 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The p53 tumor-suppressor protein is a cellular phosphoprotein and a negative regulator of cell growth. Most p53 mutations occur in exons 5–8 within the DNA-binding domain. Therefore, p53 can potentially be targeted with novel drugs designed to bind to a mutation and restore its stability or wild-type conformation. For the current study, Hartree–Fock calculations were used to investigate the solvent-induced effects of five different solvent media (acetone, ethanol, methanol, dimethyl sulfoxide, and water) on the thermochemical parameters and relative energies, and on the multinuclear nuclear magnetic resonance shielding tensors of oxygen, nitrogen, and phosphorus nuclei, of GAT. To understand how the solvent affects the mutation region (the “hot spot”) of p53, the relative energies of GAT in selected solvent media were determined. Some biological evidence suggested the structural stabilities of hot spots of GAT have the optimum temperature and solvent type for mutation. All the authors’ findings are in accordance with common biological phenomena. Another important objective of this study was to compare the hydration Gibbs free energies of CUA and GAT in water using two different approaches where the solvent was treated as a continuum of the constant at different levels of Hartree–Fock theory. The Gibbs hydration energy values obtained in water with the polarized continuum model directly applied on the isolated CUA and GAT sequences were compared with those determined from the hydrated models with four, six, and eight water molecule clusters around the hot spots uracil and adenine. The clustered structures of water molecules around the hot spots of GAT (in DNA level) and CUA (in transcriptional level) were found to be energetically favored. The results of this study provide a reliable insight into the nature of mutation processes, which is of utmost importance for the study of biochemical structures, and provide a basis for drug design.
Introduction
The p53 tumor-suppressor protein is a cellular phosphoprotein and a negative regulator of cell growth. These functions make p53 a key factor in protection against cancer. More than half of all human cancers harbor p53 mutations and have no functioning p53 protein. More than half of human tumors contain a mutation or deletion of the TP53 gene. These mutations mostly occur in exons 5–8 and they make p53 a potential candidate for gene therapy. Most of the cancerous mutations are point mutations where a base pair is substituted by another with distributions along the DNA sequence. It should be noted that p53 is considered a significant contender for use in gene therapy targeting tumor cells.Citation1,Citation2
The tridimensional, dynamic structure of biological macromolecules influences all processes of molecular diagnosis. In recent years, significant improvement has been made in the development of theoretical methods in studies of structural and dynamic nucleic acid. Other recent successes include the application of quantum chemistry for solving biological problems. One of the applications of nucleic acid modeling is the possibility of attaching a drug to DNA.Citation3 A large part of the challenge of this is due to determination of DNA structure of all the common biological molecules. Direct observation of these phenomena is not possible at the atomic level. Some methods such as X-ray crystallography and nuclear magnetic resonance (NMR) provide useful information at the atomic level but are faced with many practical challenges.
According to research reports, p53 plays an important role in different types of human tumors, including pancreatic tumors, and changes in this gene can increase or decrease the sensitivity of tumor cells to anticancer drugs. The authors studied GAT codon in exon 5 of p53, which undergoes point mutation in pancreatic cancers. It has been found that the frequency and spectra of p53 mutation and its function depend on the cell-type development stage and the tissue origin of the cancer.Citation4 In other cases, mutations in the gene have been observed in 60%–80% of pancreatic cancers.Citation1
A statistical analysis of mutation-induced charge transfer modifications was performed.Citation5–Citation8 In contrast to noncancerous mutations, hot-spot mutations tend to result in significantly weaker changes of transmission properties.Citation9 The theoretical chemical shifts of these systems have been reported. Theoretical calculations predicted a significant downfield shift for protons involved in intermolecular hydrogen bonding between N–H and dimethyl sulfoxide (DMSO). The solvent-induced effects on chemical shifts of other nuclei have also been reported at the Hartree–Fock theory level.Citation10–Citation14
Until now, most of the investigations in this field have focused on conformational aspects of biological sequences.Citation15–Citation20 Solvation effects could be considered in drug metabolism, as the kinetics and thermodynamics of enzymatic biotransformations of drugs may be dependent on solvation effects. Therefore, extensive data related to solvation are probably essential.Citation21
In this current research, the authors have conducted theoretical studies of solvent effects on NMR tensors and on the thermochemical parameters of GAT codon; they have coupled this with consideration as to whether the polarity of solvent can have an influence on these physicochemical quantities. Structural changes introduced by various solvents were monitored by chemical-shielding changes. The selected solvents exhibited a wide range of hydrogen-bonding and polarity/polarizability properties. Due to the importance of hydrogen-bonding interactions governed in biological systems, the authors’ main theoretical argument has been focused on exploring the NMR parameters of nitrogen, oxygen, and phosphorus nuclei involved in the hydrogen-bonding network structure.
To study the temperature and solvent effects on the stabilities of the hot-spot segment of the GAT sequence, thermochemical parameters of GAT such as thermal energy, entropy, and enthalpy were calculated at 300 K, 310 K, and 313 K using five different solvent media. The temperature and a suitable solvent responsible for structural instability of hot spots leading to mutation were suggested. In order to identify the most probable sequence for mutations among GAU and CUA for RNA and GAT for DNA, all energy values of these systems were calculated in vacuum at RHF/6-31G theoretical level and a logical trend was revealed. To justify the solvent-induced effects, the energy values of the GAT system were determined in different solvent media with ethanol as a reference solvent; their relative energy (ΔErelative) values were analyzed.
The current study covers a theoretical background concerning thermochemical and solvent effects equations defined for the analysis of theoretical assumptions.
Theoretical background
An accurate knowledge of the magnitude of NMR tensors has been found to be valuable in indentifying biomolecular structure and dynamics with NMR spectroscopy.Citation18 Therefore, for reliable structural investigations, it is necessary to determine each nucleus of interest involved in the hydrogen-bonding network. Quantum chemical calculations are increasingly being used to rationalize the relationship between shielding tensors and biological structures.Citation22,Citation23
The following quantities are often used to describe NMR shielding tensors – namely, the isotropic, anisotropic shielding, and the asymmetry parameters:
The isotropic value (σiso) of the shielding tensor, which is defined as
(1)
The anisotropy parameter (Δσ), which is defined as
and
The asymmetry parameter (η), which is given byCitation24
For evaluating solvation free energies (ΔGhydration), the following equation can be defined as the difference between the Hartree–Fock free energies of the species in solution and the Hartree–Fock free energies of the species in the gas phase; in fact, ΔGS involve three nonelectrostatic contributions (cavitations, dispersions, and repulsion energies) asCitation25
Software and modeling
Computational biology techniques play a key role in expanding our knowledge on the mode of behavior of solvents in normal biological processes. Physics-based computations such as biomolecular simulation simulate biomolecular motion according to the laws of physics; they provide quantitative information on biomolecular dynamics and energetics, as well as help with the interpretation of biophysical data.Citation26
This research is an example of how quantum mechanical techniques can be successfully applied to biologically relevant problems in rather large and complex systems. For this purpose, the electronic structure and solvent-effects calculations are performed on GAT codon using Gaussian 98 software (Gaussian, Inc, Wallingford, CT).Citation27
First, the authors considered the geometry optimizations of GAT existing in DNA and coupled with it, carrying out the Hartree–Fock level of theory using the STO-3G, 3–21G, and 6–31G basis set in the gas phase as well as in the five different solvent media (acetone, ethanol, methanol, dimethyl sulfoxide, and water) by assuming a polarized continuum model (PCM) solvation method. Before starting PCM calculations, the cavity radius obtained for incorporation of solvent effects was determined. After optimization in different solvents, the authors calculated NMR shielding parameters for some nuclei involved in the GAT hydrogen-bonding network.
Second, the authors considered just the adenine section of GAT sequence, regarded as a hot spot for mutation, for performing a frequency calculation in order to obtain thermochemical parameters at three different temperatures (300 K, 310 K, and 313 K) to find out important structural stability factors.
Third, due to the key role of the number of water molecules on mutation and other biological phenomena, the authors constructed the hydrated complexes of four, six, and eight water molecules bonded directly to uracil and adenine as the hot spots of CUA and GAT sequences through several hydrogen bonds to be used in calculation of ΔGhydration (as defined in Equationequation 4(4) ). This parameter has been used to investigate the effect of hydration of the CUA and GAT sequences to predict its actual structure in aqueous solution at finite temperature.
Hence, the interaction energies of studied systems were calculated as the difference between the total energy of a whole system and the energies of subsystems. Solvent effects in water were calculated by employing the PCM method in which the solvent is considered a continuum dielectric, recognized by its constant permittivity. Indeed, an important objective of this study was to compare the solvation energies calculated by adopting the PCM method directly from the data obtained by equation 6. The energy values of CUA and GAT affected by the hydration were studied by comparing the energetic features of the isolated sequences and their hydrated model.
Results and discussion
Solvent effects on multinuclear NMR parameters
The theoretical values of σiso, Δσ, and η of oxygen, nitrogen, and phosphorus atoms of GAT sequence in different solvent media are shown in and .
Table 1 Nuclear magnetic resonance shielding tensors (ppm) of nitrogen and oxygen nuclei involved in the hydrogen-bonding network of GAT codon in different solvent media at the level of RHF/6–31G theory
Table 2 Relative thermochemical parameters of GAT obtained in different solvent media at three different temperatures
For prorogated nitrogen atoms such as N1 the minimum fluctuations in asymmetry parameter were observed. It was shown that all solvents had no significant effect on the variation of charge density of proton-donor atoms or among all atoms of GAT involved in the hydrogen-bonding network. The highest values of asymmetry parameter were observed in different solvents. For GAT sequence the maximum and minimum values of δiso for N11, N32, and N47 were observed in water and acetone, respectively.
Temperature and solvent effects on thermochemical functions
There have been numerous reports on the analysis of thermochemical parameters of the isolated uracil and its hydrated model.Citation28–Citation30 However, there are no experimental data on the relative energies or enthalpies of the above model systems.Citation31
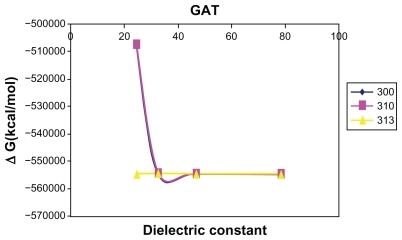
In the current study, the authors’ emphasis was placed on the variations of thermochemical parameters due to the temperature effect in different solvents. According to thermochemical functions of GAT reported in at three different temperatures (300 K, 310 K, and 313 K) and different solvent media, the most negative value was obtained at 300 K and the highest stability in water solvent at 313 K.
In view of the solvent and temperature effects on GAT model, the negative Gibbs free energy of GAT in water with the highest polarity at 313 K reveals the most stable condition and the least probability for mutation to occur. However, these results suggest that the polar solvents – specifically, a highly polar system solvent such as water – provide the most suitable condition for mutation of GAT located in DNA.
The graph of the relative Gibbs free energy versus dielectric constant of GAT () shows that with increased dielectric constant there is a dramatic decrease in Gibbs free energy. The minimum region is observed for ethanol, and after passing this point the slope of the curve remains constant.
Figure 1 Relative Gibbs free energies (ΔGrelative) of GAT sequence in different solvent media.
Hydration models of CUA and GAT sequences
Computing the free energy solvation is essential in rational drug design for pharmokinetic and pharmodynamic studies. Some recent research has been devoted to modeling the first hydration shell of nucleic acid base using electronic structure methods.Citation32,Citation33 Also, based on the reviewing literature the uracil-water interaction has been explored by means of the natural bond orbital analysis that provides supplementary information of the relative stability ordering.Citation31
In two CUA, nH2O and GAT, nH2O local minima structures, water molecules are bonded to the uracil of CUA as well as to the adenine of GAT by several hydrogen bonds. The oxygen and hydrogen atoms of water that are involved in forming the hydrogen bonds are in the same plane of uracil and adenine, while the free water hydrogen atoms are outside the plane.
Two different approaches were compared in this research by considering solvent effects on Gibbs solvation energies and finding the most proper hydrated model (). According to the hydrated models of CUA, nH2O and GAT, nH2O, the most negative energy values were obtained considering quantized number of water molecules (n 4, 6, 8) rather than the PCM model, which assumes the solvent as a continuum medium. A notable result is that these energy values (ECUA, nH2O and EGAT, nH2O) led to more negative values through adding water molecules from n = 4 up to n = 8. In the case of CUA, nH2O and of GAT, nH2O, the most negative energy value, referred to as the most probable model, was observed with four water molecules.
Table 3 Hydration Gibbs free energies and energy values of CUA and GAT obtained in different solvent media (kcal/mol)
Therefore, based on reported results, the authors conclude that hydration of CUA codon as well as GAT codon tends to yield stabilization energies for a more negative nonhydrated complex.
Solvent effects on the relative structural stabilities of hot spots
Analysis of the total energy values of a biological system is the first critically important step for evaluation of a wide range of structural electronic properties. It is now well accepted that the solvent plays a key role in the stabilization of biomolecular systems in general.Citation19
Based on the energy calculations for CUA and GAT, the more negative energy value and the higher stability were found for CUA rather than GAT. Here we can see the lowest stability for GAT in DNA. According to this fact it is logical to expect mutation to occur more in GAT than in CUA sequence involved in RNA. This observation is in accordance with the common biological fact that the majority of mutations generally occur in DNA.
According to the relative energy values (ΔErelative) in the solvents with a variety of polarities, it seems that the stability of all three systems was influenced by the polarity of the solvent. Hence, the lowest ΔErelative value was found in the lowest dielectric constant and the highest value was observed in water, with the highest dielectric constant and with high polarity (). Also, there are two extremum regions in the graph of energy values of GAT versus dielectric constants of different solvent media (ɛ = 24.55 and ɛ = 46.8). In other words, the lowest point indicates the most stable GAT was found in ethanol, with the maximum region belonging to DMSO. This fact is attributed to the solvent-induced effect on the stability of GAT systems.
Figure 2 Relative energies (Erelatives) of GAT sequence versus dielectric constant (ɛ) (A) and relative energies (Erelatives) of GAT sequence versus Ln (1/ɛ) (B) in different solvent media.
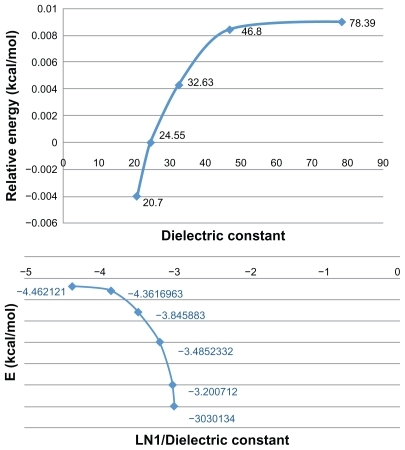
Finally, it is realized that relative energies (ΔE) of GAT in solution state were smaller and the structures were more stable than with the gas phase system, which was due to easier interactions in solution relative to the gas phase.
Conclusion
An improved understanding of the nature of the biological interactions causing the shift in NMR or thermochemical parameters may hold the key to finding the most suitable condition for the occurrence of various biological phenomena – specifically, mutation. This issue is inherently intriguing due to the relatively large number of atoms and to the complex nature of the interactions involved in the quantum hot spot-DNA system that require ab initio electronic structure methods. However, with the progress of computational techniques, the domain of application of molecular simulations is constantly expanding.
It seems likely that NMR chemical shielding tensors may play an even more important role in structural determination. It has been found that hydrogen bonding is the most important factor for deshielding of the electronic charge density around noticed nuclei. A dipole in the molecule will induce a dipole in the medium, and the electric field applied by the solvent dipole will in turn interact with the molecular dipole, leading to net stabilization.
Based on the energy calculation of GAT, it was observed that the relative energies (ΔE) of GAT in solution were smaller than in the gas phase, which is due to interactions in solution that were larger than in the gas phase. This observation is in accordance with the common biological fact that the majority of mutations generally occur in DNA.
Furthermore, considering the hot spots of CUA and GAT sequences, a structure with four water molecules around uracil and adenine segments is slightly favored. It is notable that hydration of CUA codon as well as GAT codon tends to yield the stabilization energies of more negative nonhydrated complex. Therefore, it seems likely that in the future it may be possible to directly incorporate quantum chemical results into biological structure refinement.
Disclosure
The authors report no conflicts of interest in this work.
References
- DongMNioYYamasawaKTogaTYueLHaradaTp53 alteration is not an independent prognostic indicator, but affects the efficacy of adjuvant chemotherapy in human pancreatic cancerJ Surg Oncol20038211112012561067
- IraniSMonajjemiMHonarparvarBAtyabiSMSadeghizadehMInvestigation of solvent effect and NMR shielding tensors of p53 tumor-suppressor gene in drug designInt J Nanomedicine2011621321821499418
- RiahiSGanjaliMRBagheriMTheoretical investigation of interaction between Gatifloxacin and DNA: implications for anticancer drug designMat Sci Engineer C20092918081813
- FeiPBernhardEJEl-DeiryWSTissue-specific induction of p53 targets in vivoCancer Res2002627316732712499275
- SherrCJPrinciples of tumor suppressionCell200411623524614744434
- MessiasACSattlerMStructural basis of single-stranded RNA recognitionAcc Chem Res20043727928715147168
- BrameldKDasguptaSGoddardWADistance dependent hydrogen bond potentials for nucleic acid base pairs from ab initio quantum mechanical calculations (LMP2/cc-pVTZ)J Phys Chem B199710148514859
- KalidOBen-TalNStudy of MDM2 binding to p53-analogues: affinity, helicity, and applicability to drug designJ Chem Inf Model20094986587619323449
- ShihCTRocheSRomerRAPoint-mutation effects on charge-transport properties of the tumor-suppressor gene p53Phys Rev Lett200810001810518232825
- KurinovichMALeeJKThe acidity of uracil from the gas phase to solution: the coalescence of the N1 and N3 sites and implications for biological glycosylationJ Am Chem Soc200012262586262
- HocquetAGhomiMThe peculiar role of cytosine in nucleoside conformational behaviour: hydrogen bond donor capacity of nucleic basesPhys Chem Chem Phys200025351
- PodolyanYGorbLLeszczynskiJProtonation of nucleic acid bases: a comprehensive post-Hartree–Fock study of the energetics and proton affinitiesJ Phys Chem A200010473467352
- MillerTMAronoldSTViggianoAAStevens MillerAEAcidity of a nucleobase: uracilJ Phys Chem A200410834393446
- ChandraAKNguyenMTHuyskensTZTheoretical study of the interaction between thymine and water: protonation and deprotonation enthalpies and comparison with uracilJ Phys Chem A199810260106016
- BeveridgeDLMcConnellKJNucleic acids: theory and computer simulation, Y2KCurr Opin Struct Biol20001018219610753816
- YansonIKTeplitskyABSukhodubLFExperimental studies of molecular interactions between nitrogen bases of nucleic acidsBiopolymers19791811491170435611
- CornellWDCieplakPBaylyCIA second generation force field for the simulation of proteins, nucleic acids, and organic moleculesJ Am Chem Soc199511751795197
- KupkaTKolaskiMPasternaGRuudKTowards more reliable prediction of formaldehyde multinuclear NMR parameters and harmonic vibrations in the gas phase and solutionJ Mol Struct19994676378
- AuffingerPHashemYNucleic acid solvation: from outside to insightCurr Opin Struct Biol20071732533317574833
- XuLDingYTengQTheoretical research on effects of substituents and the solvent on quadruple hydrogen bonded complexesBull Chem Soc Ethiop200721419426
- LeppertJHeiseBRamachandranR15N chemical shift tensor magnitude and orientation in the molecular frame of uracil determined via MAS NMRJ Magn Reson200014530731410910699
- DiosACOldfieldERecent progress in understanding chemical shiftsSolid State Nucl Magn Reson199661011258784950
- MonajjemiMHaddadiAHonarparvarBIraniSMollaaminFTahanANMR and solvent effect study on the thymine-adenine-thymine sequence: a theoretical investigation on the chemical behavior of nucleotides in solutionEgypt J Biochem Mol Biol20082683100
- PeculMSadlejJ15N chemical shift tensor magnitude and orientation in the molecular frame of uracil determined via MAS NMRJ Chem Phys1998234111119
- DiezNMSenentMLGarciaBAb initio study of solvent effects on the acetohydroxamic acid deprotonation processesJ Chem Phys2006324350358
- AutschbachJThe calculation of NMR parameters in transition metal complexesKaltsoyannisNMcGradyJEPrinciples and Applications of Density Functional Theory in Inorganic Chemistry I, vol 112, Structure and BondingHeidelberg, GermanySpringer Verlag2004148
- FrischMJTrucksGWSchlegelHBScuseriaGERobbMAGaussian 98, Revision A7Gaussian, IncPittsburgh, PA1998
- GaigeotMPSprikMAb initio molecular dynamics computation of the infrared spectrum of aqueous uracilJ Phys Chem B200310710344
- NguyenMTZhangRBNamPCCeulemansASinglet-triplet energy gaps of gas phase RNA and DNA bases: a quantum chemical studyJ Phys Chem A200410865546561
- ZhangRBCeulemansANguyenMTA theoretical study of uracil and its tautomers in their lowest-lying triplet stateMol Phys2005103983994
- ZhangRHuyskensdTZCeulemeansANguyenMTInteraction of triplet uracil and thymine with waterJ Chem Phys20053163544
- ShishkinOVGorbLLeszczynskiJModeling of the first hydration shell of uracil and thymineInt J Mol Sci2000117
- KimSWheelerSESchaeferHFMicrosolvation effects on the electron capturing ability of thymine: thymine-water complexesJ Chem Phys2006124204310