 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
We report an analysis of in vitro and in vivo drug release from an in situ formulation consisting of triamcinolone acetonide (TR) and poly(d,l-lactide-co-glycolide) (PLGA) and the additives glycofurol (GL) and hydroxyapatite nanoparticles (HA). We found that these additives enhanced drug release rate. We used the Taguchi method to predict optimum formulation variables to minimize the initial burst. This method decreased the burst rate from 8% to 1.3%. PLGA-HA acted as a strong buffer, thereby preventing tissue inflammation at the injection site caused by the acidic degradation products of PLGA. Characterization of the optimized formulation by a variety of techniques, including scanning electron microscopy, X-ray diffraction, differential scanning calorimetry, and Fourier transform near infrared spectroscopy, revealed that the crystalline structure of TR was converted to an amorphous form. Therefore, this hydrophobic agent can serve as an additive to modify drug release rates. Data generated by in vitro and in vivo experiments were in good agreement.
Introduction
Injectable drug delivery systems have improved significantly.Citation1–Citation3 These improvements make them easier to use, enable targeted delivery,Citation4–Citation6 optimize delivery periods, minimize side effects by reducing drug dose, and increase patient comfort and compliance.Citation7 Creating in situ implants has become a frequently used technique, by which the drug and a biodegradable polymer are dissolved in an organic solvent and administered by subcutaneous injection, generating a semi-solid depot. This drug delivery system is a good alternative to other implant devices for the following reasons: the process causes much less pain in comparison to implants, which require local surgery; localized or systemic drug delivery can be maintained for months; ‘infusion-like’ plasma level time profiles minimize side effects. Other advantages include dose reduction to avoid peaks and valleys of drug concentration in blood, and enhanced patient compliance by reducing application frequency. In situ forming depot systems using polymers are relatively simple to produce and cost less than microspheres, which have to be washed and isolated.Citation8
There are two different techniques for delivering in situ forming implants and in situ forming microparticles, depending on depot type. An in situ forming implant consists of a drug-containing biodegradable polymer solution in a biocompatible organic solvent. The solvent diffuses in the body’s aqueous environment, leading to polymer precipitation and formation of an implant at the injection site. The most popular and biocompatible solvent used is N-methyl-2-pyrrolidone (NMP).Citation9–Citation13 Poly(d,l-lac-tide-co-glycolide) (PLGA) is the most successfully used biodegradable and biocompatible polymer because of its two biodegradable hydrolysis products, lactic and glycolic acids ().Citation14
Figure 1 Poly(d,l-lactide-co-glycolide) hydrolysis.
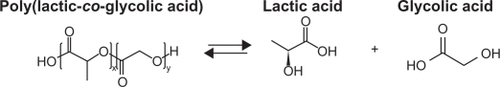
The properties of PLGA that affect drug release, including its molecular weight (MW), lactide/glycolide ratio, and thermal properties, have been thoroughly investigated. Using lower MW PLGA increases the rates of polymer degradation and drug release. Decreasing the lactide content results in slower polymer degradation and slower drug release.Citation15
Experimental design involves defining and investigating all factors thought to affect the outcome,Citation16 and experiments are executed together or sequentially to optimize the variable parameters and define the optimum conditions. In situations involving numerous variables, a method called factorial design is used to discover the best combination of control factors to increase efficiency and reduce costs.Citation17 However, this may not be possible in cases of high complexity.Citation18 In the present study, we evaluated the parameters affecting the release rate of triamcinolone acetonide (TR) loaded on PLGA in the presence of other additives. Process parameters were optimized using the robust Taguchi method, which provides a comprehensive system for improving and optimizing drug release by considering equal contributions of variables.Citation19 The resulting optimized formulation was characterized using scanning electron microscopy (SEM), X-ray diffraction (XRD), differential scanning calorimetry (DSC), and Fourier transform near infrared spectroscopy (FTNIR).
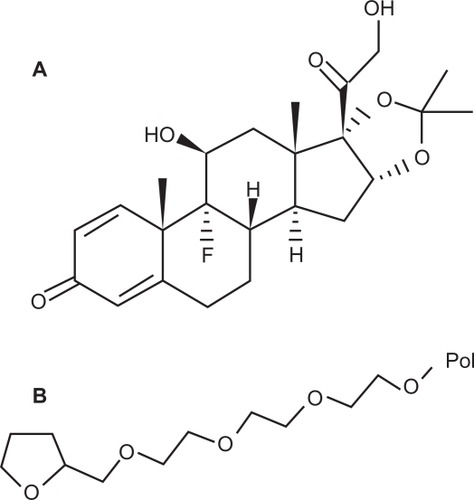
TR is a steroidal drug usually administered by the parenteral route (). Its release from a biodegradable PLGA polymer can be evaluated by the two key parameters of burst release rate and release duration. These parameters are affected by drug loading, which is considered the main focus for in vivo burst release optimization.
Figure 2 Structures of A) triamcinolone acetonide and B) polyethylene glycol ether.
Materials and methods
Chemicals
The following chemicals were used as received from the suppliers: PLGA, Resomer® RG 504H (MW = 48,000 kDa) (Boehringer Ingelheim, Ingelheim, Germany), TR (Farmabios, Gropello Cairopoli, Italy), NMP (Merck KGaA, Darmstadt, Germany), methanol (HPLC Gradient Grade, Merck KGaA), acetonitrile (HPLC Gradient Grade, Merck KGaA), NaOH (Merck KGaA), tetrahydrofurfuryl alcohol, polyethylene glycol ether (GL) (glycofurol, Merck KGaA), and hydroxyapatite (HA) nanoparticles (Sigma-Aldrich, Munich, Germany).
Methods and characterization
Casting solution preparation
Homogeneous casting solutions (33% w/w) were prepared from PLGA in NMP. Varying amounts of drug were added and completely dissolved before introducing additives (). To study drug release patterns, experiments were carried out in glass vials type I (15 mL) (Schott Duran®, Mainz, Germany), because they possess the lowest available drug absorption properties. Vials were charged with the casting solutions, weighed immediately, and then filled with 10 mL degassed phosphate buffer (PB) (0.03 M, pH = 7.4) to minimize solvent exchange with air. All vials were incubated at 37 ± 0.5°C. The entire receptor phase (10 mL) was withdrawn at predetermined intervals and replaced by the same amount of freshly prepared receptor medium.
Table 1 Selected parameters and their levels for experimental design
Release measurements
Formulation performance was evaluated by measuring TR release kinetics using a reverse phase high performance liquid chromatography (HPLC) method (C-18 column, 4.6 × 250 mm; NovaPak®; Waters, Milford, MA). The isocratic mobile phase consisted of a mixture of deionized water, acetonitrile, and methanol (62:33:5). Ultraviolet light absorbance was measured at 254 nm, and the column temperature was maintained at 25°C. Each experiment was performed in triplicate.
Formulation optimization
The experiments were designed using the Taguchi method. In order to determine the best formulations, three parameters, including TR, HA nanoparticles, and GL, as well as three levels for each parameter () and a fractional factorial design, namely, a standard L9 orthogonal array,Citation20 were employed (). This orthogonal array was selected to reduce interaction among the parameters. Each row of the matrix represents one run. The sequence of these runs is randomized. As shown in , three different levels related to each parameter are represented as ‘1’, ‘2’, and ‘3’. QUALITEK-4 software (Nutek, Bloomfield Hills, MI) was used for experimental design and data analysis.Citation21 Results are expressed as the average of the three analyses.
Table 2 L9 orthogonal array
SEM
Formulation surfaces were studied with a scanning electron microscope, VEGA 3 SB (TESCAN, as, Brno, Czech Republic). Gold-plated samples were examined at room temperature.
XRD
The XRD patterns of the formulations were measured using a PANalytical X-ray diffractometer (XPERT PRO, PW 3040/60 generator, PIXcel Detector and X’Pert Data Collector software for automatic powder diffraction, v 2.2i; PANalytical, Almelo, the Netherlands). The X-ray diffractometer was operated with an anode current of 40 mA and an accelerating voltage of 40 kV. Samples were slightly pressed on an aluminium sample tray using a glass slide (75 × 25 mm) and exposed to CuKα radiation at diffraction angles (2θ) from 5° to 40° (step size, 0.02°; time per step, 40.01 seconds). X’Pert HighScore Plus (v 2.2d; PANalytical) software was used to locate the peaks in XRD diffractograms by detecting second derivative minima. Intensity maxima are expressed as Kα1 net peak height in counts at Kα1 position in degrees.
DSC
A Perkin–Elmer DSC 7 (PerkinElmer, Skovlunde, Denmark) with Pyris software was used to determine the glass transition temperature (Tg). The sample (1.5–3.0 mg) was transferred to an aluminium pan in which a pinhole was punched in the pan lid, and the sides of the lid were crimped. A similar empty pan was used as reference. Samples were scanned from 10–70°C at a rate of 10°C/minute under nitrogen gas (20 mL/minute). Independent triplicate measurements are expressed as their means. The instrument was calibrated using an indium standard.
FTNIR
Infrared spectra were determined using a Bruker IR-spectrometer Tensor 37 (Bruker, Ettlingen, Germany) in single beam transmittance mode with optical probe. Each spectrum consisted of 32 scans collected in single beam at a resolution of 4 cm−1 at 25°C. Spectra were obtained on a straight baseline in the near infrared (NIR) region.
Hot plate method
The hot plate test was performed in mice to assess the release response of triamcinolone. The hot plate test was performed by placing mice on an aluminum plate maintained at 51 ± 1°C. The latency to an analgesic response, identified as licking of either hind paw, or lifting of either hind paw from the heated surface, was measured.
Results and discussion
Data analysis
shows the release profiles of TR from different formulations of PLGA-based implants. In the present study, to reduce the burst release associated with PLGA-based delivery systems formed in situ, we prepared nine different formulations incorporating TR and the additives described in . Experiments were executed based on the Taguchi design ().
Figure 3 Triamcinolone acetonide release kinetics in poly(d,l-lactide-co-glycolide) in phosphate buffer.
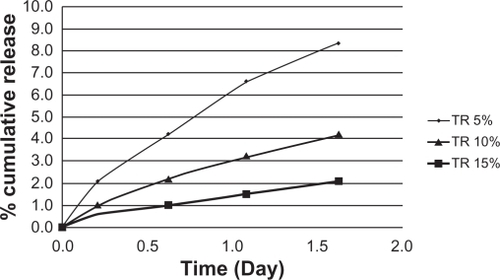
presents the release rates of a variety of formulations containing different levels of additives. All additives, especially HA, improved release rates. HA naturally occurs in mineral form known as calcium apatite, whose formula, Ca5(PO4)3(OH), is usually written as Ca10(PO4)6(OH)2 to denote that the crystal unit cell includes 2 entities. HA is the hydroxyl-end member of the complex apatite group. The SEM image of HA () revealed that these non-sintered nanoparticles are nanocrystal clusters with equally spherical shape.Citation22 Most of the drug-loaded formulations showed a biphasic release pattern () exhibiting an initial burst followed by sustained release.Citation23,Citation24 The high initial release may be due to the presence of free and weakly bound drug on the surface of carriers.
Table 3 Cumulative release after 1.5 days
Figure 4 Hydroxyapatite nanoparticles.

Figure 5 Cumulative triamcinolone acetonide release kinetics in poly(d,l-lactide-co-glycolide)-hydroxyapatite in phosphate buffer.
Prediction, optimization and characterization
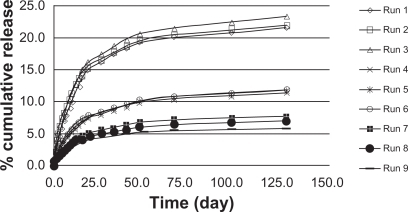
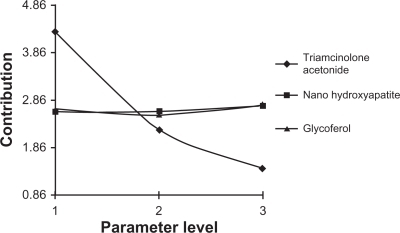
To identify the main parameters’ influence on burst release, calculations were based on release data over 1.5 days (). We used QUALITEK-4 software for this analysis (). In the Taguchi method, the main effect of control parameters indicates the trend of a parameter’s influence. The key effects were calculated using average release. The results indicate the drug loading effect on the release profile (). Another technique for optimization of the results suggested by the Taguchi method is analysis of variance (ANOVA). presents the analysis of variance (ANOVA) values, which indicate the relative influence of factor and interaction to the variation of results. ANOVA is similar to regression analysis, which is used to investigate and determine the relationship between a response variable and one or more independent variables.Citation21 Furthermore, Bese et alCitation25 state that optimal combination of process parameters can be predicted from the performance characteristics and ANOVA analyses. The contributions of each parameter to burst release optimization and expected optimum condition are listed in . According to the Taguchi method, TR had the highest contribution to the decrease of burst release. The highest percentage for control parameters was obtained with the following composition: TR (15%), HA (1%), and GL (3%).
Table 4 ANOVA for cumulative release after 1.5 days
Table 5 Optimum conditions for the burst release reduction
Figure 6 Cumulative TR release kinetics in poly(d,l-lactide-co-glycolide)-hydroxyapatite in phosphate buffer (after 1.5 days).
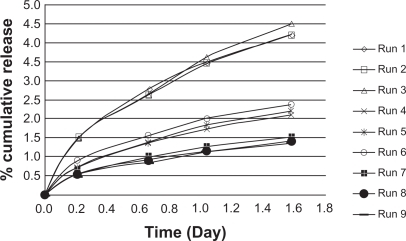
Figure 7 Effects of variables vs parameter levels.
At optimal conditions, burst release decreased to 1.3%. The drug contents in PLGA and PLGA-HA based formulations were 5%, 10%, and 15% (drug/PLGA). These starting values of TR for PLGA and PLGA-HA formulations were taken as the 100% starting values of the released drug. TR release from PLGA () and PLGA-HA () was conducted in PB (pH 7.4). As can be seen, the release profiles of the two formulations are different ( and ). The PLGA release profile is characterized by a biphasic drug release typically exhibited by biodegradable polymers: an initial burst followed by a sustained release. However, PLGA-HA displayed a triphasic drug release. During the first 1.5 days, a small ‘burst’ was observed for both PLGA (8%) and PLGA-HA (4.5%) based on the total amount of loaded drug. The initial burst was caused by the drug’s rapid release near the surface. The second plateau phase for PLGA-HA lasted around 20 days with a low dose of TR released. The plateau phase was followed by a third stage with decreasing drug release lasting for over 100 days. A gradually increased release during the first 20 days was followed by a slow release at a nearly constant rate of 0.05% per day for PLGA-HA. The cumulative drug release profile of TR (15%) from PLGA was similar to that of PLGA-HA. These release rates were 1.5 and 2 times that of 10% and 5% TR, respectively ( and ). These results show that at 15% TR, its release is independent of additive. In contrast, HA controlled the release rate of 5% and 10% TR.
Triphasic drug release follows the following stages: initial burst, followed by drug diffusion, with polymer degradation occurring at the third phase. The second phase, which continues for about 20 days, is governed by swelling of the PLGA in situ forming implant. TR is released slowly from the in situ gel, and is replaced by water. The precipitation of PLGA follows a biphasic mechanism. After injection, a thin PLGA membrane is immediately formed at the injection site. Dissolved NMP and TR diffused through this polymer shell into the aqueous environment, and water diffused into the polymer, simultaneously. As hardening of the PLGA membrane increases due to further polymer precipitation, the diffusion distances are increased. According to the Einstein–Smoluchowski Equationequation (1)(1) , where D, d, and t are the diffusion coefficient, distance and time, respectively, as diffusion distances inside the implant are increased, the diffusion times for NMP outside, and water inside, are increased.Citation26,Citation27 This is represented by the following equation:
During the formation of implants of PLGA and PLGA-HA in situ in PB, the pH decreased to approximately 5 and 6, respectively. In the presence of HA, the pH decreased more slowly (). Lactic and glycolic acids are two products that are generated by PLGA biodegradation and cause tissue inflammation at the injection site. Compared to PLGA, PLGA-HA demonstrated significant buffer capacity. This anti-inflammatory property, which was expected from this formulation, is in accordance with other studies.Citation28,Citation29 After 20 days, while the PLGA formulation was extensively degraded (), PLGA-HA was only partially disrupted (). Slower degradation of PLGA-HA is due to the presence of HA. Meanwhile, this additive caused a low burst and controlled the overall rate of drug release as shown in . HA as a bisphosphonated and chelating component can be encapsulated into the PLGA matrix. This non-mutually exclusive hypothesis can be proposed as a reason for the slow degradation of PLGA-HA. In addition, HA degradation in pH < 4.2 has been investigated previously.Citation29 This biodegradation releases Ca2+ and the result is pH buffering capacity. This can be combined with the anti-inflammatory properties of TR for better control of inflammation due to PLGA hydrolysis at the injection site, using this optimized formulation.
Figure 8 Effects on pH of triamcinolone acetonide (5%) with PLGA or PLGA-HA.
Abbreviations: PGLA, poly(d,l-lactide-co-glycolide); PGLA-HA, poly(d,l-lactide-co-glycolide) with hydroxyapatite.
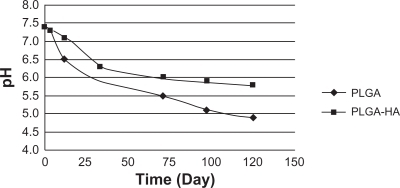
Figure 9 SEM image of membranes after immersion in phosphate buffer for 20 days: A) poly(d,l-lactide-co-glycolide) and B) PGLA-HA: poly(d,l-lactide-co-glycolide) with hydroxyapatite.

Glycofurol is a water-miscible solvent which is used in the preparation of parenteral pharmaceutical dosage forms and with PLGA for in situ gel forming.Citation7,Citation30 This non-toxic solvent enhances the solubility and permeability of water-insoluble drugs.Citation31,Citation32 XRD data evaluation leads to the conclusion that TR forms a crystalline structure, which becomes somewhat degraded in the PLGA matrix. This structure was converted to amorphous TR in the presence of 3% GL. This transition results from hydrogen bonding between TR and GL (). The solubility of amorphous TR in intrinsically amorphous PLGA is greater than that of crystalline TR. This increases the density of the membrane structure () and leads to lower burst release. In the absence of GL, upon phase inversion, TR remains crystalline, and some of these crystals escape from the membrane () causing a burst release. GL increases the solubility of TR, increasing its bioavailability, consistent with studies by Bakarat.Citation33
Figure 10 Diffractograms of A) triamcinolone acetonide B) PLGA and triamcinolone acetonide (15%), C) PLGA and triamcinolone acetonide (15%) and polyethylene glycol ether (3%).
Abbreviation: PLGA, poly(D,L-lactide-co-glycolide).
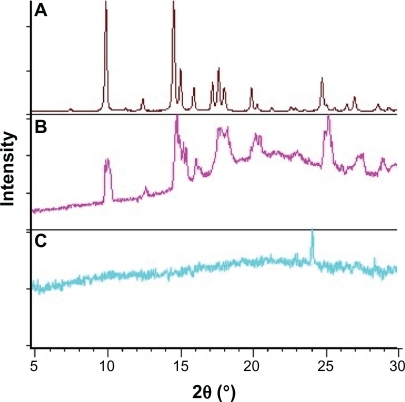
Figure 11 Scanning electron microgram of poly(d,l-lactide-co-glycolide) membranes: A) 3% and B) 0% polyethylene glycol ether.

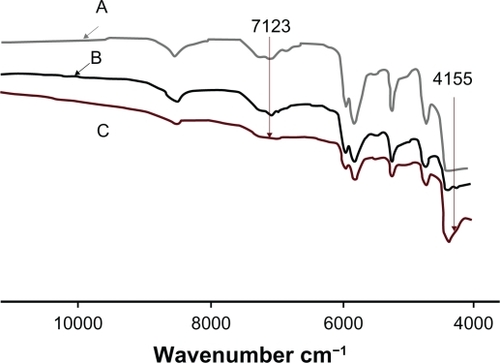
The structure of polymeric materials can be identified by combinations and overtone vibration of X–H bonds using near infrared (NIR) absorption. Hydrogen bonds in O–H groups give rise to sharp peaks in infrared (IR) spectroscopy.Citation34 The Fourier Transform infrared spectroscopy (FTIR) spectrum revealed interaction between PLGA and amorphous TR, which decreases burst release ().
Figure 12 Fourier transform near infrared spectra of poly(d,l-lactide-co-glycolide) membranes in the presence of A) 0%, B) 1%, and C) 3% polyethylene glycol ether.
DSC analysis of TR shows endothermic peaks in the 242–280°C range, which is characteristic of polymorph A and B.Citation35,Citation36 The glass transition temperature (Tg) of PLGA in situ forming gels, including TR (15%) and GL (3%), was 49.8°C. This is also evidence for the transition of TR from a crystalline to an amorphous form in the presence of GL, and the existence of elastic properties in the PLGA membrane.Citation31 Solubilization by GL causes this, decreases the burst release, and makes a dense structure of the PLGA implant.
In vivo studies
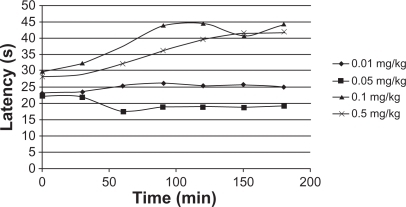
Drug release through PLGA has been studied in detail.Citation37–Citation41 The purpose of the present study is to systematically incorporate specific excipients into TR-loaded PLGA in situ gel formulations in order to evaluate the burst effect in vivo. The hot-plate test was administered to mice to assess their response to TR release. The latency of an analgesic response was identified as either licking or lifting of a hind paw from the heated surface. A preliminary study of the time course of TR’s analgesic activity in mice was performed to determine the time to peak analgesic activity and effective dose 50 (ED50). Eight groups of mice (five mice per group), designated as group A and B, were selected. TR (0.01 mg/kg, 0.05 mg/kg, 0.1 mg/kg and 0.5 mg/kg) with and without PLGA was injected intramuscularly into members of group A and B, respectively. Latency time was measured at 30, 60, 90, 120, 150, and 180 minutes after administration. Injections without PLGA caused a significant increase in reaction times (), while PLGA injections caused only a small change (). The increase in reaction time was dose-dependent and differed significantly among the groups of mice receiving different TR doses. The data demonstrate that the burst release of TR from the PLGA matrix was very low and that a sustained release of the drug was achieved. Overall evaluation of these results shows that the data are quite comparable to those acquired in vitro. The burst release was decreased and the TR concentration had the greatest effect on control of the burst effect. Thus, we conclude that PLGA injected into the body acts as a depot for the sustained release of TR.
Figure 13 Hot-plate response of mice (n = 5) administered triamcinolone acetonide.
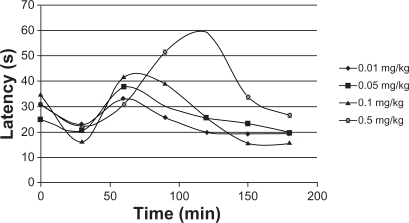
Figure 14 Hot-plate response of mice (n = 5) administered triamcinolone acetonide with poly(d,l-lactide-co-glycolide).
Conclusion
We applied the Taguchi method to predict and optimize in situ gel formulation of TR and PLGA polymer in the presence of HA nanoparticles and GL additives. Drug release followed a linear pattern throughout dissolution after 1.5 days using PLGA and PLGA-HA. The amount of TR released after 1.5 days, irrespective of the type of formulation, was less than 8%. Different formulations were achieved using the Taguchi method and we found that burst release decreased to 1.3% using TR (15%), HA (1%), and GL (3%) formulations.
Analysis of in vitro and in vivo studies showed that TR concentration contributes most significantly to the control of burst release. The study of drug release continued over 125 days in vitro. This prolonged delivery period will enable decreased drug dosage and possibly reduce the TR’s undesirable side effects. The PLGA-HA combination exhibited significant buffering capacity that prevented tissue inflammation at the injection site, which is the result of acidic degradation products of the PLGA membrane.
The structure of TR changed from crystalline to amorphous in the presence of GL, which increases TR’s solubility in PLGA, resulting in low burst release. The drug release pattern observed during this study can be considered the result of developing a novel drug delivery system based on optimizing water-insoluble drug release, decreasing tissue inflammation and minimizing burst release. This system promises to be useful for routine drug delivery research particularly in regard to in situ gel formulation and burst control for high potency drugs.
Acknowledgements
The authors thank Exir Pharmaceutical Co. (Boroujerd, Iran) and Copenhagen University for financial support, and the latter for generously providing equipment.
Disclosure
The authors report no conflicts of interest in this work.
References
- KakinokiSTaguchiTAntitumor effect of an injectable in-situ forming drug delivery system composed of a novel tissue adhesive containing doxorubicin hydrochlorideEur J Pharm Biopharm20076767668117493793
- LeeFChungJEKurisawaMAn injectable hyaluronic acid-tyramine hydrogel system for protein deliveryJ Control Release200913418619319121348
- FraylichMRLiuRRichardsonSMThermally-triggered gelation of PLGA dispersions: towards an injectable colloidal cell delivery systemJ Colloid Interface Sci2010344616920070971
- HollisterLESite-specific drug delivery to CNS: Old and newNeurobiol Aging198910628631
- LevyRJVinodLStrickbergerSAUnderwoodTDavisJControlled release implant dosage forms for cardiac arrhythmias: review and perspectivesDrug Delivery19963137142
- TiptonAJDunnRLIn situ gelling systemsSeniorJRadomskyMSustained Release Injectable ProductsEnglewood, COInterpharm Press200071102
- HatefiAAmsdenBBiodegradable injectable in situ forming drug delivery systemsJ Control Release20028092811943384
- PackhaeuserCBSchniedersJOsterCGKisselTIn situ forming parenteral drug delivery systems: an overviewEur J Pharm Biopharm20045844545515296966
- DunnRLEnglishJPCowsarDRVanderbiltDP inventorsBiodegradable in-situ forming implants and methods of producing the samePatent57339501998331
- RavivarapuHBMoyerKLDunnRLSustained suppression of pituitary-gonadal axis with an injectable, in situ forming implant of leuprolide acetateJ Pharml Sci200089732741
- RavivarapuHBMoyerKLDunnLParameters affecting the efficacy of a sustained release polymeric implant of leuprolideInt J Pharm200019418119110692642
- LambertWJPeckKDDevelopment of an in situ forming biodegradable poly-lactide-co-glycolide system for the controlled release of proteinsJ Control Release199533189195
- WangLKleinerLVenkatramanSStructure formation in injectable poly(lactide-co-glycolide) depotsJ Control Release20039034535412880701
- KumariAKumarYSYadavSCBiodegradable polymeric nanoparticles based drug delivery systemsColloids Surf B Biointerfaces20107511819782542
- LuanXBodmeierRInfluence of the poly(lactide-co-glycolide) type on the leuprolide release from in situ forming microparticle systemsJ Control Release200611026627216300851
- RoyRKA Primer on the Taguchi MethodNew YorkVan Nostrand Reinhold1990
- FisherRAThe Design of Experiments8th edNew YorkHafner Press1966
- AshrafizadehSNKhorasaniZAmmonia removal from aqueous solutions using hollow-fiber membrane contactorsChem Eng J2010162242249
- KuoHCJengMCEffects of part geometry and injection molding conditions on the tensile properties of ultra-high molecular weight polyethylene polymerMater Des201031884893
- ParkSHRobust design and Analysis for Quality EngineeringLondon, UKChapman and Hall1996
- MadaeniSSKoochekiSApplication of taguchi method in the optimization of wastewater treatment using spiral-wound reverse osmosis elementChem Eng J20061193744
- JosephRMcGregorWJMartynMTTannerKECoatesPDEffect of hydroxyapatite morphology/surface area on the rheology and processability of hydroxyapatite filled polyethylene compositesBiomaterials2002234295430212194532
- BirnbaumDTKosmalaDHenthornBPeppasBControlled release of beta-estradiol from PLAGA microparticles: the effect of organic phase solvent on encapsulation and releaseJ Control Release20006537538710699296
- OtsukaMUdenodanHMatsudaYTherapeutic effect of in vivo sustained estradiol release from poly (lactide-co-glycolide) microspheres on bone mineral density of osteoporosis ratsBiomed Mater Eng20021215716712122239
- BeseAVBoruluNCopurMColakSNuri AtaOOptimization of dissolution of metals from Waelz sintering waste (WSW) by hydrochloric acid solutionsChem Eng J2010162718722
- AtkinsPWPhysical Chemistry4th edOxford, UKOxford University Press1990
- KempeSMetzHMaderKDo in situ forming PLG/NMP implants behave similar in vitro and in vivo? A non-invasive and quantitative EPR investigation on the mechanisms of the implant formation processJ Control Release200813022022518611421
- XuQCzernuszkaJTControlled release of amoxicillin from hydroxyapatite-coated poly(lactic-co-glycolic acid) microspheresJ Control Release200812714615318325617
- LinPLFangHWTsengTLeeWHEffects of hydroxyapatite dosage on mechanical and biological behaviors of polylactic acid composite materialsMater Lett20076130093013
- EliazREWallachDKostJDelivery of soluble tumor necrosis factor receptor from in-situ forming PLGA implants: in-vivoPharm Res2000171546155011303966
- BarakatNSOptimization of physical characterization, skin permeation of naproxen from glycofurol-based topical gelAsian J Pharm20104154162
- AllhennDLamprechtAMicrosphere preparation using the untoxic solvent glycofurolPharm Res20111156357121049282
- BarakatNEvaluation of glycofurol-based gel as a new vehicle for topical application of naproxenAAPS Pharm Sci Tech20101111381146
- IwamotoRAmiyaSSaitoYSamuraHFT-NIR spectroscopic study of OH groups in ethylene-vinyl alcohol copolymerAppl Spectrosc200155864870
- SuitchmezianVJessINatherCInvestigations on the polymorphism and pseudopolymorphism of the glucocorticoid triamcinolone: new findings for a well-known drugCryst Growth Des200776974
- da SilvaAAde MatosJRFormarizTPThermal behavior and stability of biodegradable spray-dried microparticles containing triamcinoloneIntl J Pharm20093684555
- YoungTHChuangWYWeiCWTangCYInvestigation of the drug distribution and release characteristics from particulate membranesJ Memb Sci2001191199205
- StamatialisDFPapenburgBJGironesMMedical applications of membranes: drug delivery, artificial organs and tissue engineeringJ Memb Sci2008308134
- MaDMcHughAJThe interplay of phase inversion and membrane formation in the drug release characteristics of a membrane-based delivery systemJ Memb Sci2007298156168
- XiangAMaDMcHughAJThe interplay of phase inversion, polymer membrane formation, and drug release in a membrane-based delivery systemJ Memb Sci20103588592
- McHughAJThe role of polymer membrane formation in sustained release drug delivery systemsJ Control Release200510921122116288815