 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Multiscale computational modeling of drug delivery systems (DDS) is poised to provide predictive capabilities for the rational design of targeted drug delivery systems, including multi-functional nanoparticles. Realistic, mechanistic models can provide a framework for understanding the fundamental physico-chemical interactions between drug, delivery system, and patient. Multiscale computational modeling, however, is in its infancy even for conventional drug delivery. The wide range of emerging nanotechnology systems for targeted delivery further increases the need for reliable in silico predictions. This review will present existing computational approaches at different scales in the design of traditional oral drug delivery systems. Subsequently, a multiscale framework for integrating continuum, stochastic, and computational chemistry models will be proposed and a case study will be presented for conventional DDS. The extension of this framework to emerging nanotechnology delivery systems will be discussed along with future directions. While oral delivery is the focus of the review, the outlined computational approaches can be applied to other drug delivery systems as well.
Introduction
Nanotechnology-based drug delivery systems (DDS) are a primary component of nanomedicine’s potential to improve human health. Over 70% bionanotechnology scientific articles focus on DDS (CitationWebster 2006). Multifunctional nanoparticle drug carriers and nanostructured responsive drug matrices, in particular, have gained increasing attention (CitationKawasaki and Player 2005). Whether through nanotechnology or conventional technologies, advancements in drug delivery aim to improve patient treatment by enabling the administration of complex new drugs, improving the bio-availability of existing drugs, and providing spatial and temporal targeting of drugs in order to dramatically reduce side effects and increase effectiveness. Through these achievements, patients and physicians could benefit from personalized prescriptions, increased ease of administration, increased patient compliance, and reduced dosage frequency.
This envisioned future of medicine demands quantitative, mechanistic models as tools to predict the complex interactions between drug, drug delivery system, and patient physiology. These models, by definition, must span broad scales of space and time. The purpose of this review is to present a multiscale framework for integrating existing computational approaches at the different scales of the drug delivery problem for both conventional and nanoparticles-based drug delivery. As oral delivery remains dominant over other possible routes (intravenous, oral, subcutaneous, nasal, vaginal, rectal or ocular), this review will focus on oral DDS. For this reason, interaction between the drug and the gastrointestinal (GI) tract becomes common to all problems considered and will receive particular focus. While the emphasis is on oral delivery, the multiscale computational approaches and framework outlined could be applied to other delivery modes as well.
Oral drug delivery systems
Defining the problem
Assessment of past drug development activities has estimated that ~39% of failures could be attributed to poor pharmacokinetics and ~11% to toxicity (CitationKennedy 1997; Citationvan de Waterbeemd and Gifford 2003). For this reason, the in silico prediction of the absorption, distribution, metabolism, excretion and toxicity (ADMET) of potential new drugs in early stages of development has become a critical area of research. The success of high throughput screening, proteomics, genomics, combinatorial chemistry and biotechnology to create increasing numbers of novel therapeutic candidates further amplifies the need for modeling. Despite the potential, the contribution of in silico computational approaches to the design of conventional drug delivery systems has so far been limited. Some commercial software such as Gastroplus™ (CitationAgoram et al 2002) and iDEA™ (CitationGrass 1997) for ADMET prediction and others for toxicity prediction exist (see Green (CitationGreene 2002) for a more complete list of commercial software). A good number of the models used in these tools are mostly rule-based limiting their application. In addition, the multi-functionality, complexity, and emergent properties of nanoparticle-based systems create additional and unique challenges.
In their review (Citationvan de Waterbeemd and Gifford 2003), as well as others, assert a need for a new generation of mechanism-based models to provide sufficient knowledge to successfully predict and simulate ADMET and drug action. The key mechanisms that govern the drug system occur across a wide range of scales in both space and time (). DDS design requires the quantification and prediction of the nano and microscale molecular interactions, microscale cellular mechanisms and macroscopic physiological effects in the targeting, entry and delivery processes at multiple scales. Each of these processes may be best modeled by different mathematical strategies. The grand challenge is to seamlessly integrate different model types across wide scales of space and time, and even allow multiple scales to be simulated concurrently.
Figure 1 The drug delivery process as is spans across multiple spatial scales.
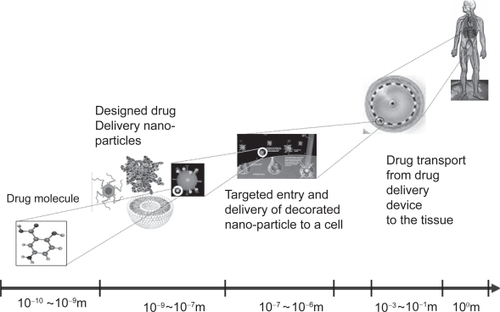
Defining the drug delivery system
Both conventional and nanoparticle DDS aim to target specific cellular/tissue/organ sites, control the delivery of the active component at the desired rate, and to provide solutions to important problems of drug solubility, protection, side effects, toxicity, and efficacy. For purposes of this paper, the term ‘conventional’ or ‘traditional’ DDS includes both immediate and controlled release systems achieved by incorporating drugs within bulk materials such as polymers or by altering the chemistry of the drug for example by prodrug formation. Examples of nanoparticles for DDS or medical diagnostics include but are not limited to dendrimers (CitationKitchens et al 2005), shell cross-linked nanoparticles (CitationSun et al 2005; CitationThurmond et al 1996), functionalized polymer nanoparticles (CitationFarokhzad et al 2006), inorganic nanocrystals (CitationAkerman et al 2002), and RNA nanoparticles (CitationKhaled et al 2005).
The power of nanoparticles for oral drug delivery derives from two major advantages: an ability to form multi-functional particles on size scale relevant to cells and an ability to design unique and emergent properties compared to bulk materials. Multi-functionality refers the integration of multiple capabilities or properties onto a single particle. These functions may include targeting by recognition (eg, cell type specific ligands-receptor interaction) (CitationPopielarski et al 2005), targeting by external intervention (eg, photosensitization) (CitationMcCarthy et al 2005), properties for handling and sorting (eg, magnetic or chemical separation) (CitationDresco et al 1999), imaging and identification (optical properties, quantum dots etc) (CitationLewis et al 2006), increased bioavailability, solubility, or circulation time of drug (CitationGref et al 1994; CitationManjunath and Venkateswarlu 2005), and selective delivery of drug payload. Multi-functionality can be achieved by the conjugation at the surface, layering (CitationProw et al 2005), or by the design and synthesis of the nanoparticle core itself. Nanoparticles from this point of view are a composite of molecules and materials each having at least one advantageous property or function. The nanoscale size of the particles also means that the resulting novel properties of materials at this scale can be utilized. Quantum dots, gold core/shell particles and superparamagnetic particles are all good examples.
Establishing a framework for multiscale modeling of DDS
Complete models need to consider key physical, chemical, and the often omitted, biological processes at the spectrum of scales in temporal and spatial dimensions. The framework should address the processes and interactions involved during delivery process at different scales using three fundamental elements:
Macroscale changes of the drug delivery vehicle. As the delivery vehicle transits the gastrointestinal (GI) tract or the systemic circulation, change and deformation, will impact drug availability. For conventional delivery and for nanoparticles delivered in a bulk carrier, this is a macroscale problem. Prediction of these changes requires continuum based modeling (eg, represented as differential equations), which address the different processes such as diffusion, dissolution, degradation and swelling. The application of a moving boundary and free boundary approaches are essential.
Nano and microscale molecular interactions between drug, vehicle, GI tract, and target site. Along the path of the drug, particle and carrier, many molecular processes and interactions are critical. These include the specific interaction of the delivery polymer/system with the drug, enzymatic action on the drug/particle/carrier in the GI tract, drug/particle/carrier interactions with the epithelial interface at the absorption site, receptor/transporter/membrane at the target site either in the GI tract or other organs after absorption at the nano and micro levels. This requires computational chemistry approaches such as molecular dynamics (MD) and mesoscale modeling (modeling at length and time scales between MD and continuum mechanics) for the problems beyond reach of MD.
Biological and delivery system variability at all scales. Prediction of the effect of variability that arises from biological sources such as inter/intra subject variability or inherent variability in the delivery system such as polymer coating non-uniformity both at macro and micro/ nano levels. These issues can be addressed by employing stochastic approaches.
Linking and modeling across the scales
In order to simulate the entire system from drug to patient, the three elements presented above must be integrated and linked. depicts a multiscale computational framework for model-based simulation of the entire system from drug design to patient physical and pathological conditions. In this framework, the system consists of three components: the drug, the delivery system and the patient. Information from these components is gathered and compiled by means of mechanistic approaches into a multiscale mathematical model. The challenge of multiscale modeling is the transition from one space-time scale to another. With respect to spatial scale this can be from nano to micro or micro to macro and with respect to time from picoseconds (eg, such as in MD) to seconds or minutes, for instance. For mechanistic models, the nature of coupling of the physical, chemical and biological processes at each scale determines how the computational methods should be linked.
Figure 2 A framework for multiscale modeling of entire drug delivery systems using information from drug and vehicle properties, disease pathology, and patient characteristics.
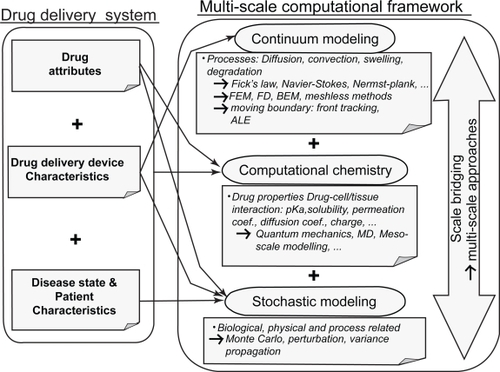
Of particular interest to the interaction of the delivery system at the biological interface is discrete-to-continuum coupling including two main categories: information-passing (sequential, serial, hierarchical, parameter passing) and concurrent (embedded, integrated, hand-shake) (CitationFish and Chen 2004a).
In the information-passing approach, the information gained in the lower scale is transferred on to the higher scale. For example, molecular dynamics may be used to predict physical parameters, such as diffusivity (eg, of a drug molecule in the carrier material or across a lipid bi-layer) or solubility (eg, of the carrier or drug at the biological interface as a function of physiological pH) that may be employed to compute higher level or scale continuum transport models. It is noteworthy that this approach is valid as long as the information obtained from the lower space-time scale can be summarized into a finite set of parameters and represent the rigorous reduction of the enormous degrees of freedom of the lower length (space or time) of scale. In the context of discrete-to-continuum coupling, the Generalized Mathematical Homogenization (GMH) theory (CitationChen and Fish 2006) can be mentioned among the information passing bridging techniques. The GMH constructs an equivalent continuum description directly from discrete (such as MD) equations. In general the mathematical homogenization theory makes the assumption that the fine scale is locally periodic and is composed of four steps (CitationChen and Fish 2006): (i) solving a sequence (various orders) of unit cell problems; (ii) computing effective coarse-scale properties; (iii) solving the coarse-scale problem; and (iv) localizing (or post-processing) the fine scale data.
In the concurrent or embedded approach, the multiple scales are resolved simultaneously in different portions of the domain of interest. More theoretical and computational effort is required in this approach than the former. The information between the different hierarchical models is communicated via the domain decomposition methods where the system response is separated into local (discrete) and global (continuum) effects. Among the concurrent bridging techniques the space-time multilevel method (CitationFish and Chen 2004b) can be mentioned. In the context of drug delivery this approach can be applied, for example, to the prediction of biodegradable polymer delivery devices where the polymer is degraded until a critical molecular weight is reached before solvation begins. The bond breaking at the solvent-polymer interface can be described by quantum mechanical model of bonding while the rest of the domain is described with empirical potential.
Given that mechanisms at each level in the drug delivery design process form a vast and complex network of dynamically interacting heterogeneous components, the multiscale modeling can involve other multiscale approaches such as continuum-to-continuum coupling between different time scales.
The next sections will introduce and summarize three basic modeling approaches and their applicability to the different scales relevant to oral drug delivery, including examples of nanoparticle processes. This review is by no means exhaustive, but rather representative of the variety of approaches at each scale, which can be integrated into the multiscale framework. A case study will then provide a specific example of this integration.
Continuum approaches at the micro and macro scales
Polymer systems for controlled or triggered release
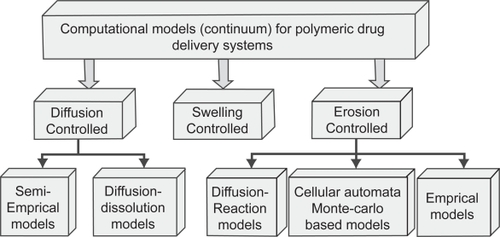
A wide range of polymers have demonstrated controlled or triggered release using different rate controlling mechanisms such as diffusion (CitationLanger and Peppas 1983; CitationCosta et al 2001), swelling (CitationSiepmann and Peppas 2001), erosion (CitationSiepmann and Gopferich 2001), ion exchange (Jeong et al 2006) or any combination of these in response to environmental stimuli. As a result a large number of mathematical, typically continuum, models and theories have been developed to describe the mass transport mechanisms and chemical processes that occur within polymer systems. (CitationKanjickal and Lopina 2004) have compiled an excellent review on the continuum based computational approaches for polymeric drug delivery systems. The classification of the models is summarized in .
Figure 3 Classification of computational approaches of polymeric drug delivery systems.
In general, the mechanistic models can be represented by a single equation incorporating reaction-transport, ion exchange and a source term with pertinent different initial and boundary conditions.
This model contains a large number of parameters and non-linearities associated with the different chemical and physical processes. Moreover, as the dosage form transits in the GI tract its physical geometry continually deforms due to swelling and erosion, which affect the drug release kinetics. These changes create a moving boundary problem, as the change in geometry should be accounted for as the simulation progresses. These factors generally pose difficulty in solving the system of equations using analytical techniques. For such detailed models, one turns to numerical techniques such as Finite element method (FEM) (CitationReddy 2005), Finite volume method (FVM) (CitationLeVeque 2002), Finite difference, boundary element methods (CitationKane 1993) and recently meshless methods (CitationAtluri and Shen 2002). Of these methods, FEM is the preferred due to its flexibility in handling irregular geometric shapes and boundary conditions. In addition, general purpose computer programs can be developed easily to analyse various kinds of problems.
In FEM, a geometrical model of the delivery system is discretized into a finite number of elements, interconnected by a finite number of nodes. The continuous concentration fields in the domain is approximated by a polynomial that interpolate in the nodal values. Applying approximation techniques such as the Galerkin weighted residual method a system of first order differential equations is obtained (CitationSegerlind 1984).
The classical problems in partial differential equations involve solving an equation within a fixed spatial region. When attempting to model the changing and deforming dosage form as it transits the GI tract, however, the domain will also vary and must be determined. Such problems, for which the solution of a partial differential equation is sought in a region which is itself varying in an unknown manner, are referred to as moving boundary problems. (CitationLi et al 2005) classify numerical solutions of moving boundary problems into two categories, moving grid methods and the fixed grid methods. Each method has its own advantages and disadvantages. Methods in the context of fixed grid approach, such as the front-tracking (CitationTryggvason and Unverdi 1990, Citation1992), surface tracking (CitationHirt and Nichols 1981; CitationGueyffier et al 1999; CitationScardovelli and Zaleski 1999), and volume tracking (the volume of fluid (VOF) method) (CitationPopinet and Zaleski 1999; CitationRenardy and Renardy 2002) methods are capable of simulating very complex interface motion. New algorithms have also been developed to improve the accuracy in the treatment of boundary conditions (CitationRyskin and Leal 1984; CitationMaury 1999; CitationFast and Shelley 2004). Moving grid methods employ a so-called boundaryfitted grid system (CitationDonea et al 2004; CitationKnobbe 2004; CitationLi et al 2005; CitationNithiarasu 2005). This method has the tremendous advantage that the boundary condition is treated neatly and resolved accurately, because the moving boundary coincides with one line of the numerical grid.
One example from the latter category is the Arbitrary Lagrangian-eulerian approach. This boundary tracking method attempts to secure the best features of the Lagrangian (where the computational grid follows the material movement, thus unable to follow large distortions) and Eulerian (in which the computational mesh is fixed and the continuum moves with respect to the grid; though allows large distortions in the continuum but at the expense of precise interface definition) approaches while minimizing their respective drawbacks. This allows the spatial configuration the material configuration move and deform independently over a fixed referential configuration which gives this approach the ability to follow large deformations without recourse to frequent remeshing operations reducing the computational cost substantially. For details of the finite element analysis – Arbitrary lagrangian-eulerian method and implementation, the reader is referred to (CitationDonea et al 2004; CitationKnobbe 2004; CitationLi et al 2005; CitationNithiarasu 2005).
Kinetic and biophysical models of nanoparticle transport
Experimental results suggest that endocytosis is a primary transport mechanism for particle uptake (CitationRussell-Jones et al 1999; CitationKenneth et al 2005; CitationKitchens et al 2005,). As a result, endocytotic uptake has been a focus of several continuum models. These models can generally be classified as kinetic models and biophysical models. The kinetic models describe the internalization of particles based on classic kinetic theory of biochemical binding and reaction, while biophysical models attempt to describe the biophysical aspects of uptake and transport.
Kinetic models with varying degree of details were proposed from early 80’s for bare and surface functionalized nanoparticles (CitationWiley and Cunningham 1981, Citation1982; CitationZigmond et al 1982; CitationGex-Fabry and DeLisi 1984; CitationShimizu and Kawashima 1989; CitationLauffenburger and Linderman 1993; CitationNunes-Correia et al 1999; CitationEliaz et al 2004). A standard kinetic model of ligand-induced endocytosis, such as that by Lauffenburger and Linderman, (CitationLauffenburger and Linderman 1993) can be applied to nanoparticle uptake.
The primary limitation of kinetic models used to describe processes at the cellular level is the number of parameters that need to be estimated simultaneously from limited experimental data (quantitative data for parameter estimation) and a lack of time resolved data for model validation. The Lauffenburger and Linderman model, for example, contains seven kinetic constants to be determined experimentally. A need to enhance the information content of the experiments through optimal design of experiments using statistical techniques (CitationMunack 1989; CitationNahor et al 2001) or many experimental permutations is therefore, needed. For instance, Tzafriri and coworkers (CitationTzafriri et al 2004) attempted to estimate the involved parameters and analyze the base model (CitationLauffenburger and Linderman 1993) by designing three different experimental protocols.
Fewer cell mechanics models of nanoparticle uptake exist. Gao and coworkers (CitationGao et al 2005) modeled the uptake of cylindrical and spherical nanoparticles to simulate receptor mediated endocytosis by adopting a mathematical framework developed by Freund and Lin (CitationFreund and Lin 2004). The Gao model considers a flat cell membrane containing diffusive mobile receptors that wrap around cylindrical or spherical nanoparticles presenting compatible ligands. The ligands on the particle are assumed immobile and uniformly distributed whereas the receptors are mobile and undergo rapid diffusion in the plane of the cell membrane (). As the receptors of the cell membrane diffuse to the wrapping site (adhesion front), the binding of the receptors around the particle lowers the free energy of the interaction.
Figure 4 An illustration of receptor mediated endocytosis process of particle uptake as modeled by Gao and co-workers (CitationGao et al 2005) (a) Ligands on the particle bind to receptors on initially uniformly distributed receptors on the membrane causing diffusive movement of the mobile receptors (b) The receptor density distribution, ξ(s,t), in the membrane becomes nonuniform upon ligand-receptor binding; the receptor density is depleted in the near vicinity of the binding area and induces diffusion of receptors toward the binding site. 2*a(t) is the contact area between the particle and the membrane. CitationGao H, Shi W, Freund LB. 2005. Mechanics of receptor-mediated endocytosis. PNAS 102: 9469–74. Copyright © 2005 National Academy of Sciences, USA.
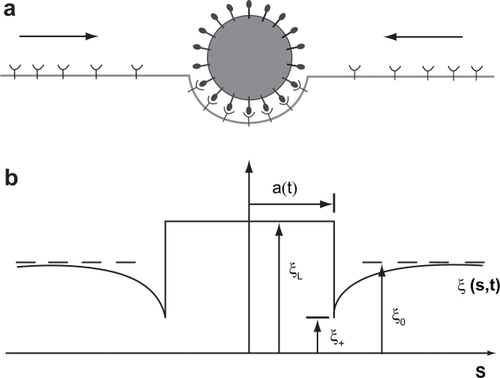
Considering the global conservation of the receptors, their diffusive flux was treated as a diffusion problem in a moving front (Stephan problem). A simplified free energy function was employed to account for the energy fluctuations as the particle enters or leaves the host cell. The model predicted a maximum, optimum and minimum particle sizes for receptor-mediated endocytosis, which was in agreement with experimental observations. Below the minimum radius the wrapping causes an increase in free energy and cannot proceed. This minimum for a spherical particle can be derived as EquationEquation 5(5) :
Although this model is still greatly simplified relative to the real system, the mechanistic approach provides a solid theoretical framework upon which a true engineering design approach to particle based targeted cellular delivery can be applied.
Computational chemistry approaches at the micro and nano scales
Methods for conventional DDS
The characterization of the different interactions such as drug-carrier, carrier-medium (biological) and drug-medium are a critical consideration and are often expressed as model parameters at the higher space-time scales. Molecular modeling and computational chemistry provide a number of tools including quantum mechanical ab initio methods, molecular dynamics, free energy perturbation and docking to quantify these interactions (CitationNeumann et al 2004). Due to the extreme complexity of the interactions, however, the relationship between chemical and physical properties is poorly described.
The development of empirical formulae, commonly referred to as quantitative structure-property/activity relationships (QSPR/QSAR), is an attractive approach for drug and drug delivery design (CitationKarelson et al 1999). These relationships are derived from empirical correlation of physico-chemical properties of the drug or carrier molecules such as molecular descriptors, lipophilicity, electronic properties (eg, steric properties molar refractivity), quantum chemical or structural data. These properties can be divided into several categories such as constitutional, topological, geometric, electrostatic, quantum-chemical, thermodynamic and salvation descriptors, depending on the compound and property and activity to predict. The origin and definitions of these descriptor types and others is extensively described in (CitationKatritzky et al 1994, Citation1996, Citation1997). Application of QSAR/ QSPR range from prediction of drug pharmacologic activity (CitationGozalbes et al 2002), drug biological activity (CitationKarelson et al 1996; CitationKarelson 2000;), drug solubility (CitationRytting et al 2004), to diffusion coefficients for polymers (CitationClement et al 2004).
The use of empirical molecular descriptors that rely on experimental data is limited by the availability of the necessary experimental data and can result in complicated artificial parameters that are difficult to interpret. (CitationKubinyi 2004) argues that prediction by using QSAR models from empirical molecular descriptors remains uncertain. An alternative is the use of molecular descriptors derived using only the information encoded in the chemical structure of the compound. (CitationKarelson et al 1999) outline three steps for the derivation of the theoretical molecular descriptors from the chemical structure of a compound with the aid of computer software: 1) input of molecular geometry; 2) optimization preliminary geometry using molecular mechanics; and 3) refinement of the 3-D molecular structure and calculation of electronic properties of compounds using semi-empirical quantum mechanical methods.
Quantum mechanical ab initio methods are the most complex and time-consuming. The density functional theory can be mentioned as one of the successful methods to simulate few hundred atoms without any experimental input. This method together with higher level methods can be a very useful tool to investigate and predict the properties of emerging nano-based drug delivery systems. The results of ab initio calculations are often taken as a reference to verify results from other computational methods or to calibrate these methods. In contrast to the ab initio methods, semi empirical calculations are much less time consuming as they try to approximate time consuming components of the quantum mechanics calculations by simple empirical models. These models are based on experimental data (bond length, binding energy, electronegativity) and thus semi empirical methods have problems describing unusual compounds reliably.
The classical molecular mechanics calculations are even less time consuming than semi empirical ones. Here, the potential energy of the system is calculated as the sum of a set of simple equations. Each so-called force field uses its own values for van der Waals radii, optimal bond lengths, etc to describe the molecular mechanics of molecules. When compared to quantum mechanical methods, classical molecular mechanics is computationally cheap and can thus be employed to simulate the dynamic behavior of molecules for larger time spans.
An overview of the ab initio, semi empirical and molecular mechanics methods is well summarized in (CitationGhoniem and Cho 2002). An extension of the molecular dynamics simulation, which is used to quantify relative binding free energies of ligands to receptors, is the free-energy perturbation. In addition to the kinetic influences, this method primarily considers thermodynamic factors to characterize selective binding of molecules to yield different molecular complexes in non-equilibrium conditions such as biological (cellular) environments. This enables to rank the binding free energy in the complex formation of two chemically similar and biologically active ligands and thus of particular importance in the design of targeted drug delivery systems. Detailed information on the method and some applications on drug design can be found in the book edited by (CitationReddy and Erion 2001). Moreover, detailed information for the implementation of the method in molecular dynamics program for the computation of free energies is described in Reddy et al (CitationReddy et al 2000).
A computationally less expensive method for predicting ligand-receptor interaction is the molecular docking. Docking is a molecular process of searching for a ligand that is able to fit both geometrically and energetically the binding site. Usually the best geometries resulting from this process are energy minimized and the conformation with the lowest energy is further evaluated. The results of molecular docking are less accurate compared to the above mentioned methods but usually give good idea in ranking the level of interaction of ligands with a given receptor (CitationNeumann et al 2004).
Ab initio, semi empirical and molecular mechanics methods are usually not suited for modeling large systems like proteins and polymers. For these systems often mesoscale modeling techniques that deal with length scales (10–100 nm) between the atomistic and engineering scales are required (CitationMaiti et al 2004). In this technique subunits are defined instead discarding atomistic detail. The selection of the subunits depends on the system being modeled such as, for instance the monomer of a polymer as opposed to molecular model, which builds the monomer atom by atom. This method has been applied to drug delivery systems and drug diffusivity through cell membrane predictions by Maiti et al (CitationMaiti et al 2004).
Application of molecular level prediction to nanoparticle DDS
Naylor et al (CitationNaylor et al 1989) pioneered the study and characterization of dendrimers using molecular level modeling and simulation. In this work, the morphological and topological structures of dendrimers as a function of generation were investigated using molecular dynamics to calculate the moment of inertia. Simulations indicated that dramatic changes in morphology occur with generation number; generations 1–3 are highly asymmetric whereas generations 5–7 are nearly spherical with the transition between the two forms occurring at generation 4. In addition, average structures for the early generations are open, domed shapes, as compared to more dense, spheroid-like topologies for the latter generations with solvent-filled interior hollows connected by channels that run the entire length of the macromolecule. Generations 4–7, therefore, appear capable of encapsulating guest molecules. Bhalgat and Roberts (CitationBhalgat and Roberts 2000) expanded this work in 2000 to evaluate the effect of chemical modification on the interaction of the dendrimers with the biological target site. Similar efforts carried out using Monte Carlo simulation by Mansfield and Klushin (CitationMansfield and Klushin 1993) resulted in equilibrium values. Although the latter lack atomistic details, the longer simulation times allow equilibrium values to be obtained, which is very difficult with MD simulations.
The application of molecular level simulation for the design and synthesis of nanoparticle drug delivery device was carried out by Quintana et al (CitationQuintana et al 2002). They employed molecular dynamics simulation to evaluate the biologic function of dendrimer-folate nanodevice by applying different capping groups to enhance selective binding with the receptor. The primary amines on the surface of the dendrimer normally result in charge-based non-specific interaction diminishing the targeting. Modeling showed that capping the primary amines with carboxy groups produced local branch aggregation implying intermolecular branch interaction. On the other hand capping with 2, 3-dihydroxy propyl or acetamide yielded neutral surfaces resulting in the overall relaxation of the molecular structure due to absence of repulsive forces. Evaluation of the biological activity of the dendrimer folate nanodevices experimentally with KB (epidermoid carcinoma) cells confirmed the model prediction with the acetamide capping group showing the highest binding ability. The hydroxyl-surfaced nanodevice showed an overall binding capacity similar to the acetamide surfaced nanodevice at 2 fold concentration of the acetamide-surfaced nanodevice.
Stochastic approaches to macroscale (tissue, organ) parameter variability
Human subject variability due to age, pathological state, nutritional state, and intrinsic diversity can result in large differences in tissue and organ level parameters and lead to unacceptable error if models are based on mean parameters only. Sources of the inter/intra subject variability in the GI tract include the inherent variability of residence time of contents in the different GI tract segments, varying conditions of fed/fast state, or varying or progressing pathological state of the GI tract reflected as varying pH, osmolarity and residence time. There are several approaches to deal with the stochastics of this nature: the Monte carlo (MC) method, stochastic perturbation analysis and variance propagation algorisms.
In the Monte carlo method, samples of the random parameters are generated by means of a random generator and the corresponding model equations are solved (CitationFishman 1996). This procedure is repeated several times and finally mean values and variances as well as higher order moments are estimated. While the Monte Carlo method yields a rather complete picture of the stochastic system, the typically slow convergence properties require a large number of runs to obtain results with acceptable accuracy.
The stochastic perturbation method is based on the computation of the propagation of an infinitesimal perturbation of the (stochastic) parameters during the process. In this method the random field of the stochastic parameters can be represented in several ways such as Taylor expansion, Neuman expansion methods and mean values and (co)variances of the process variables can then easily be evaluated. In the case where large perturbation are present, the spectral expansion methods (CitationGhanem and Kruger 1996) that focus on higher order statistics such as the Karhunen-Loeve and the polynomial chaos expansion are employed. The method can only be applied to problems with random parameters that do not change with time. Often the stochastic perturbation equations are solved using numerical techniques such as FEM. This method is applied in transport problems in heat and mass transfer (CitationNicolai and Baerdemaeker 1997), porous media flow (CitationAguirre and Haghighi 2002) and coupled heat and mass transfer (CitationScheerlinck et al 2001).
The variance propagation algorithm computes the mean values and variances of the variable of interest (drug concentrations in drug delivery systems) under processes and environmental parameters that fluctuate transiently in a random way. Because a complete probabilistic description of the variable of interest is difficult to obtain, a first order approximate expression for the mean vector and the covariance matrix is provided from basic stochastic systems theory (CitationMelsa and Sage 1973). This method is also of interest because it is based on numerical methods such as FEM that can be applied where no analytical solutions can be obtained. Although successfully applied to similar problems in other fields, such as transport problems of heat and mass transfer (CitationNicolai and Baerdemaeker 1999), the stochastic perturbation method and the variance propagation algorithm are not yet exploited in the mathematical modeling of drug delivery systems.
Case study: Multiscale modeling of targeted oral drug delivery to the colon
Oral drug delivery targeted to the colon is important for both the treatment of local disorders of the colon and for the potential delivery of protein and peptide drugs into the body via the colon. In both cases the drug must survive the harsh environment of the upper GI tract with a maximal payload delivery at the colon. Many approaches to this problem are based on the utilization of the varying physiological conditions such as pH along the GI tract to prevent release until arrival at the colon. Variability between patients, in disease progression, in fed-fasted state, in tablet properties and in tablet geometry, all complicate the practice of these approaches.
In an effort to provide clinically relevant predictions using a more realistic computational model for the delivery to the colon, we developed a drug release model for targeted oral drug delivery systems based on the multiscale framework presented in section 3 (CitationHaddish-Berhane et al 2006b). The commercially available targeted delivery system, Asacol®, which delivers 5-aminosalicyclic acid (5-ASA) to the colon, served as the drug subject. The Asacol® tablet contains a core of 400 mg of mesalamine coated with pH-sensitive anionic copolymers of methacrylic acid and methyl methacrylate called Eudragit®-S (Röhm GmbH & Co. KG, Pharma Polymers, Darmstadt, Germany), which delay release of mesalamine until the tablet reaches an environment of pH 7 or above.
A continuum approach was pursued to model the drug diffusion and coating erosion by considering the relevant geometry of the delivery device using the mathematical description given in EquationEquation (1)(1) . The source term in this equation results from drug dissolution in this particular case and was expressed by the Whitney-Noyes equation (EquationEquation 6
(6) ).
The interaction of the drug molecule (5-amino, 2-hydroxybenzioc acid) with water was investigated using MD simulation to predict the diffusion properties of the drug. After obtaining the topology of the involved molecules using PRODRG (CitationSchuttelkopf and van Aalten 2004) MD simulations were carried out using GROMACS (CitationLindahl and van der Spoel 2001). To compute the diffusion coefficients, the Einstein relation based upon Mean Square Displacement was used. Parameters that characterize the interaction of the polymeric coating with the medium were derived from free volume theory (CitationFlory 1953).
Prediction of coating dissolution dynamics
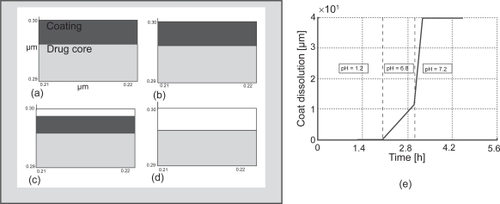
Coating dissolution of the tablet as it transits the GI tract is a key factor on timely and targeted release of the payload at the delivery site. Thus, prediction of the coating dissolution during transit in the GI tract provides important information on the performance of the delivery system. show the predicted dissolution of the polymeric coating in different intestinal buffers qualitatively and quantitatively, respectively as the tablet transits through the stomach (pH =1.2), proximal intestine (pH = 6.8) and distal intestine (pH = 7.2). The simulations confirmed that dissolution of the coating was almost non-existent at low pH environments (stomach) and takes place at a high rate in the distal segment of the intestine as the pH progressively increases. 72% of the coating dissolved in approximately 15 min as the tablet reached the distal intestine. Experimental in vitro data validated the predicted drug release kinetics with favorable agreement with in 5% mean square error.
Figure 5 Simulation of the polymeric coating (EudraGI tract® S-100) of Asacol® (a) enlarged section of the interface between the drug core and coating at t = 0 h; (b) after 2 h in the stomach fluid at pH = 1.2; (c) after 1 h in the intestinal fluid I (proximal) at pH = 6.8; (d) after 15 min in the intestinal fluid II (distal) at pH = 7.2; (e) Coating thickness dissolution in pH 1.2, 6.8 and 7.2 gastrointestinal buffers.
Drug release kinetics and biological variability
Often, the efficacy of the delivery systems for the topical treatment of colonic diseases is hampered by inter/intra-subject variability in the GI tract environment. Theoretical investigation using the model considered experimentally observed variability in healthy and diseased (Ulcerative colitis) subjects obtained from literature. To demonstrate the importance of the multiscale tool to obtain clinically relevant predictions, here results from diseased subjects will be summarized. Details can be found in Haddish-Berhane et al (CitationHaddish-Berhane et al 2006a).
Subjects with ulcerative colitis (UC)
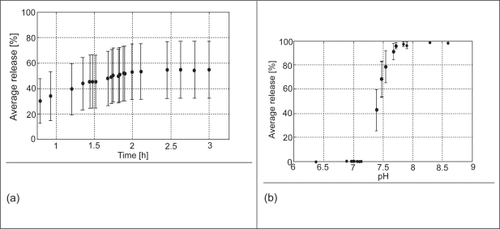
In the case of diseased subjects, three drug release patterns also resulted from the numerical experiment (400 MC runs) where drug release occurred in i) the distal small bowel only; ii) the distal small bowel and colon; and iii) the colon or excreted intact. The variation in drug release ranged from 0% to 100% at the target site (distal intestine/proximal colon) as a result of variation in pH and transit time. The overall average drug release in the distal part of the intestine stands at 48% with 95% confidence interval of 20%. The relatively larger confidence interval associated with the diseased subjects is as a result of the higher observed variability in the pH and transit time in the distal intestine. For the pH range considered, the average drug release varies from 30–55% (). This variation shows the spread expected due to distal intestine transit time variability. The drug release due to distal intestine pH variability ranges from 0–100% (). Here, the confidence intervals, which show the uncertainty due to variability in transit time, are smaller than that of the pH. This implies that pH is a dominant factor that influenced the drug release at the target site. The payload that was delivered to the colon and that entered the systemic circulation, calculated from the predicted release, agreed favorably with reported clinical results.
Figure 6 (a) The average drug release in the distal intestine versus the distal transit time for subjects with UC. Each data point is the average of the cumulative drug release of all the pH values randomly sampled at the end of the gastric and intestinal residence time. (b) The average drug release versus pH in the distal intestine for UC patients. The error bars represent the 95% confidence interval with respect to the pH and intestinal transit time variability, respectively.
The characteristic curves generated from the model results () can be produced for other delivery systems. These characteristic curves can be used for design purposes to improve the performance of the dosage forms. Moreover, physicians can use these characteristic curves and the pH and transit time profile of their patient to determine and prescribe a dosage form and treatment regimen that would work best for that specific patient. Even though similar evaluation results for a range of oral targeted delivery systems exist from in-vivo (clinical) evaluations, this example demonstrates that computational modeling could provide clinically relevant information, which can be used for the rational design of the delivery system.
Current state of DDS multiscale modeling
Computational approaches to evaluate, design and optimize the conventional drug delivery systems have not yet gained widespread acceptance and confidence by the pharmaceutical and research community. Although several factors have contributed to this current reality, including infrastructural and organizational issues (CitationGrasela et al 2005), the limited predictive ability and accuracy of existing mathematical models plays a significant role. (CitationRhodes and Porter 1998) and similarly (CitationBoobis et al 2002) argue that many models are often insufficient to predict the in vivo dissolution and release rates reliably and precisely. This limited predictive ability, we believe, can largely be attributed to either one of two problems. Comprehensive approaches that strive to model the entire system typically rely on the use of simple empirical or semi-empirical models, which are not truly sufficient to predict the essential behavior of the interaction of drug and its carrier. On the other hand, more complex, mechanistic theories and models focus on one particular process, but are not yet coupled to other physical, chemical or biological processes that are critical in the real system. The establishment of multiscale methods that can integrate processes across spatial scales is required to describe the complete system mechanistically.
Certainly multiscale modeling has been applied to nanoparticle problems such as prediction of dendrimer interactions with different solvents (CitationCagin et al 2001). We are unaware of any multiscale modeling studies that have integrated mechanistic models across the scales to predict both the uptake and transport process of the nanoparticles (nano/micro to macro level) and the response of the involved organs (atomic to organ level scales). Fundamental mechanisms at each isolated scale are still new territories themselves. As progress is made in understanding the mechanisms of nanoparticle interaction with cells and tissues and in the multiscale modeling of conventional drugs, the application of this and similar frameworks to nanoparticle DDS is sure to follow.
Future directions and implications
Need for measurement techniques and experimental paradigms
Mechanistic approaches to modeling depend upon the quantity, quality and form of existing experimental data as well as the possible experimental paradigms available for estimating parameters and validating models. Acquisition of temporally and spatially resolved data to track levels of molecules and nanoparticles in living cells and organisms remains an important technological challenge along side the modeling challenges that are highlighted in this review. In general four types of experimental data and systems are needed for the further advancement of the computational modeling: 1) time-resolved spatial data in living cells, tissues and organisms; 2) data sets from experiments designed to optimally extract parameter estimates from the measurable variables; 3) novel in vitro paradigms for simulating more realistic physiological conditions in controlled environments; and finally 4) statistical distribution data characterizing physiological and delivery system variability.
The ability to track detailed physiological and transport events in single cells or whole tissues over time is critical for building and testing realistic models. For example, methods such as multiple particle tracking have enabled the real measurement of nanoparticle transport rates and diffusion coefficients in gastric mucosal layers (CitationDawson et al 2004). Sensing systems for spatially and temporally resolved cellular data, however, are not widespread and much more development is needed. This is an area in which novel cytomic and physiological sensing capabilities can contribute significantly to computational models.
Novel in vitro paradigms that can simulate the effects of physiological parameters under controllable conditions are needed to compliment the time resolved measurement systems. A recent example of such an experimental paradigm uses a microfluidic device built by Farokhzad et al (CitationFarokhzad et al 2005) to study the effects of shear stress on the specific and non-specific binding of polymeric nanoparticles to cells expressing the cancer associated prostate specific membrane antigen. The advantage of such a system is that physical conditions such as flow in the microvasculature can be simulated to measure model parameters in vitro that would be more difficult and expensive to measure in vivo. By estimating parameters for use in larger scale model components and validating model predictions from smaller scale model components, the multiscale modeling can then provide a quantitative and testable link between less expensive cell culture studies and more resource associated in vivo biodistribution studies.
Establishing methods for the rigorous control of nanoparticle properties such as size, composition, and morphology are a primary goal for developing new nanomaterials and methods (CitationRolland et al 2005). Understanding the effects of variability and distribution of nanoparticles as well as conventional delivery vehicles, however, will continue to be important if such particle technologies intend to advance to clinical stages and industrial production. Certainly stochastic modeling components within multiscale models and statistical distribution data of experimentally measured delivery vehicle properties and physiological conditions will be critical.
Application to safety
Nanoparticles are designed for biological interactions but we have little experience or data (relative to single molecule drugs) regarding those interactions in organisms. The safety of producing, handling, and using nanoparticles is therefore an issue of major importance to the field (CitationOberdorster et al 2005). As the diversity of new nanoparticle materials increases, the ability to experimentally test all particles in an in vivo system becomes unrealistic. The use of mechanistic models, such as those proposed here, that are based on nanoparticle physiochemical properties could be a powerful tool, in addition to in vitro and in vivo models, for classifying nanoparticles in terms of safety. Models of oral delivery will be particularly applicable to safety as the GI tract is one of the major exposure points for nanoparticles to enter the body.
Evolution of existing and development of novel multiscale approaches
The projection that by 2016 about 30% of pharmaceutical R&D expenditure will be on computer simulation (CitationAnderson 2002) connote that we will see more and more mechanistic models developed. Multiscale computational modeling approach for rational design of drug delivery systems, however, is in its infancy even for the conventional drug delivery systems. Due to the permutations of the confounding factors that complicate the traditional experimental (wet lab) approach, the need for multiscale computational models is even higher for the design of drug delivery systems of nanoparticles that involve multiscale processes both in the spatial and temporal dimensions. Thus, a pathway to future designs of smart delivery systems will be integration of the different elements at various levels of scales involving the physico-chemical and biological phenomena.
The multiscale problem involved in DDS is that of the drug delivery device and that of the biological environment with which it interacts. Modeling efforts concerning the device so far have focused on bringing the disparate scales using purely empirical, not easily understood relationships in the translation from one level to the other. Such approach is pursued in the QSAR models. This arises from the time and length limitations in terms of obtaining adequate information for prediction of macroscale properties from atomistic simulations. Development of advanced multiscale approaches will thus investigate the intermediate levels above and below the disparate scales in order to bridge the gap using mechanistic approach.
On the other hand, the biological environment and its interaction with the xenobiotics should be represented from subcellular to organ level which involves disparate length and time scales. Besides connecting scales with parameters and coupling coefficients efforts towards bridging this scale gap will focus on merging the discrete atomistic level processes with the continuum at the higher levels and the coupling of the processes at different time scales within the different scales. The challenge here arises from the non-linear relationship between the scales. That is, the average behavior exhibited at the macroscales dramatically differs from the subscales (eg, metabolism). This will take development of advanced coarsening and refinement schemes both for time and length scales that must take key microscale phenomena such as bifurcation and seamless meshing/discretization techniques at the interface of the scales.
Other mathematical related problems that need to be addressed to produce fully predictive models will be the propagation of errors as the different scales are bridged. In case of multiscale simulation the sources of error include models selected at each scale, parameters used in each model, solution transfer errors, equation discretization and nonlinear equation solution errors (CitationShephard 2004). Hence, the development of useful error estimators correction indicators for the various components of the multiscale framework using existing adaptive approaches need to be accelerated.
Finally, multiscale approaches are not as widely applied in the drug delivery field as in other fields such as materials science and engineering. The reason for this is that each level in the mechanisms of drug delivery () is vast, involving complex network of dynamically interacting heterogeneous components. The design of drug delivery devices can utilize and benefit from the application of the multiscale modeling for the rational design of smart delivery systems.
Abbreviations
| ADMET | = | absorption, distribution, metabolism, excretion and toxicity |
| DDS | = | drug delivery system |
| FEM | = | finite element method |
| GI | = | gastrointestinal |
| MC | = | monte carlo |
| MD | = | molecular dynamics |
| PAMAM | = | polyamidoamine |
| QSPR | = | quantitative structure-property relationships |
| QSAR | = | quantitative structure-activity relationships |
Acknowledgements
The authors are grateful to Prof. Joseph DiStefano, III and the UCLA Biocybernetics Laboratory for extraordinarily useful feedback and comments during the writing of the manuscript.
References
- AgoramBWoltoszWSBolgerBB2002Predicting the impact of physiological and biochemical processes on oral drug bioavailabilityAdv Drug Deliver Rev50
- AguirreCGHaghighiK2002Finite Element Analysis of Transient Contaminant Transport: A Stochastic ApproachT ASAE45204959
- AkermanMEChanWCWLaakkonenPBhatiaSNRuoslahtiE2002Nanocrystal targeting in vivoPNAS %R 10.1073/pnas.152463399991261721
- AndersonRJ200220/20 Vision: A brave new world of drug developmentCurr Drug DiscovAugust259
- AtluriSNShenS2002The meshless local petrov-galerkin (MLPG) method: A simple and less-costly alternative to the Finite Element and Boundary Element methodsComput Modeling Eng Sci31152
- BhalgatMKRobertsJC2000Molecular modeling of polyamidoamine (PAMAM) Starburst(TM) dendrimersEur Polym J3664751
- BoobisAGundert-RemyUKremersP2002In silico prediction of ADME and pharmacokinetics: Report of an expert meeting organised by COST B15Eur J Pharm Sci171839312453607
- CaginTWangGMartinR2001Multiscale modeling and simulation methods with applications to dendritic polymersComput Theor Polym, S1134556
- ChenWFishJ2006A generalized space-time mathematical homogenization theory for bridging atomistic and continuum scalesInternational Journal for Numerical Methods in Engineering6725371
- ClementRJonquieresASartiI2004Original structure-property relationships derived from a new modeling of diffusion of pure solvents through polymer membranesJ Membrane Sci23214152
- CostaPSousaLJoseM2001Modeling and comparison of dissolution profilesEur J Pharm Sci131233311297896
- DawsonMKraulandEWirtzD2004Transport of polymeric nanoparticle gene carriers in gastric mucusBiotechnology Progress20851715176891
- DoneaJHuertaAPonthotJ-P2004Arbitrary lagrangian–eulerian methods (Chapter 14)Encyclopedia of Computational Mechanics, Volume 1: FundamentalsSteinERdBTJRHNew YorkJohn Wiley & Sons, Ltd
- DrescoPAZaitsevVSGambinoRJ1999Preparation and properties of magnetite and polymer magnetite nanoparticlesLangmuir15194551
- EliazRENirSSzokaJFrancisC2004Interactions of hyaluronan-targeted liposomes with cultured cells: modeling of binding and Endocytosis. Methods in EnzymologyLiposomes, Part DDuzgunesN1633Academic Press
- FarokhzadOCChengJTeplyBA2006Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivoPNAS10363152016606824
- FarokhzadOCKhademhosseiniAJonS2005Microfluidic system for studying the interaction of nanoparticles and microparticles with cellsAnal Chem775453916131052
- FastPShelleyMJ2004A moving overset grid method for interface dynamics applied to non-newtonian hele-shaw flowJ Comput Phys19511742
- FishJChenW2004aDiscrete-to-continuum bridging based on multigrid principlesComput Method Appl M1931693711
- FishJChenW2004bDiscrete-to-continuum bridging based on multigrid principlesComputer Methods in Applied Mechanics and Engineering1931693711
- FishmanGS1996Monte Carlo: Concepts, Algorithms and applicationsNew YorkSpringer Verlag698
- FloryPJ1953Principles of Polymer ChemistryIthaca, NYCornell University
- FreundLBLinY2004The role of binder mobility in spontaneous adhesive contact and implications for cell adhesionJ Mech Phys Solids52245572
- GaoHShiWFreundLB2005Mechanics of receptor-mediated endocytosisP Natl Acad Sci102946974
- Gex-FabryMDeLisiC1984Receptor-mediated endocytosis: a model and its implications for experimental analysisAm J Physiol247R768R796149699
- GhanemRGKrugerRM1996Numerical solution of spectral stochastic finite element systemsComputer Methods in Applied Mechanics and Engineering129289303
- GhoniemNMChoK2002The emerging role of multiscale modeling in nano- and micro-Mechanics of materialsComput Modeling Eng Sci3
- GozalbesRDoucetJPDerouinF2002Application of Topological Descriptors in QSAR and Drug Design: History and New TrendsCurr Drug Targets: Infect Disord29310212462157
- GraselaTFiedler-KellyJWalawanderCOwenJCirincioneB2005Challenges in the Transition to Model-Based DevelopmentAAPS J07E488E9516353926
- GrassGM1997Simulation models to predict oral drug absorption from in vitro dataAdv Drug Deliver Rev23199219
- GreeneN2002Computer systems for the prediction of toxicity: an updateAdv Drug Deliver Rev5441731
- GrefRMinamitakeYPeracchiaMT1994Biodegradable Long-Circulating Polymeric NanospheresScience263160038128245
- GueyffierDLiJNadimA1999Volume-of-fluid interface tracking with smoothed surface stress methods for three-dimensional flowsJ Comput Phys15242356
- Haddish-BerhaneNFarhadiANyquistC2006aBiological variability and targeted delivery of therapeutics for Inflammatory Bowel Diseases: an in silico approachInflammation & Allergy - Drug Targets Submitted
- Haddish-BerhaneNNyquistCHaghighiK2006bA multi-scale stochastic drug release model for polymer-coated targeted drug delivery systemsJ Control Release1103142216288814
- HirtCWNicholsBD1981Volume of fluid (VOF) method for the dynamics of free boundariesJ Comput Phys3920125
- KaneJH1993Boundary element analysis in engineering continuum mechanicsEnglewood Cliffs, NJPrentice-Hall
- KanjickalDGLopinaST2004Modeling of drug release from polymeric delivery systems - a reviewCrit Rev Ther Drug21245386
- KarelsonM2000Molecular Descriptors in QSAR/QSPRNew YorkJ. Wiley & Sons
- KarelsonMLobanovVSKatritzkyAR1996Quantum-Chemical Descriptors in QSAR/QSPR StudiesChem Rev9610274411848779
- KarelsonMMaranUWangY1999QSPR and QSAR models derived using large molecular descriptor spaces. A review of CODESSA applicationsCollect Czech Chem Commun64155171
- KatritzkyARIgnatchenkoESBarcockRA1994Prediction of gas chromatographic retention times and response factors using a general qualitative structure-property relationships TreatmentAnal Chem17991807
- KatritzkyARMaranUKarelsonM1997Prediction of melting points for the substituted benzenes: A QSPR ApproachJ Chem Inf Comput Sci379139
- KatritzkyARRachwalPLawKW1996Prediction of Polymer Glass Transition Temperatures Using a General Quantitative Structure-Property Relationship TreatmentJ Chem Inf Comput Sci3687984
- KawasakiESPlayerA2005Nanotechnology, nanomedicine, and the development of new, effective therapies for cancerNanomedicine1101917292064
- KennedyT1997Managing the drug discovery/development interfaceDrug Discovery Today243644
- KennethDCharlesSKevinL2005Epithelial internalization of superparamagnetic nanoparticles and response to external magnetic fieldBiomaterials2620617215576180
- KhaledAGuoSLiF2005Controllable Self-Assembly of Nanoparticles for Specific Delivery of Multiple Therapeutic Molecules to Cancer Cells Using RNA NanotechnologyNano Letters5179780816159227
- KitchensKMEl-SayedMEHGhandehariH2005Transepithelial and endothelial transport of poly (amidoamine) dendrimersAdv Drug Deliver Rev57216376
- KnobbeEM2004A FEM-ALE approach for continuous domains with arbitrary moving boundariesPresented at European congress on computational methods in applied sciences and engineering ECCOMAS 2004Jyvaskyla
- KubinyiH2004Validation and predictivity of QSAR modelsPresented at 15th EURO-QSAR SymposiumIstanbul, Turkey
- LangerRPeppasNA1983Chemical and physical structure of polymers as carriers for controlled release of bioactive agents: a reviewJ Macromol Sci Rev Macromol Chem PhysC2361126
- LauffenburgerDALindermanJJ1993Receptors: Models for binding, Trafficking and signalingNew YorkOxford University Press (Chapter 3)
- LeVequeJR2002Finite volume methods for hyperbolic problemsCambridgeCambridge University Press578
- LewisJDDestitoGZijlstraA2006Viral nanoparticles as tools for intravital vascular imagingNature Medicine1235406
- LiJHesseMZieglerJ2005An arbitrary lagrangian eulerian method for moving-boundary problems and its application to jumping over waterJ Comput Phys208289314
- LindahlEBHvan der SpoelD2001GROMACS 3.0: a package for molecular simulation and trajectory analysisJ Mol Model730617
- MaitiAWescottJGoldbeck-WoodG2004Mesoscale modelling: recent developments and applications to nanocomposites, drug delivery and precipitation membranesInt J Nanotechnology2198214
- ManjunathKVenkateswarluV2005Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduedenal administrationJournal of Controlled Release1072152816014318
- MansfieldMLKlushinLI1993Monte carlo studies of dendrimer macromoleculesMacromolecules2642628
- MauryB1999Direct simulations of 2D fluid-particle flows in biperiodic domainsJ Comput Phys15632551
- McCarthyJRPerezJMBrucknerC2005Polymeric nanoparticle preparation that eradicates tumorsNano Letters52552616351214
- MelsaJLSageAP1973An introduction to probability and stochastic processesNew JersyPrentice-Hall
- MunackA1989Optimal feeding strategy for identification of monod-type models by fed-batch experimentsComputer applications in fermentation technology, modelling and control of biotechnological processFishNMFoxRIThomhillNF195204AmsterdamElsevier
- NahorHBScheerlinckNVerniestR2001Optimal experimental design for the parameter estimation of conduction heated foodsJournal of Food Engineering4810919
- NaylorAMGoddardWAIIIKieferGE1989Starburst dendrimers. Part V: Molecular shape controlJ Am Chem Soc111233941
- NeumannDLehrC-MLenhofH-P2004Computational modeling of the sugar-lectin interactionAdv Drug Deliver Rev5643757
- NicolaiBMBaerdemaekerJD1997Finite element perturbation analysis of non-linear heat coduction with random field parametersInt J Numer Method H752544
- NicolaiBMBaerdemaekerJD1999A variance propagation algorithm for the computation of heat conduction under stochastic conditionsInt J Heat Mass Transfer42151320
- NithiarasuP2005An arbitrary lagrangian eulerian (ALE) formulation for free surface flows using the characterstic-based split (CBS) schemeInt J Numer Meth Fl48141528
- Nunes-CorreiaIRamalho-SantosJNirS1999Interactions of influenza virus with cultured cells: Detailed kinetic modeling of binding and endocytosisBiochemistry3810951019894006
- OberdorsterGMaynardADonaldsonK2005Principles for characterizing the potential human health effects from exposure to nanomaterials: elements of a screening strategyParticle and Fibre Toxicology213515813962
- PopielarskiSRHu-LieskovanSFrenchSW2005A nanoparticle-based model delivery system to guide the rational design of gene delivery to the Liver 2. In Vitro and In Vivo Uptake ResultsBioconjugate Chem16107180
- PopinetSZaleskiS1999A front-tracking algorithm for accurate representation of surface tensionInt J Numer Meth Fl3077593
- ProwTWRoseWAWangN2005Biosensor-Controlled Gene Therapy/Drug Delivery with Nanoparticles for NanomedicineProc SPIE5692199208
- QuintanaARaczkaEPiehlerL2002Design and function of a den-drimer-based therapeutic nanodevice targeted to tumor cells Through the folate receptorPharm Res191310612403067
- ReddyJN2005An introduction to the finite element methodNew YorkMcGraw-Hill Book Co
- ReddyMRErionMD2001Free energy calculations in rational drug designNew YorkSpringer–Verlag.
- ReddyMRErionMDAA2000Free energy calculations: Use and limitations in predicting ligand binding affinitiesReviews in Computational Chemistry217304New YorkVCH Publishers
- RenardyYRenardyM2002PROST: A parabolic reconstruction of surface tension for the volume-of-fluid methodJ Comput Phys18340021
- RhodesCTPorterCS1998Coatings for controlled release drug delivery systemsDrug Dev Ind Pharm241139549876571
- RollandJPMaynorBWEulissLE2005Direct fabrication and harvesting of monodisperse shape-specific nanobiomaterialsJournal of the American Chemical Society1271009610016011375
- Russell-JonesGJVeitchHArthurL1999Lectin-mediated transport of nanoparticles across Caco-2 and OK cellsInt J Pharm1901657410547456
- RyskinGLealLG1984Numerical solution of free-boundary problems in fluid mechanics. Part 2: Buoyancy-driven motion of a gas bubble through a quiescent liquidJ Fluid Mech1481936
- RyttingELentzKAChenX-Q2004A quantitative structure-property relationship for predicting drug solubility in PEG 400/Water cosolvent systemsPharm Res212374415032304
- ScardovelliRZaleskiS1999Direct numerical simulation of free surface and interfacial flowAnn Rev Fluid Mech31567604
- ScheerlinckNVerbovenPStigterJD2001A variance propagation algorithm for stochastic heat and mass transfer problems in food processesInt J Numer Meth Eng5196183
- SchuttelkopfAWvan AaltenDMF2004PRODRG: a tool for high-throughput crystallography of protein-ligand complexesActa Crystallogr D6013556315272157
- SegerlindLJ1984Applied finite element analysisNew YorkWiley
- ShephardMS2004Adaptive discretization and modelingFinal report: second DOE workshop on multiscale problemsEstepDShadidJNZachmannD52Broomsfield, ColoradoDepartment of energy
- ShimizuAKawashimaS1989Kinetic study of internalization and degradation of I-labeled follicle-stimulating hormone in mouse Sertoli cells and its relevance to other systemsJ Biol Chem2641363282503503
- SiepmannJGopferichA2001Mathematical modeling of bioerodible, polymeric drug delivery systemsAdv Drug Deliver Rev4822947
- SiepmannJPeppasNA2001Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC)Adv Drug Deliver Rev4813957
- SunXRossinRTurnerJL2005An assessment of the effects of shell cross-linked nanoparticle size, core composition, and surface PE-Gylation on in vivo biodistributionBiomacromolecules625415416153091
- ThurmondKBIKowalewskiTWooleyKL1996Water-Soluble Knedel-like Structures: The preparation of shell-cross-linked small particlesJ Am Chem Soc118723940
- TryggvasonGUnverdiSO1990Computations of three-dimensional rayleigh-taylor instabilityPhys Fluids A-Fluid26569
- TzafririARWuDEdelmanER2004Analysis of compartmental models of ligand-induced endocytosisJ Theor Biol2291273815178191
- UnverdiSOTryggvasonG1992A front-tracking method for viscous, incompressible, multi-fluid flowsJ Comput Phys1002537
- van de WaterbeemdHGiffordE2003ADMET in silico modeling: Towards prediction paradise?Nat Rev Drug Discov219220412612645
- WebsterTJ2006Welcome to the international journey to improving human healthInt J Nanomedicine11217722257
- WileyHSCunninghamDD1981A steady state model for analyzing the cellular binding, internalization and degradation of polypeptide ligandsCell25433406269748
- WileyHSCunninghamDD1982The endocytotic rate constant. A cellular parameter for quantitating receptor-mediated endocytosisJ Biol Chem257422296279628
- ZigmondSSullivanSLauffenburgerD1982Kinetic analysis of chemo-tactic peptide receptor modulationJ Cell Biol9234436276415