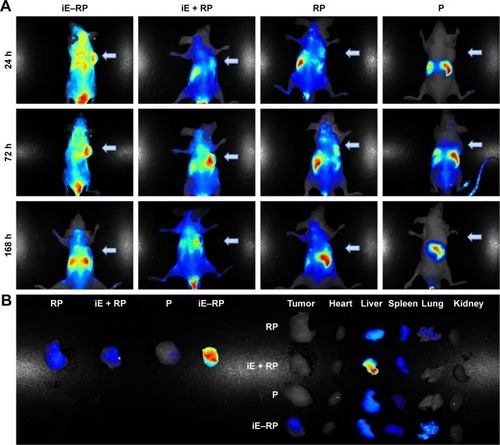
The biodistribution of four groups NPs (iE–RP, iE + RP, RP, and P) were monitored via real-time NIR fluorescence imaging (). At 24 hours postinjection, different fluorescence signals in intensity appeared at the tumor site of iE–RP, iE + RP, and RP groups except P group and iE–RP group showed the strongest fluorescence signal among them. The result demonstrated that iE–RBCm–PLGA NPs have enhanced tumor-targeting ability than RBCm–PLGA NPs in vivo. At 72 h postinjection, the intensity of the fluorescent signal continued to increase, which reflected the “long circulating” advantage of biomimetic nanocarriers. During the process, the fluorescence intensity of iE + RP and RP were similar and weaker than that of iE–RP group. It meant that free anti-EGFR-iRGD proteins cannot guide RBCm–PLGA NPs to accumulate efficiently at tumor site. At 168 h postinjection, the fluorescence intensity at tumor site decreased compared with the previous time point. Then, the mice of all groups were sacrificed, and ex vivo fluorescence intensity images were obtained for tumor tissues as well as other major organs including hearts, lungs, livers, spleens, and kidneys. As shown in , the fluorescence signal intensity of subcutaneous tumor in iE–RP group was significantly higher than the rest of the three groups. Moreover, compared with the high intensity of fluorescence signal of liver and spleen, the fluorescence signal of subcutaneous tumor in the iE–RP group was also visible. This matched with above results in vivo.
Figure 5 Real-time NIR fluorescence images of iE–RBCm–PLGA NPs.
Notes: (A) Images of Caco-2 tumor-bearing mice following intravenous injection of DiR-labeled iE–RBCm–PLGA NPs (iE–RP), RBCm–PLGA NPs with free anti-EGFR-iRGD protein (iE + RP), RBCm–PLGA NPs (RP), and PLGA NPs (P) for 24, 72, and 168 h. (B) Images of various organs at 168 h postadministration with four different NPs (iE–RP, iE + RP, RP, and P).
Abbreviations: DiR, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide; NIR, near-infrared; NPs, nanoparticles; PLGA, poly(lactic-co-glycolic acid); RBCm, red blood cell membrane.
Antitumor efficiency in colorectal cancer
MTT assay was used to evaluate the cytotoxicity of iE–RBCm–GA/PLGA NPs against HT-29 cells in vitro. For 12, 24, and 48 h incubation, both iE–RBCm–GA/PLGA NPs and GA showed a dose-dependent cytotoxicity against HT-29 cells (). In addition, compared with respective cell viability, iE–RBCm–GA/PLGA NPs and free GA almost had no difference against HT-29 at all the time points.
Figure 6 Antitumor activity of iE–RBCm–GA/PLGA NPs in vitro.
Notes: (A) In vitro cytotoxicity of iE–RBCm–GA/PLGA NPs in comparison with free GA against HT-29 colorectal cancer cells after 12, 24, and 48 h of incubation (n=4, P>0.05). Data are given (graph bars) as the mean ± SD (n=4). Statistical significance. (B) Apoptosis rate of HT-29 incubated with iE–RBCm–GA/PLGA NPs in increasing concentration for 12, 24, 48, and 72 h. (C) HT-29 cell apoptosis detected by Annexin V-FITC/PI double staining image of iE–RBCm–GA/PLGA NPs and free GA in 1.5 µg/mL GA concentration at 12, 24, 48, and 72 h.
Abbreviations: GA, gambogic acid; NPs, nanoparticles; PLGA, poly(lactic-co-glycolic acid); RBCm, red blood cell membrane.
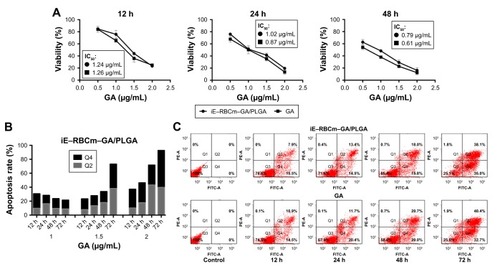
To illustrate the possible mechanism of cytotoxicity in vitro, the further analysis of apoptosis that may lead to cytotoxicity was also conducted. When HT-29 cells were exposed to different concentrations of iE–RBCm–GA/PLGA NPs from 12 to 72 h, obvious increase in apoptotic rate (Q2 + Q4) was observed (). And when the concentration of iE–RBCm–GA/PLGA NPs and GA was >1.5 µg/mL, the cell apoptosis of HT-29 cells was apparent. Therefore, flow cytometric analysis was conducted to assess apoptosis induced by iE–RBCm–GA/PLGA NPs and GA at certain concentration (1.5 µg/mL) for 12, 24, 48, and 72 h (). The result reflected that the rate of both early (Q4) stage and late (Q2) stage apoptotic HT-29 cells were increasing with the time. At all the time points, both two groups showed similar apoptosis rate of HT-29 cells. It matched with the results of MTT assay and suggested that both iE–RBCm–GA/PLGA NPs and free GA have the same efficient antitumor effect in vitro.
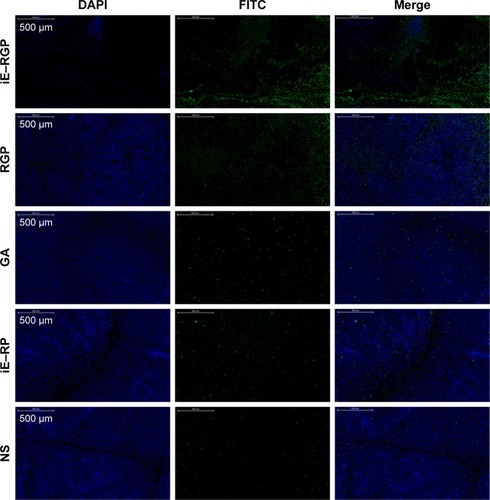
In vivo, we sequentially investigated whether the anti-tumor activity of iE–RBCm–GA/PLGA NPs could act as effective therapy against tumor growth. BALB/c nude mice-bearing subcutaneous inoculated Caco-2 tumors, acted as colorectal cancer model, were used to observe both the apoptosis at tumor site and the long-term tumor burden in vivo. The treatments were administrated via tail vein with the solution of iE–RBCm–GA/PLGA NPs, RBCm–GA/PLGA NPs, GA, iE–RBCm–PLGA NPs, and NS. Apoptotic cells were visualized as green fluorescence signal, and all the tumor cells were visualized as blue fluorescence signal in the image of subcutaneous tumor tissue slice (). The result showed a large amount of apoptotic cells and a small number of tumor cells in the visual field of slice in the iE–RGP group, which indicated that iE–RBCm–GA/PLGA NPs with active targeting ability accumulated and penetrated efficiently at tumor site within a short time to lead significant cell apoptosis in vivo. The presence of fluorescence signal in the RGP group suggested that RBCm–GA/PLGA NPs also could passively target and accumulate at tumor site to lead apoptosis due to the stability, biocompatibility and “long circulating” characteristic in vivo. However, it still was slightly inferior than iE–RBCm–GA/PLGA NPs. In addition, the green fluorescence signal that represents apoptotic cells was not obvious in the GA group compared with the control group (iE–RP and NS) within 48 h. It reflected that GA-loaded NPs achieved better bioavailability than free GA in vivo, and the NPs with active targeting ability could expand this advantage.
Figure 7 Apoptosis at tumor site following intravenous injection of iE–RBCm–GA/PLGA NPs (iE–RGP), RBCm–GA/PLGA NPs (RGP), free GA, iE–RBCm–PLGA NPs (iE–RP), and NS.
Notes: Green apoptotic cells and blue active cancer cells are shown in the images of tumor sections. Scale bar =500 µm.
Abbreviations: GA, gambogic acid; NPs, nanoparticles; NS, normal saline; PLGA, poly(lactic-co-glycolic acid); RBCm, red blood cell membrane.
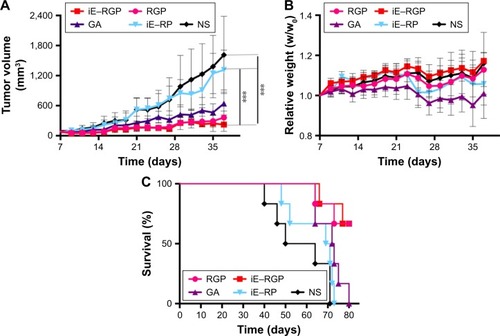
Antitumor efficacy of iE–RBCm–GA/PLGA NPs in vivo was evaluated via the long-term subcutaneous tumor burden in the colorectal cancer model of mice. As shown in , compared with the control group (iE–RP and NS), the tumor volumes of iE–RGP treatment group grew quite slowly (***P≤0.001, n=6), which showed an expected result of iE–RBCm–GA/PLGA NPs in terms of tumor growth inhibition. RGP treatment group also showed satisfactory antitumor efficacy at GA dose of 6 mg/kg, reflecting the advantage of biomimetic nanocarriers for tumor therapy. Moreover, the fact that the weights of mice in GA-loaded NPs groups were increasing gradually rather than remained as in control groups, indicating that mice of these groups benefited from antitumor efficiency and did not suffer from the increasing tumor burden (). GA group showed certain tumor inhibition effect, but the weight of mice in GA group was decreased, which indicated that mice suffered from the toxicity of free GA. It also reflected in the result of survival (); mice in control group with increasing tumor burden began to die first (from day 40). In contrast, mice in the iE–RGP group began to die after 38 days (from day 66) after the last administration and extended the median survival time at least 27 days more compared with the control group. Simultaneously, mice in the GA group died within 17 days (from days 64 to 80) due to cumulative toxicity during the treatment.
Figure 8 In vivo antitumor efficacy of iE–RBCm–GA/PLGA NPs.
Notes: (A) Tumor growth inhibition in mice treated with iE–RBCm–GA/PLGA NPs (iE–RGP), RBCm–GA/PLGA NPs (RGP), free GA, iE–RBCm–PLGA NPs (iE–RP), and NS by tail vein injection (n=6). Tumor size of mice in each group was measured on day 7 postimplantation and every other day after injection until the appearance of dead (37 days in total). Data are given (graph bars) as the mean ± SD (n=6). Statistical significance. (B) Relative weights of tumor-bearing mice treated with iE–RBCm–GA/PLGA NPs (iE–RGP), RBCm–GA/PLGA NPs (RGP), free GA, iE–RBCm–PLGA NPs (iE–RP), and NS. Weight of mice in each group was measured on day 7 postimplantation and every other day after injection until appearance of dead (n=6, P>0.05). Data are given (graph bars) as the mean ± SD (n=6). Statistical significance. (C) Survival of mice treated with iE–RBCm–GA/PLGA NPs (iE–RGP), RBCm–GA/PLGA NPs (RGP), free GA, iE–RBCm–PLGA NPs (iE–RP), and NS. ***P≤0.001.
Abbreviations: GA, gambogic acid; NPs, nanoparticles; NS, normal saline; PLGA, poly(lactic-co-glycolic acid); RBCm, red blood cell membrane.
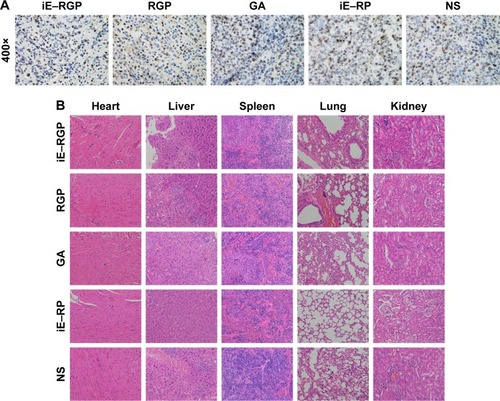
In Ki67 staining images, the nuclei of positive cells were stained brown, indicating high proliferation ability. The result showed that there were less positive cells in the tumor tissue of the iE–RGP group compared with the control group and so was of RGP group (). Moreover, H&E-stained section images of major organs in each group showed no noticeable signal of organ damage (). It suggested that iE–RBCm–GA/PLGA NPs had optimal antitumor efficiency and safety in vivo.
Figure 9 Antiproliferative effect and safety of iE–RBCm–GA/PLGA NPs in vivo.
Notes: (A) Ki67 staining images magnification 400× of cell proliferation in tumor issues of iE–RBCm–GA/PLGA NPs (iE–RGP), RBCm–GA/PLGA NPs (RGP), free GA, iE–RBCm–PLGA NPs (iE–RP), and NS. (B) Representative H&E-stained slices image of major organs in each group (iE–RGP, RGP, GA, iE–RP, and NS).
Abbreviations: GA, gambogic acid; H&E, hematoxylin and eosin; NPs, nanoparticles; NS, normal saline; PLGA, poly(lactic-co-glycolic acid); RBCm, red blood cell membrane.
Discussion
On the basis of the RBC-NP platform with good stability and biocompatibility, the modification of anti-EGFR-iRGD recombinant protein with two target antigens endowed the active bispecific targeting ability for this synthetic biomimetic nanocarrier. The physical insertion into the RBCm avoided damaging the existing surface proteins and remained the integrality and stability of structure. Moreover, potential antitumor drug GA that loaded into iE–RBCm–PLGA NPs brought the synthetic nanocarrier antitumor efficacy. The resulting iE–RBCm–GA/PLGA NPs not only lifted restrictions on the application of GA with poor water solubility but also delivered GA to tumor site efficiently to improve its bioavailability in vivo.
The targeting ability of iE–RBCm–PLGA NPs and antitumor efficiency of GA loaded iE–RBCm–GA/PLGA NPs were taken as the object to investigate the effect on the colorectal cancer in vitro and in vivo. In terms of the targeting and penetrating efficiency of iE–RBCm–PLGA NPs, we evaluated it at the level of MCS and nude mice bearing subcutaneous inoculated tumors. Compared with RBCm–PLGA NPs, more iE–RBCm–PLGA NPs could be taken up by MCS and showed better penetration into MCS in vitro. iE–RBCm–PLGA NPs also showed better targeting efficiency in nude mice model of colorectal cancer with obvious accumulation at tumor site in a certain time. It indirectly reflected that anti-EGFR-iRGD protein successfully modified on the surface of NPs via lipid insertion, and iE–RBCm–PLGA NPs with the modification of anti-EGFR-iRGD protein achieved satisfactory targeting and penetrating efficiency in vitro and in vivo. Besides, RBCm–PLGA NPs with free anti-EGFR-iRGD protein showed a little advantage in targeting and penetrating ability when compared with RBCm–PLGA NPs, suggesting that the targeting and penetrating ability of free anti-EGFR-iRGD to guide more RBCm–PLGA NPs to tumor site were limited. In consideration of anti-EGFR-iRGD recombinant protein with dual targeting of αvβ3 and EGFR, we emphatically studied specific EGFR target in colorectal cancer in future work. Colorectal cancer models with the high expression of EGFR and low or no expression of EGFR were established to explore the target differentiation of iE–RBCm–PLGA NPs between different colorectal cancer models.
In terms of the antitumor efficiency of iE–RBCm–GA/PLGA NPs, we also evaluated it in vitro and in vivo, which contained cytotoxicity, apoptosis, and tumor inhibition. iE–RBCm–GA/PLGA NPs and free GA were taken up via different mechanisms – endocytosis and pure diffusion – and free GA could diffuse into the cancer cells more easily compared with NPs. However, iE–RBCm–GA/PLGA NPs modified with active targeting ligands enhanced the transport capacity across cellular membrane. As a result, iE–RB-Cm–GA/PLGA NPs showed similar cytotoxicity compared with free GA at the same dose in vitro. Simultaneously, the cell apoptosis of both iE–RBCm–GA/PLGA NPs and GA were almost the same in vitro, but the apoptosis of cancer cells at subcutaneous tumor site in vivo seemed to be different. Considering complex and changeable biological environment in vivo, free GA did not induce obvious cell apoptosis as shown in in vitro. Nevertheless, iE–RBCm–GA/PLGA NPs showed significant cell apoptosis within 48 h benefiting from the active targeting ability. Difference was also observed in subsequent experiment of tumor growth inhibition. Compared with control group, iE–RBCm-GA/PLGA NPs inhibited the growth of subcutaneous tumor efficiently with safety and nontoxicity. Although GA exerted certain effect on tumor inhibition, it accompanied with serious toxicity, which appeared as decreasing weight and death within a short time. In addition, RBCm–GA/PLGA NPs with good stability and biocompatibility achieved a significant inhibition of tumor growth at a dose of 6 mg/kg, indicating the superiority of biomimetic nanocarrier in cancer therapy. However, it did not objectively show that iE–RBCm-GA/PLGA NPs possess the ability of active targeting delivery. Subsequently, we repeated and adjusted this experiment with the dose of GA intended to be reduced accordingly to observe whether iE–RBCm–GA/PLGA NPs with active and efficient tumor targeting ability could improve the bioavailability of GA in vivo at a dose that is lower than effective dose for free GA.
Conclusion
In our study, recombinant protein anti-EGFR-iRGD was modified on the surface of RBCm-coated NPs to build bispe-cific targeting biomimetic NPs, combining biocompatibility, stability, and active target. In regard to targeting ability, when compared with RBCm–PLGA NPs, more iE–RBCm–PLGA NPs could be taken up by MCS and penetrate into MCS in vitro and more iE–RBCm–PLGA NPs targeted and accumulated at tumor site in vivo. It was confirmed that the modification of anti-EGFR-iRGD protein significantly improved the targeting ability of iE–RBCm–PLGA NPs. In regard to antitumor efficacy, despite the similar antitumor effect of iE–RBCm–GA/PLGA NPs and free GA that shown in vitro, the antitumor efficacy of GA weakened in vivo, along with considerable toxicity. In contrast, iE–RBCm–GA/PLGA NPs still retained antitumor efficiency via short-term cell apoptosis at tumor site and long-term tumor inhibition in vivo. Moreover, iE–RBCm–GA/PLGA NPs reduced the side effects of GA and ensured safety in vivo. Therefore, the bispecific targeting biomimetic NPs reinforce the advantage of drug delivery. We expected that the new nanocarrier could extend the clinical application of many other potential anti-tumor drugs similar to GA in the future.
Acknowledgments
This study was supported by grants from Provincial Natural Science Foundation of Jiangsu (no BK20161107), National Natural Science Foundation (no 81602106 and 81502037), Jiangsu Health and Family Planning Commission development program (no H2817042) and Nanjing Health and Family Planning Commission key program (no ZKX17012).
Disclosure
The authors report no conflicts of interest in this work.
References
- ShiDBedfordNMChoHSEngineered multifunctional nanocarriers for cancer diagnosis and therapeuticsSmall20117182549256721648074
- MahmoodiNOGhavidastAAmirmahaniNA comparative study on the nanoparticles for improved drug delivery systemsJ Photochem Photobiol B201616268169327498233
- ChengZAlZAHuiJZMuzykantovVRTsourkasAMultifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilitiesScience2012338610990391023161990
- GunatillakePAAdhikariRBiodegradable synthetic polymers for tissue engineeringEur Cell Mater20035116 discussion 1614562275
- AryalSHuCMZhangLPolymeric nanoparticles with precise ratio-metric control over drug loading for combination therapyMol Pharm2011841401140721696189
- AcharyaSSahooSKPLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effectAdv Drug Deliv Rev201163317018320965219
- GaoWHuCMFangRHZhangLLiposome-like nanostructures for drug deliveryJ Mater Chem B201314818
- LukBTZhangLCell membrane-camouflaged nanoparticles for drug deliveryJ Control Release2015220pt B60060726210440
- FangRHLukBTHuCMZhangLEngineered nanoparticles mimicking cell membranes for toxin neutralizationAdv Drug Deliv Rev201590698025868452
- ZhangJGaoWFangRHDongAZhangLSynthesis of nanogels via cell membrane-templated polymerizationSmall201511344309431326044721
- CoppJAFangRHLukBTClearance of pathological antibodies using biomimetic nanoparticlesProc Natl Acad Sci U S A201411137134811348625197051
- WickiAWitzigmannDBalasubramanianVHuwylerJNanomedicine in cancer therapy: challenges, opportunities, and clinical applicationsJ Control Release201520013815725545217
- FangRHKrollAVZhangLNanoparticle-based manipulation of antigen-presenting cells for cancer immunotherapySmall201511415483549626331993
- VoonSHKiewLVLeeHBIn vivo studies of nanostructure-based photosensitizers for photodynamic cancer therapySmall201410244993501325164105
- MinelliCLoweSBStevensMMEngineering nanocomposite materials for cancer therapySmall20106212336235720878632
- HuCMZhangLAryalSCheungCFangRHZhangLErythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platformProc Natl Acad Sci U S A201110827109801098521690347
- AryalSHuCMFangRHErythrocyte membrane-cloaked polymeric nanoparticles for controlled drug loading and releaseNanomedicine (Lond)2013881271128023409747
- PiaoJGWangLGaoFYouYZXiongYYangLErythrocyte membrane is an alternative coating to polyethylene glycol for prolonging the circulation lifetime of gold nanocages for photothermal therapyACS Nano2014810104141042525286086
- GuoYWangDSongQErythrocyte membrane-enveloped polymeric nanoparticles as nanovaccine for induction of antitumor immunity against melanomaACS Nano2015976918693326153897
- FangRHHuCMZhangLNanoparticles disguised as red blood cells to evade the immune systemExpert Opin Biol Ther201212438538922332936
- RaoLBuLLXuJHRed blood cell membrane as a biomimetic nanocoating for prolonged circulation time and reduced accelerated blood clearanceSmall201511466225623626488923
- LukBTFangRHHuCMSafe and immunocompatible nanocarriers cloaked in RBC membranes for drug delivery to treat solid tumorsTheranostics2016671004101127217833
- RaoLXuJHCaiBSynthetic nanoparticles camouflaged with biomimetic erythrocyte membranes for reduced reticuloendothelial system uptakeNanotechnology201627808510626820630
- LukBTHuCMFangRHInterfacial interactions between natural RBC membranes and synthetic polymeric nanoparticlesNanoscale2014652730273724463706
- TsaiRKRodriguezPLDischerDESelf inhibition of phagocytosis: the affinity of ‘marker of self’ CD47 for SIRPalpha dictates potency of inhibition but only at low expression levelsBlood Cells Mol Dis2010451677420299253
- FangRHHuCMChenKNLipid-insertion enables targeting functionalization of erythrocyte membrane-cloaked nanoparticlesNanoscale20135198884888823907698
- FuQLvPChenZProgrammed co-delivery of paclitaxel and doxorubicin boosted by camouflaging with erythrocyte membraneNanoscale2015794020403025653083
- ZhouHFanZLemonsPKChengHA facile approach to functionalize cell membrane-coated nanoparticlesTheranostics2016671012102227217834
- BellGIModels for the specific adhesion of cells to cellsScience19782004342618627347575
- HuCMFangRHWangKCNanoparticle biointerfacing by platelet membrane cloakingNature2015526757111812126374997
- DehainiDWeiXFangRHErythrocyte-platelet hybrid membrane coating for enhanced nanoparticle functionalizationAdv Mater201729161606209
- ParodiAQuattrocchiNvan de VenALSynthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functionsNat Nanotechnol201381616823241654
- SunHSuJMengQCancer-cell-biomimetic nanoparticles for targeted therapy of homotypic tumorsAdv Mater201628439581958827628433
- FangRHHuCMLukBTCancer cell membrane-coated nanoparticles for anticancer vaccination and drug deliveryNano Lett20141442181218824673373
- RaoLBuLLCaiBCancer cell membrane-coated upconversion nanoprobes for highly specific tumor imagingAdv Mater201628183460346626970518
- ZhuJYZhengDWZhangMKPreferential cancer cell self-recognition and tumor self-targeting by coating nanoparticles with homotypic cancer cell membranesNano Lett2016165895590127513184
- SugaharaKNTeesaluTKarmaliPPTissue-penetrating delivery of compounds and nanoparticles into tumorsCancer Cell200916651052019962669
- SugaharaKNTeesaluTKarmaliPPCoadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugsScience201032859811031103520378772
- MantisCKandelaIAirdFReproducibility Project: Cancer Biology. Replication study: coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugsElife20176e1758428100395
- TeesaluTSugaharaKNKotamrajuVRRuoslahtiEC-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetrationProc Natl Acad Sci U S A200910638161571616219805273
- HaspelNZanuyDNussinovRTeesaluTRuoslahtiEAlemanCBinding of a C-end rule peptide to the neuropilin-1 receptor: a molecular modeling approachBiochemistry201150101755176221247217
- ChenRBraunGBLuoXSugaharaKNTeesaluTRuoslahtiEApplication of a proapoptotic peptide to intratumorally spreading cancer therapyCancer Res20137341352136123248118
- ZhangJWangLFaiCHCo-delivery of paclitaxel and tetrandrine via iRGD peptide conjugated lipid-polymer hybrid nanoparticles overcome multidrug resistance in cancer cellsSci Rep201774605728470171
- GaoFZhangJFuCiRGD-modified lipid-polymer hybrid nanoparticles loaded with isoliquiritigenin to enhance anti-breast cancer effect and tumor-targeting abilityInt J Nanomedicine2017124147416228615942
- HuangYLiXShaHsTRAIL-iRGD is a promising therapeutic agent for gastric cancer treatmentSci Rep20177157928373646
- ZhuZXieCLiuQThe effect of hydrophilic chain length and iRGD on drug delivery from poly(ε-caprolactone)-poly(N-vinylpyrrolidone) nanoparticlesBiomaterials201132359525953521903260
- CeccarelliCPiazziGPateriniPConcurrent EGFr and Cox-2 expression in colorectal cancer: proliferation impact and tumour spreadingAnn Oncol200516suppl 4iv74iv7915923435
- ShaHLiRBianXA tumor-penetrating recombinant protein anti-EGFR-iRGD enhance efficacy of paclitaxel in 3D multicellular spheroids and gastric cancer in vivoEur J Pharm Sci201577607225998561
- ShaHZouZXinKTumor-penetrating peptide fused EGFR single-domain antibody enhances cancer drug penetration into 3D multicellular spheroids and facilitates effective gastric cancer therapyJ Control Release201520018820025553823
- WardESGüssowDGriffithsADJonesPTWinterGBinding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coliNature198934162425445462677748
- TeillaudJLFrom whole monoclonal antibodies to single domain antibodies: think smallMethods Mol Biol201291131322886242
- MuyldermansSNanobodies: natural single-domain antibodiesAnnu Rev Biochem20138277579723495938
- HarmsenMMDe HaardHJProperties, production, and applications of camelid single-domain antibody fragmentsAppl Microbiol Biotechnol2007771132217704915
- BianXWuPShaHAnti-EGFR-iRGD recombinant protein conjugated silk fibroin nanoparticles for enhanced tumor targeting and antitumor efficiencyOnco Targets Ther201693153316227313461
- XinXShaHShenJZhangBZhuBLiuBCoupling Gd-DTPA with a bispecific, recombinant protein anti-EGFR-iRGD complex improves tumor targeting in MRIOncol Rep20163563227323527035336
- WangTWeiJQianXDingYYuLLiuBGambogic acid, a potent inhibitor of survivin, reverses docetaxel resistance in gastric cancer cellsCancer Lett2008262221422218248784
- WangLHLiYYangSNGambogic acid synergistically potentiates cisplatin-induced apoptosis in non-small-cell lung cancer through suppressing NF-κB and MAPK/HO-1 signallingBr J Cancer2014110234135224300974
- YinDYangYCaiHWangFPengDHeLGambogic acid-loaded electrosprayed particles for site-specific treatment of hepatocellular carcinomaMol Pharm201411114107411725290462
- WenCHuangLChenJGambogic acid inhibits growth, induces apoptosis, and overcomes drug resistance in human colorectal cancer cellsInt J Oncol20154751663167126397804
- ZhangZQianHYangMGambogic acid-loaded biomimetic nanoparticles in colorectal cancer treatmentInt J Nanomedicine2017121593160528280328
- ChouSHShettyAVGengYPalmitate-derivatized human IL-2: a potential anticancer immunotherapeutic of low systemic toxicityCancer Immunol Immunother201362359760323124508
- KimSAPeacockJSThe use of palmitate-conjugated protein A for coating cells with artificial receptors which facilitate intercellular interactionsJ Immunol Methods1993158157658429217
- HuangAHuangLKennelSJMonoclonal antibody covalently coupled with fatty acid. A reagent for in vitro liposome targetingJ Biol Chem198025517801580187410345
- ChenAZhengGTykocinskiMLHierarchical costimulator thresholds for distinct immune responses: application of a novel two-step Fc fusion protein transfer methodJ Immunol2000164270571110623813
- LiuSBreiterDRZhengGChenAEnhanced antitumor responses elicited by combinatorial protein transfer of chemotactic and costimulatory moleculesJ Immunol200717853301330617312181
- ZhengGChenASternerREInduction of antitumor immunity via intratumoral tetra-costimulator protein transferCancer Res200161228127813411719441
- LiuSFosterBAChenTZhengGChenAModifying dendritic cells via protein transfer for antitumor therapeuticsClin Cancer Res200713128329117200367
- ChenAZhengGTykocinskiMLQuantitative interplay between activating and pro-apoptotic signals dictates T cell responsesCell Immunol2003221212813712747954
- ZhuJTangCKottke-MarchantKMarchantREDesign and synthesis of biomimetic hydrogel scaffolds with controlled organization of cyclic RGD peptidesBioconjug Chem200920233333919191566