
Abstract
Background
PARP inhibitors, such as Olaparib, have advanced the treatment of ovarian cancer by providing patients with an effective and molecularly-targeted maintenance therapy. However, all orally-administered drugs, including Olaparib, must undergo first-pass metabolism. In contrast, a nanoparticle delivery system has the advantage of administering Olaparib directly into the peritoneal cavity for local treatment. Consequently, we sought to optimize the sustained-release formulation NanoOlaparib, previously deemed effective as an intravenous solid tumor treatment, for the local treatment of disseminated disease via intraperitoneal (i.p.) therapy.
Methods
The tumor cell line 404, which was derived from a Brca2−/−, Tp53−/−, Pten−/− genetically engineered mouse model, exhibited high sensitivity to Olaparib in vitro. It was chosen for use in developing an i.p. spread xenograft for testing nanotherapy efficacy in vivo. NanoOlaparib as a monotherapy or in combination with cisplatin was compared to oral Olaparib alone or in combination using two different dose schedules. A pilot biodistribution study was performed to determine drug accumulation in various organs following i.p. administration.
Results
Daily administration of NanoOlaparib reduced tumor growth and decreased the variability of the treatment response observed with daily oral Olaparib administration. However, systemic toxicity was observed in both the NanoOlaparib and vehicle (empty nanoparticle) treated groups. Scaling back the administration to twice weekly was well tolerated up to 100 mg/kg but reduced the effect on tumor growth. Biodistribution profiles indicated that NanoOlaparib began accumulating in tissues within an hour of administration and persisted for at least 72 hours after a single dose, exiting the peritoneal cavity faster than expected.
Conclusion
NanoOlaparib must be modified for use against disseminated disease. Future avenues to develop NanoOlaparib as an i.p. therapy include a modified surface-coating to retain it in the peritoneal cavity and prevent entry into systemic circulation, in addition to targeting moieties for localization in tumor cells.
Introduction
Poly-ADP-ribose polymerase (PARP) inhibitors are a new class of targeted therapy for the treatment of ovarian cancer. PARPs are a family of enzymes heavily involved in DNA repair and maintaining genomic integrity.Citation1,Citation2 A prominent mechanism of action for PARP inhibitors exploits the concept of synthetic lethality, in which cells deficient in homologous recombination (HR) repair, such as those with BRCA1/2 mutations, cannot repair DNA damage when the PARP enzyme is also blocked.Citation3,Citation4 The complexity of the role of PARP in DNA repair pathways has resulted in a number of models of synthetic lethality, with the most notable halting the repair of single-strand breaks (SSBs) through the base excision repair pathway.Citation5,Citation6 If these SSBs are not repaired prior to DNA replication, the replication fork stalls leading to double-strand breaks (DSBs), which must be repaired by HR or non-homologous end joining, a lower-fidelity repair mechanism that results in genomic instability.Citation7 Most importantly, PARP inhibition does not affect healthy cells, which have intact DNA repair mechanisms. More recently, PARP inhibitor function has expanded to include other mechanisms not reliant on HR deficiencies, including PARP trapping, in which the PARP complex is trapped at the site of DNA damage and inhibits DNA repair by stalling the replication forks.Citation8,Citation9
Olaparib, one of three Food and Drug Administration (FDA) approved PARP inhibitors, is used for the treatment of germline BRCA1/2 mutant recurrent ovarian cancer patients who have been treated with three or more prior lines of chemotherapy. The high morbidity of ovarian cancer is associated with relapsed disseminated disease, and the gradual development of acquired chemoresistance leads to low response and a high death rate.Citation10–Citation12 Olaparib gained FDA approval after a clinical trial showed enhanced progression-free survival (PFS) from 4.8 to 8.4 months in relapsed ovarian cancer patients, with a greater PFS increase in the BRCA1/2 patient population from 4.3 to 11.2 months.Citation13,Citation14 Preclinical studies have shown PARP inhibitors to be effective at potentiating DNA damaging agents, including temozolomide, cisplatin, carboplatin, and cyclophosphamide.Citation15 Olaparib specifically has been found to be synergistic with cisplatin in growth inhibition of BRCA2-deficient mammary tumor cells but additive in BRCA2 wild-type cells.Citation16 Clinical trials combining chemotherapy and PARP inhibitors have been plagued by dose reduction and delays due to myelosupression.Citation17–Citation21 Thus, while Olaparib in combination with carboplatin was shown to be efficacious, hematologic toxicity did not allow for the continuous dosing of Olaparib with the traditional carboplatin regimen.Citation22 Reduction of the Olaparib dose in combination studies is problematic as there is clinical evidence for greater activity with higher doses.Citation23,Citation24 So far, combination trials involving chemotherapy and PARP inhibitors have not shown enough clinical impact over single-agent treatment to warrant FDA approval.
Nanoparticle formulations of drugs are frequently utilized to reduce toxicity while maintaining therapeutic efficacy.Citation25 Typically, nanoformulations exploit the leaky vasculature of tumors to preferentially accumulate at the tumor site, an effect referred to as enhanced permeability and retention (EPR).Citation26 For the local treatment of metastatic ovarian cancer, intraperitoneal (i.p.) delivery is preferred as recurrent disease is usually disseminated throughout the peritoneal cavity. Furthermore, in a landmark clinical trial, i.p. chemotherapy was shown to enhance both PFS and overall survival when compared to intravenous chemotherapy.Citation27 A nanoformulation for deliver of Olaparib, an orally administered PARP inhibitor, offers a means to specifically target the peritoneal cavity while bypassing first pass metabolism. In addition, due to poor drug solubility, patients have to take two tablets twice per day to sustain an effective dose of Olaparib and reach clinical efficacy. By contrast, a nanoformulation has the advantage of slow drug release over days, thus reducing the burden on patients.Citation28
Previously, we have shown that Olaparib can be encapsulated in lipid nanoparticles and that prostate cancer cells pretreated with NanoOlaparib exhibit more DNA damage after exposure to radiation than those pretreated with free Olaparib.Citation29 We hypothesized that utilizing NanoOlaparib for i.p. delivery would be more effective at treating metastatic ovarian tumors compared to orally administered Olaparib in a tumor xenograft that mimics disseminated peritoneal disease. We also hypothesized that the dose of NanoOlaparib could be further reduced compared to oral Olaparib and still be effective since 100% of the drug would be delivered directly to the affected disease site. Consequently, we anticipated that the i.p. delivery of NanoOlaparib would still be effective even in cases where dose reduction was required.
Materials and methods
Materials
1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol, 1,2-distearoyl-sn-glycero-3 phosphoethanolamine-N-[amino(polyethyleneglycol)-2000] (DSPE-PEG-2000 Amine), and 1,2-dioleoyl-3-tri methyl-ammonium-propane (chloride salt) (DOTAP) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Olaparib was purchased from Selleck Chemicals (Houston, TX, USA). 2-Hydroxyl-propyl-β-cyclodextrine and α-cyano-4-hydroxycinnamic acid were purchased from Sigma -Aldrich Co. (St Louis, MO, USA). All solvents used were HPLC grade from Sigma -Aldrich Co. DMEM, PBS, trypsin–EDTA solution, and FBS were purchased from Gibco (Gaithersburg, MD, USA). d-Luciferin was purchased from PerkinElmer Inc. (Waltham, MA, USA). Cisplatin was purchased from Patterson Veterinary Supply Inc. (Devens, MA, USA) as a 1 mg/mL saline solution (volume 50 mL). Oasis HLB 1 cc Vac Cartridges were purchased from Waters (Milford, MA, USA).
NanoOlaparib synthesis
NanoOlaparib was prepared using the thin film hydration method as previously described.Citation29,Citation30 Briefly, a fixed ratio of DPPC/DOTAP/cholesterol/DSPE-PEG-2000 Amine in chloroform was mixed with Olaparib and dried on a rotary evaporator overnight. The film was hydrated at 50°C and underwent five cycles of heating, vortexing, and cooling. The crude particles were sized via sonication in a bath sonicator for 10 minutes. Unbound drug was removed by dialysis for 4 hours.
NanoOlaparib characterization
The size and zeta potential of the nanoparticle were measured with a Brookhaven 90Plus analyzer equipped with ZetaPALS. The particle size was further confirmed via transmission electron microscopy with a negative stain of 1.5% phosphotungstic acid. The concentration of Olaparib inside nanoparticles was measured by HPLC after particle lysis via methanol. HPLC was performed on an Agilent Technologies 1100 or 1260 series instrument (Santa Clara, CA, USA) with a Supelco C18 column (Sigma-Aldrich Co.). The mobile phase was 64:36 methanol:water with a flow rate of 0.4 mL/min. Olaparib was detected at 207 nm with a retention time of ~2.3 minutes. The kinetics of Olaparib release from both the nanoparticles and the oral formulation (10% 2-hydroxyl-propyl-β-cyclodextrine/PBS) were measured via static release into PBS at 37°C under sink conditions to ensure release media were not saturated with Olaparib at any time. Briefly, 250 µL of each formulation was placed in Slide-A-Lyzer MINI Dialysis devices (Thermo Fisher Scientific, Waltham, MA, USA) and placed over 11.4 mL PBS in a 37°C water bath. At each time point, the PBS was removed and replaced with fresh PBS at 37°C. The PBS release media were analyzed by HPLC at each predetermined time to determine the concentration of Olaparib. All experiments were done in triplicate.
In vitro therapeutic efficacy studies
The 403 and 404 tumor lines were derived from conditional Brca2−/−, Pten−/−, Tp53−/− mice while the 4306 and 4412 lines were developed from conditional K-rasLSL-G12D/+; Pten−/− mice.Citation31,Citation32 The 403 and 404 tumor lines were lentivirally transduced to express luciferase. All lines were maintained in DMEM + 10% FBS. The doubling time was measured for each cell line via MTS assay prior to performing dose–response analysis. Cell lines were exposed to various concentrations of Olaparib (in DMSO) or NanoOlaparib ranging from 0 to 100 µM. Each cell line was treated for a total of four doubling cycles to ensure that the percent viability for each cell line was comparable.Citation33 Cell viability was ascertained by measuring the metabolic activity of the cells via MTS assay. The dose–response was plotted and fitted using a four-parameter logistic equation constrained at 0 and 100. All experiments were done in triplicate.
Animals
All animal studies and procedures were conducted in accordance with the Institutional Animal Care and Use Committee (IACUC) protocol #04187 reviewed and approved by the Harvard Medical Area Standing Committee on Animals.
Biodistribution profiles
Tissue collection and processing
To calculate the biodistribution profiles, healthy mice were injected with 50 mg/kg NanoOlaparib i.p. and sacrificed at 1, 3, 6, 24, 48, and 72 hours post injection (n=3/time point). The kidneys, liver, spleen, lungs, heart, gastrointestinal (GI) tract, and plasma were collected and weighed before homogenization. Olaparib was extracted from the tissues and plasma using methanol, and tissue extracts were further purified using OASIS solid phase extraction. Eluents were dried and reconstituted in α-cyano-4-hydroxycinnamic acid matrix before deposition of each sample in triplicate onto the matrix-assisted laser desorption/ionization (MALDI) plate.
MALDI mass spectrometry
Analysis was done on a Model 5800 MALDI-TOF/TOF-MS in MS2 mode. Each sample spot was surveyed and then 400 laser pulses collected. Each spectrum included 400 pulses for the triplicate spots of each sample for a total of 1,200 laser pulses averaged per sample. The area under the curve was used for further analysis and comparison of samples.
Gross toxicity and Olaparib accumulation
Three healthy mice were injected with 50 mg/kg NanoOlaparib or 10 µL/g empty nanoparticles (vehicle), a volume equivalent to NanoOlaparib administration, twice weekly for 4 weeks (n=3/group). The physical and behavioral condition of the animals was monitored according to IACUC guidelines, and all animals were observed for humane endpoint conditions, such as bodyweight loss. Mice were sacrificed 72 hours after the last NanoOlaparib treatment, and the tissues were harvested following the protocol described above. Samples were analyzed via MALDI mass spectrometry as detailed above.
In vivo therapeutic efficacy studies
Daily i.p. administration of NanoOlaparib
Drug Preparation
NanoOlaparib was prepared as described above. To have the proper concentration to inject, the suspension was concentrated via ultrafiltration with 100K MWCO Pall Macrosep devices (Port Washington, NY, USA) for 45 minutes at 3,200 rcf. The supernatant was resuspended in PBS at a final concentration of 5.0 mg/mL Olaparib. Oral Olaparib for injection was prepared by diluting 50 mg/mL stocks in DMSO with 10% 2-hydroxyl-propyl-β-cyclodextrine/PBS to create a 5.0 mg/mL solution. Cisplatin was purchased as a 1 mg/mL cisplatin solution diluted in saline.
Model generation
Female NOD/scid mice from Taconic (Rensselaer, NY, USA) were engrafted i.p. with 5.0×106 cells (404 tumor line, Brca2−/−, Tp53−/−, Pten−/−) to mimic late stage, disseminated disease. One week after engraftment, the animals underwent bioluminescence imaging (IVIS Lumina II In Vivo Imaging System, PerkinElmer Inc.) to determine a baseline of tumor burden, followed by separation into six groups.
Treatment and imaging
One group of control mice was left untreated while a second control group was administered the vehicle daily i.p. at 10 µL/g bodyweight, a volume that was equivalent to the dosing of NanoOlaparib. Olaparib was administered daily at a dose of 50 mg/kg by oral gavage while 50 mg/kg NanoOlaparib was given daily i.p. Cisplatin was administered i.p. twice weekly at a dose of 3 mg/kg. For the combination group, animals received i.p. injections of 50 mg/kg NanoOlaparib five times weekly and 3 mg/kg cisplatin twice weekly 30 minutes after the NanoOlaparib treatment.Citation34 Each treatment group consisted of five animals. The summary of the various treatment regimens and control groups is provided in . Animals were treated for 4 weeks. Tumor burden was monitored via bioluminescence imaging at the beginning of each week. Gross toxicity was assessed via daily weighing and by observing the general physical and behavioral condition of the animals consistent with IACUC guidelines.
Table 1 Overview of treatment doses and schedule for oral Olaparib, NanoOlaparib, and cisplatin
Twice weekly i.p. administration of NanoOlaparib
Female NCr nude mice from Taconic were engrafted with 404 tumor cells at 3.0×106 cells per animal in the i.p. cavity to generate an i.p. spread model. The drugs were prepared as described above. Each treatment group consisted of four to five animals. The doses were the same as described above, except each treatment was only given twice a week. Animals were not responding to 50 mg/kg twice weekly, therefore, after 3 weeks of treatment the dose was escalated to 75 mg/kg for the remaining 2 weeks. Doses for the vehicle and oral Olaparib were increased to match NanoOlaparib. An additional group of animals was treated with NanoOlaparib twice weekly at a dose of 100 mg/kg for 6 weeks. The summary of the various treatment regimens and control groups is provided in . Tumor burden was monitored via bioluminescence imaging at the beginning of each week. Gross toxicity was monitored via bodyweight measurements and by observing the physical and behavioral condition of the mice. At animal sacrifice, tumors were harvested and weighed to determine the total tumor weight.
Statistical analysis
All values are shown as mean ± SD (in vitro) or mean ± SEM (in vivo). Normality of all data tested with Shapiro–Wilk test with P<0.05 considered not to be a normal distribution. Statistical significance of data following a normal distribution was assessed via Student’s t-tests or one-way ANOVA followed by Tukey’s test for multiple comparisons at α=0.05. Statistical significance of data that did not follow a normal distribution was assessed via Mann–Whitney U tests or Kruskal–Wallis one-way ANOVA followed by Dunn’s correction for multiple comparisons at α=0.05. Survival significance was assessed via log-rank test with Bonferroni correction for multiple comparisons. All statistical testing computed with Prism 7.
Results
NanoOlaparib characterization
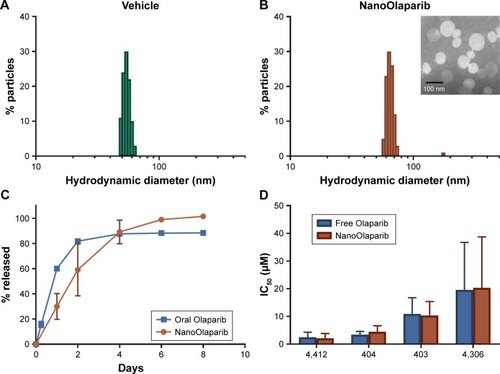
The size of NanoOlaparib was estimated at 72.8±5.8 nm with a zeta potential of −30.5±9.0 mV. The formulation consists predominantly of ~70 nm particles with a small population of larger particles resulting in a polydispersity index of 0.218, while the vehicle is slightly smaller at ~60 nm (). The formulation was concentrated via ultrafiltration to consist of 5.0 mg Olaparib/mL of formulation. Under static release conditions, the nanoformulation displayed a first-order drug release profile with 100% of the drug being released over the course of 8 days (). The oral Olaparib formulation released about 90% of the drug within 4 days.
Figure 1 In vitro characterization of NanoOlaparib reveals that the sustained release and efficacy profiles are comparable to those of free Olaparib.
Notes: Size of (A) empty nanoparticles as measured by dynamic light scattering and (B) NanoOlaparib, (inset) transmission electron micrograph of NanoOlaparib stained with 1.5% phospotungstic acid depicts particles with an approximate size of 70 nm. (C) The release of NanoOlaparib at 37°C indicates that 100% of NanoOlaparib is released within 8 days. (D) IC50 values comparing the treatment efficacy of NanoOlaparib vs free Olaparib.
In vitro efficacy
Murine cell lines developed from two different genetically engineered mouse models were used to test the sensitivity to Olaparib and NanoOlaparib over four doubling cycles. Doubling times for all tested cell lines ranged from 18 to 24 hours (data not shown). All the tested lines were sensitive to Olaparib and NanoOlaparib with no significant differences in their IC50 values (). The IC50 values ranged from 2.15±0.99 to 20.31±10.63 µM ().
Table 2 Olaparib and NanoOlaparib IC50 values for cell lines derived from genetically engineered mouse models of ovarian cancer
Daily i.p. administration of NanoOlaparib in vivo
Bioluminescence decreased in the animals treated with NanoOlaparib, cisplatin, and the combination of the two drugs. By contrast, the bioluminescence signals increased for Olaparib-treated and control groups (). After 1 week of treatment, the NanoOlaparib, cisplatin, and combination treated groups all showed reductions in bioluminescence that were statistically different from no-treatment controls (**P<0.01). There was variability in the response to oral Olaparib but, interestingly, this variability was not apparent in animals treated with the NanoOlaparib formulation (). The empty nanoparticles also appeared to inhibit tumor growth but only until week 3, with tumors growing unchecked after that time point. The inhibition of bioluminescence in the empty nanoparticle group resulted in a statistical difference compared to the no-treatment control group at day 7 (*P<0.05, ). NanoOlaparib alone and NanoOlaparib + cisplatin decreased tumor growth similarly or better than cisplatin alone within the first 2 weeks of treatment; however, by week 3 some of the NanoOlaparib-treated tumors began to grow again while the cisplatin-treated tumors showed little regrowth ().
Figure 2 NanoOlaparib administered daily as a monotherapy or in combination with twice weekly cisplatin in a Brca2−/−, Tp53−/−, Pten−/− intraperitoneal spread model reduces tumor growth similar to cisplatin alone.
Notes: (A) Bioluminescence measurements show the disease progression over 3 weeks of treatment. Statistically significant decreases in luminescence were seen after 1 week of treatment with vehicle (*P<0.05) and NanoOlaparib, cisplatin and the combination (**P<0.01) compared to no-treatment controls tested with ordinary one-way ANOVA followed by Tukey’s test at α=0.05. (B) Bioluminescence images of animals in the oral Olaparib and NanoOlaparib groups over the first 2 weeks of treatment. (C) Bodyweight measurements are indicative of gross toxicity. (D) The Kaplan–Meier survival curves demonstrate both treatment efficacy and premature death due to treatment-related toxicity, P<0.005 between no-treatment control and each drug treated group based on log-rank test. All data followed a normal distribution as tested by the Shapiro–Wilk test.
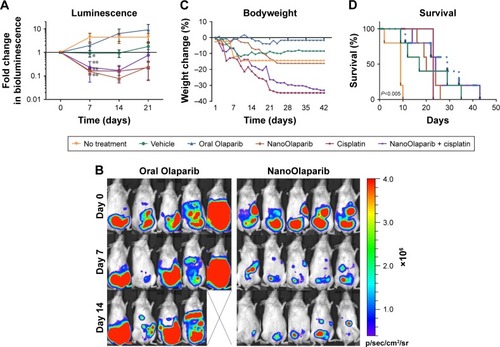
Drug toxicity was assessed both by bodyweight measurements and observing the physical and behavioral condition of the treated mice. Bodyweight profiles indicated neither PARP inhibitor monotherapy was toxic. However, observations related to the general physical condition of the mice indicated potential underlying toxicity in the nanoformulation-treated groups (vehicle and NanoOlaparib, ). Two of the vehicle-treated mice died within the first 2 weeks of treatment and two mice in each of the NanoOlaparib and NanoOlaparib + cisplatin groups died prior to week 4. The cisplatin-treated mice lost more than 20% of their bodyweight within 3 weeks of treatment and had to be sacrificed at the start of week 4 (). All drug treatments significantly extended the lifespan of treated animals compared to the no-treatment control group based on the log-rank test (**P<0.005, ). However, treatment-related toxicities were present in most groups, resulting in premature death; therefore, there were no significant differences in survival between various treatments.
Biodistribution profiles
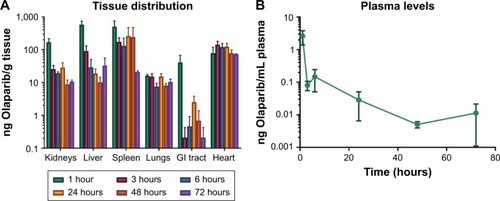
Although there was substantial variability in drug levels between animals, mass spectrometry profiles indicated that the average concentration of Olaparib detected 1 hour after i.p. injection was mainly found in kidneys, liver, and spleen (). High levels of Olaparib were detected in the plasma at the 1-hour time point but after 3 hours most of the drug was absent in plasma and only minor levels were detected at the other time points (). Most importantly, while average drug levels decreased over time in most organs, this was not the case in the heart, which may have contributed to the treatment-related toxicity and premature death. Drug was still present in tissues after 72 hours, with the highest concentrations found in the spleen and heart.
Figure 3 Drug distribution in tissues (A) and plasma (B) for Olaparib at 1, 3, 6, 24, 48, and 72 hours following intraperitoneal injection of NanoOlaparib.
Note: All data followed a normal distribution as tested by the Shapiro–Wilk test.
Abbreviation: GI, gastrointestinal.
Olaparib levels in the same tissues were compared 72 hours after a single dose, and 72 hours after animals had received treatment twice weekly for 4 weeks. In all cases, there appeared to be more accumulation in tissues and plasma of animals that had received multiple treatments (). Two of three mice showed a ~2-fold increase in drug levels in kidneys and liver as compared to a single-dose treatment while one animal had a much greater amount of drug accumulation. In the spleen, however, Olaparib levels were two orders of magnitude higher for all animals. Olaparib was detected in the plasma of two of three mice after repeated injections compared with little to no presence after a single injection (). The drug levels in the spleen, lungs, and plasma followed a normal distribution and Student’s t-tests (P≤0.05) revealed no statistical difference in accumulation in these tissues or plasma 72 hours after a single injection vs biweekly injections for 4 weeks. The drug levels in the kidneys, liver, GI tract, and heart did not follow a normal distribution, and Mann–Whitney U tests (P≤0.05) revealed no statistical difference in accumulation in these tissues 72 hours after a single injection vs biweekly injections for 4 weeks.
Figure 4 Comparison of Olaparib accumulation in various tissues at 72 hours after (A) a single 50 mg/kg dose of NanoOlaparib or (B) 50 mg/kg administered twice weekly for 4 weeks in healthy mice. (C) Comparison of Olaparib in plasma at 72 hours following a single injection or after the final dose of twice weekly injections for 4 weeks. (D) Bodyweight changes in healthy mice treated with 50 mg/kg NanoOlaparib (twice weekly for 4 weeks) or a volume equivalent of empty nanoparticles indicated no gross toxicity.
Notes: The drug levels in the spleen, lungs, and plasma followed a normal distribution while drug levels in the kidneys, liver, GI tract, and heart did not follow a normal distribution. Student’s t-tests (P≤0.05) and Mann–Whitney U tests (P≤0.05), revealed no statistical significance.
Abbreviation: GI, gastrointestinal.
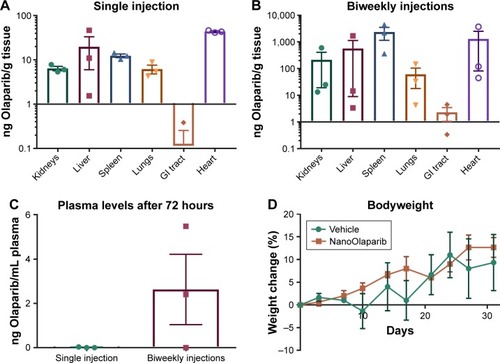
The average bodyweight of animals treated twice a week for 4 weeks with either NanoOlaparib or vehicle fluctuated as expected, and no significant weight loss was observed to suggest toxicity (). Animals did not experience any adverse effects of the treatment as they had within the first 3 weeks of daily NanoOlaparib administration.
Twice weekly i.p. NanoOlaparib administration in vivo
Tumors treated with 50 mg/kg Olaparib or NanoOlaparib twice weekly did not show decreases in bioluminescence signal compared to control groups through the first 3 weeks of treatment. In addition, the dose escalation was unable to halt tumor growth. Animals receiving 100 mg/kg NanoOlaparib twice a week showed a variable response, with the bioluminescence doubling during the first 2 weeks of treatment. The fold change increased similar to the other groups beyond week 3 (). Based on luminescence, the cisplatin-treated group showed a similar disease progression on average as the PARP inhibitor-treated groups, with no statistical significance seen between any groups (one-way ANOVA). No toxicity was observed either by measuring bodyweight changes or monitoring signs of distress in the treated animals ().
Figure 5 NanoOlaparib administered twice weekly at 50 or 100 mg/kg as a monotherapy or in combination with cisplatin in a Brca2−/−, Tp53−/−, Pten−/− intraperitoneal spread model showed no efficacy after 5 weeks.
Notes: (A) Bioluminescence measurements show the disease progression over 5 weeks of treatment. (B) Bodyweight measurements indicate no apparent gross toxicity in any of the treatment groups. (C) Final tumor weights serve as a quantitative measure of disease burden at the end of the study. The data did not follow a normal distribution therefore significance was tested with Kruskal–Wallis one-way ANOVA followed by Dunn’s test at α=0.05. Statistically significant decreases in tumor weights were seen in both cisplatin-treated tumors vs NanoOlaparib and oral Olaparib + cisplatin compared to NanoOlaparib, respectively (*P<0.05).
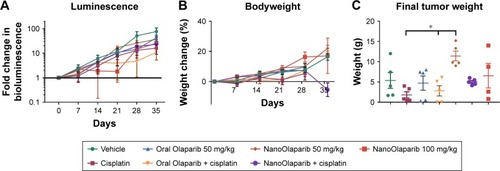
The final tumor weight was used as a more accurate quantitative measure of treatment efficacy at the endpoint and showcased the response variability in most groups (). All animals showed substantial tumor burden and the only groups with little to no visible disease were oral Olaparib, cisplatin, and the combination of the two drugs. These data did not follow a normal distribution, therefore, significance was assessed via Kruskal–Wallis one-way ANOVA followed by Dunn’s correction to compare each group to every other group. Although some groups did have animals that responded, based on final tumor weight assessments, there was no statistically significant difference between the treatment groups and vehicle; however, there were statistically significant decreases in final tumor burden seen between cisplatin-treated tumors vs 50 mg/kg NanoOlaparib and oral Olaparib + cisplatin compared to 50 mg/kg NanoOlaparib, respectively (*P<0.05).
Discussion
PARP inhibitors offer an effective means for targeting cancer cells with defective DNA repair pathways while leaving healthy cells relatively unharmed. Currently, patients are required to take two tablets of Olaparib twice daily for sustained PARP inhibition and effective tumor treatment. Previously, we demonstrated the ability to formulate Olaparib in lipid nanoparticles and increase the efficacy of radiation therapy in a radiation resistant prostate cancer model. Treatment with NanoOlaparib prior to radiation exposure resulted in increased γ-H2AX foci formation in vitro compared to free Olaparib; consequently, in vivo NanoOlaparib treatment in combination with radiation resulted in 50% of treated animals being disease free at the end of the experiment.Citation29 In this study, we explored the utility of NanoOlaparib as an i.p. therapy for ovarian cancer, a disease that spreads in the peritoneal cavity during late stages. Dose–response profiles indicated that NanoOlaparib was just as effective in vitro as free Olaparib. All the tested cell lines have conditional homozygous PTEN deletion, and loss of PTEN has been shown to lead to spontaneous DSBs, chromosomal instability, and defects in HR.Citation35,Citation36 As the first FDA approved indication of Olaparib was for germline BRCA mutant ovarian cancer patients, the 404 tumor line was chosen to generate a clinically relevant model for in vivo testing.
Oral Olaparib and NanoOlaparib were administered daily to mimic the clinical dosing of the drug. NanoOlaparib appeared to provide better efficacy than oral Olaparib; however, there was significant toxicity for the nanoformulation when using a daily dosing regimen. As a result, some of the mice treated with either NanoOlaparib or vehicle died early, indicating that there was toxicity associated with the nanoformulation. We further observed that the subset of NanoOlaparib-treated mice which died throughout the course of treatment had substantial tumor reduction compared to baseline measurements, likely due to an overdosing of the drug. The sustained release profile (as shown in vitro) suggests that daily dosing of the drug would result in a buildup of Olaparib during sequential dosing. However, it was expected that under dynamic conditions and in a complex biological environment, the in vivo release profile of both the nano- and oral formulation would be faster than the in vitro kinetics profile due to diffusion limitations seen with the in vitro setup.Citation37 The NanoOlaparib-treated animals that did not suffer from treatment-related toxicity began to show slight tumor regrowth after 3 weeks of treatment. This is consistent with the oral Olaparib-treated group, in which animals that initially responded to treatment began to show regrowth after about 2 weeks of treatment. NanoOlaparib administered i.p. extended the length of time in which animals responded to the PARP inhibitor from 2 weeks (oral formulation) to 3 weeks. A variable response to oral Olaparib was observed; however, when delivered intraperitoneally through the nanoformulation the response variability decreased noticeably.
The significant toxicity observed with the daily dosing of NanoOlaparib and vehicle suggests that carrier-associated toxicity may be a factor at this dose. A preliminary biodistribution study was performed to investigate drug accumulation. One hour after i.p. injection of NanoOlaparib, the drug was detected in all tissues as well as in plasma. By 72 hours, the heart contained the highest levels of Olaparib. This study was limited in that the treated mice were healthy and thus tumor accumulation could not be assessed. We further observed that treatment with NanoOlaparib or vehicle twice weekly for 4 weeks did not result in any signs of distress or toxicity in treated animals. Seventy-two hours after the final treatment of the twice-weekly schedule, the detected levels of drug were higher in all tissues, but not statistically significant, as compared to the single-dose treatment. This lack of statistical significance is likely due to the high variability observed within groups and small sample size (n=3).
Treatments involving NanoOlaparib and empty nanoparticles administered twice weekly showed none of the toxicity indicators that the daily treatments had previously revealed. However, modifying the dosing schedule resulted in no therapeutic efficacy for either the NanoOlaparib monotherapy or in combination with cisplatin. Both doses of 50 and 100 mg/kg NanoOlaparib twice a week were not toxic but also not effective. However, statistically significant decreases in final tumor weights were seen in both cisplatin-treated tumors vs NanoOlaparib and oral Olaparib + cisplatin compared to NanoOlaparib, respectively (*P<0.05, ). It is widely known that PARP must be 90% inhibited to suppress DNA repair.Citation38,Citation39 The pharmacodynamics of Olaparib treatments indicate that tumor reduction will only occur when doses are higher than the IC50 value for more than 13 hours and the IC90 value for more than 6 hours.Citation40 Within the first hour after injection, Olaparib is present in both plasma and tissues indicating nanoparticles are entering systemic circulation.Citation41,Citation42 This suggests that the amount of drug at the tumor site does not meet the steady-state exposure required for tumor reduction because the sustained release is occurring while the particles are circulating systemically rather than in the peritoneal cavity. Previously, NanoOlaparib administered intravenously twice weekly sensitized animals with prostate cancer to a single dose of radiation between weeks 1 and 2, with 50% of the animals showing complete response.Citation29 As it is known that the EPR effect is heterogeneous in different tumors, perhaps the EPR effect was more prominent in the prostate model than in the disseminated peritoneal disease model.Citation43,Citation44
It is possible that to increase the efficacy of NanoOlaparib when administered i.p., its surface must be modified to retain the nanoformulation in the peritoneal cavity. It has been shown that i.p. administration of bioadhesive nanoparticles both prolongs the retention of particles and increases the survival of treated animals when compared to non-adhesive, “stealthy” particles.Citation45 The addition of ligands targeting overexpressed tumor markers is also a useful strategy for localizing nanoparticles to the disease site. Potential targeting ligands include those which target mucin 1 overexpression, folate receptors, or luteinizing hormone-releasing hormone receptors.Citation46–Citation48 Targeting moieties may also increase the uptake and retention of nanoparticles by tumor cells.
Conclusion
Our results indicate that NanoOlaparib, in its current nanoformulation, is not an effective i.p. treatment for ovarian cancer. Our data further suggest that an effective dose of Olaparib cannot be achieved at the tumor site without overdosing the animals due to nanoparticles having a sustained release profile and their ability to enter systemic circulation within an hour of administration. NanoOlaparib delivered intraperitoneally for this disseminated model may not have taken advantage of the EPR effect, as was seen in our previous work. Future avenues for increasing the effectiveness of NanoOlaparib as an i.p. therapy include modifying the surface to be more adhesive, such that the release of Olaparib would occur mostly within the peritoneal cavity, which would further result in an effective Olaparib dose to be maintained at the tumor site. Surface modification could also include targeting moieties as a strategy to increase nanoparticle uptake and retention within tumor cells prior to them entering the systemic circulation.
Acknowledgments
This work was supported by IGERT grant NSF-DGE-0965843 (SS) and DOD W81XWH-14-1-0092 (SS and DMD).
Disclosure
NanoOlaparib is part subject matter of intellectual property in a patent application assigned to Northeastern University. Srinivas Sridhar and Shifalika Tangutoori are inventors. The authors report no other conflicts of interest in this work.
References
- de MurciaJMNiedergangCTruccoCRequirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cellsProc Natl Acad Sci U S A19979414730373079207086
- WangZQStinglLMorrisonCPARP is important for genomic stability but dispensable in apoptosisGenes Dev19971118234723589308963
- FarmerHMccabeNLordCJTargeting the DNA repair defect in BRCA mutant cells as a therapeutic strategyNature2005434703591792115829967
- BryantHESchultzNThomasHDSpecific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymeraseNature2005434703591391715829966
- YapTASandhuSKCardenCPde BonoJSPoly(ADP-ribose) polymerase (PARP) inhibitors: exploiting a synthetic lethal strategy in the clinicCA Cancer J Clin2011611314921205831
- DantzerFde La RubiaGMénissier-de MurciaJHostomskyZde MurciaGSchreiberVBase excision repair is impaired in mammalian cells lacking poly(ADP-ribose) polymerase-1Biochemistry200039257559756910858306
- AshworthAA synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repairJ Clin Oncol200826223785379018591545
- MuraiJHuangSYDasBBTrapping of PARP1 and PARP2 by clinical PARP inhibitorsCancer Res201272215588559923118055
- MuraiJHuangSYRenaudAStereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparibMol Cancer Ther201413243344324356813
- ArmstrongDKBundyBWenzelLGynecologic Oncology GroupIntraperitoneal cisplatin and paclitaxel in ovarian cancerN Engl J Med20063541344316394300
- YapTACardenCPKayeSBBeyond chemotherapy: targeted therapies in ovarian cancerNat Rev Cancer20099316718119238149
- IhnenMZu EulenburgCKolarovaTTherapeutic potential of the poly(ADP-ribose) polymerase inhibitor rucaparib for the treatment of sporadic human ovarian cancerMol Cancer Ther20131261002101523729402
- LedermannJHarterPGourleyCOlaparib maintenance therapy in platinum-sensitive relapsed ovarian cancerN Engl J Med2012366151382139222452356
- LedermannJHarterPGourleyCOlaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trialLancet Oncol201415885286124882434
- DonawhoCKLuoYLuoYABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor modelsClin Cancer Res20071392728273717473206
- EversBDrostRSchutESelective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatinClin Cancer Res200814123916392518559613
- DhawanMSAggarwalRRBartelinkIEfficacy and hematologic toxicity of carboplatin and talazoparib combination therapy in BRCA mutated patientsJ Clin Oncol20163415_suppl255727185839
- LandrumLMBradyWEArmstrongDKA phase I trial of pegylated liposomal doxorubicin (PLD), carboplatin, bevacizumab, and veliparib (ABT-888) in recurrent, platinum-sensitive ovarian, primary peritoneal, and fallopian tube cancer: a Gynecologic Oncology Group studyGynecol Oncol201513722325643690
- RajanACarterCAKellyRJA phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumorsClin Cancer Res20121882344235122371451
- BalmañaJTungNMIsakoffSJPhase I trial of olaparib in combination with cisplatin for the treatment of patients with advanced breast, ovarian and other solid tumorsAnn Oncol201425817117824299961
- SamolJRansonMScottESafety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I studyInvest New Drugs20123041493150021590367
- LeeJ-MHaysJLAnnunziataCMPhase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analysesJ Natl Cancer Inst20141066111
- TuttARobsonMGarberJEOral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trialLancet2010376973723524420609467
- AudehMWCarmichaelJPensonRTOral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trialLancet2010376973724525120609468
- De JongWHBormPJDrug delivery and nanoparticles: applications and hazardsInt J Nanomedicine20083213314918686775
- MaedaHThe enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targetingAdv Enzyme Regul20014118920711384745
- ArmstrongDKBundyBWenzelLIntraperitoneal cisplatin and paclitaxel in ovarian cancerN Engl J Med20063541344316394300
- MateoJMorenoVGuptaAAn adaptive study to determine the optimal dose of the tablet formulation of the PARP inhibitor olaparibTarget Oncol201611340141527169564
- van de VenALTangutooriSBaldwinPNanoformulation of olaparib amplifies PARP inhibition and sensitizes PTEN/TP53-deficient prostate cancer to radiationMol Cancer Ther20171671279128928500233
- BanghamADStandishMMWatkinsJCDiffusion of univalent ions across the lamellae of swollen phospholipidsJ Mol Biol19651312382585859039
- PeretsRWyantGAMutoKWTransformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca; Tp53; Pten modelsCancer Cell201324675176524332043
- DinulescuDMInceTAQuadeBJShaferSACrowleyDJacksTRole of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancerNat Med2005111637015619626
- BaldwinPTangutooriSSridharSGeneration of Dose -Response Curves and Improved IC50s for PARP Inhibitor NanoformulationsMethods Mol Biol2017153033734228150212
- RottenbergSJaspersJEKersbergenAHigh sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugsProc Natl Acad Sci U S A200810544170791708418971340
- ShenWHBalajeeASWangJEssential role for nuclear PTEN in maintaining chromosomal integrityCell2007128115717017218262
- Mendes-PereiraAMMartinSABroughRSynthetic lethal targeting of PTEN mutant cells with PARP inhibitorsEMBO Mol Med200916–731532220049735
- ModiSAndersonBDDetermination of drug release kinetics from nanoparticles: overcoming pitfalls of the dynamic dialysis methodMol Pharm20131083076308923758289
- RicksTKChiuHJIsonGSuccesses and challenges of PARP inhibitors in cancer therapyFront Oncol2015522226528434
- FarzanehFZalinRBrillDShallSDNA strand breaks and ADP-ribosyl transferase activation during cell differentiationNature198230058903623666292727
- Drugs@FDA, FDA Approved Drug ProductsLYNPARZA. Pharmacology Review. US Food and Drug Administration, Center for Drug Evaluation and Research Web site Available form: http://www.access-data.fda.gov/scripts/cder/drugsatfdaAccessed November 1, 2018
- BajajGYeoYDrug delivery systems for intraperitoneal therapyPharm Res201027573573820198409
- KohaneDSTseJYYeoYPaderaRShubinaMLangerRBiodegradable polymeric microspheres and nanospheres for drug delivery in the peritoneumJ Biomed Mater Res A200677235136116425240
- MaedaHToward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneityAdv Drug Deliv Rev2015913625579058
- PrabhakarUMaedaHJainRKChallenges and key considerations of the enhanced permeability and retention effect for nanomedi-cine drug delivery in oncologyCancer Res20137382412241723423979
- DengYYangFCoccoEImproved i.p. drug delivery with bioadhesive nanoparticlesProc Natl Acad Sci U S A201611341114531145827663731
- WangLMaJLiuFExpression of MUC1 in primary and metastatic human epithelial ovarian cancer and its therapeutic significanceGynecol Oncol2007105369570217368732
- KalliKRObergALKeeneyGLFolate receptor alpha as a tumor target in epithelial ovarian cancerGynecol Oncol2008108361962618222534
- QinYSongQGZhangZROvarian tumor targeting of docetaxel-loaded liposomes mediated by luteinizing hormone-releasing hormone analoguesArzneimittelforschung20115810529534