 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
This work aimed to develop a new therapeutic approach to increase the efficacy of 5-fluorouracil (5-FU) in the treatment of advanced or recurrent colon cancer. 5-FU-loaded biodegradable poly(ε-caprolactone) nanoparticles (PCL NPs) were combined with the cytotoxic suicide gene E (combined therapy). The SW480 human cancer cell line was used to assay the combined therapeutic strategy. This cell line was established from a primary adenocarcinoma of the colon and is characterized by an intrinsically high resistance to apoptosis that correlates with its resistance to 5-FU. 5-FU was absorbed into the matrix of the PCL NPs during synthesis using the interfacial polymer disposition method. The antitumor activity of gene E from the phage ϕX174 was tested by generating a stable clone (SW480/12/E). In addition, the localization of E protein and its activity in mitochondria were analyzed. We found that the incorporation of 5-FU into PCL NPs (which show no cytotoxicity alone), significantly improved the drug’s anticancer activity, reducing the proliferation rate of colon cancer cells by up to 40-fold when compared with the nonincorporated drug alone. Furthermore, E gene expression sensitized colon cancer cells to the cytotoxic action of the 5-FU-based nanomedicine. Our findings demonstrate that despite the inherent resistance of SW480 to apoptosis, E gene activity is mediated by an apoptotic phenomenon that includes modulation of caspase-9 and caspase-3 expression and intense mitochondrial damage. Finally, a strongly synergistic antiproliferative effect was observed in colon cancer cells when E gene expression was combined with the activity of the 5-FU-loaded PCL NPs, thereby indicating the potential therapeutic value of the combined therapy.
Introduction
Colorectal cancer is the third most frequently diagnosed malignancy in the world. The annual incidence of colon cancer is estimated to be around one million, with approximately 500,000 patients dying from the disease each year.Citation1 Recent advances in chemotherapy (including the use of irinotecan, oxaliplatin, and fluoropyrimidines, as well as cetuximab and bevacizumab) and radiation therapy have increased the median survival of patients. However, advanced or recurrent colon cancer remains incurable by conventional treatments,Citation2 therefore more effective treatment options are needed for the treatment of this disease.
Despite the efficacy of 5-fluorouracil (5-FU) against a wide variety of tumors,Citation3 this antitumor molecule suffers from several drawbacks. As a consequence of a very rapid in vivo metabolism, 5-FU has a short biological half-life. Furthermore, the drug displays nonuniform oral absorption (with significant patient-to-patient variations in bioavailability), and tumor cells often undergo significant development of drug resistance. These characteristics lead to the use of high doses of 5-FU and subsequent severe dose-limiting toxicity in the clinical setting.Citation3–Citation5 To enhance the concentration of anticancer molecules in the tumor mass while minimizing their biodistribution and toxicity, nanoparticle-based drug-delivery systems are currently being widely investigated.Citation6–Citation8 Biodegradable nanocarriers formulated from poly(ε-caprolactone) (PCL) can protect the loaded antitumor drug from rapid elimination in vivo through metabolism, optimizing its therapeutic effect and keeping its associated toxicity to a minimum.Citation9–Citation11
The combination of chemotherapeutic agents with gene therapy (combined therapy) is an attractive strategy to overcome the limitations of conventional cancer treatment. Suicide gene therapy, either alone or in combination with tumor irradiation or chemotherapy, has been investigated for the treatment of intractable cancers.Citation12 However, in colon cancer, gene therapy in combination with cytosine deaminase/5-fluorocytosine (CD/5-FC) and herpes simplex virus thymidine kinase (HSV-tk), the two most widely used suicide prodrug systems, has obtained only limited results. In this context, therapeutic genes that encode cytotoxic proteins may provide alternatives to classical suicide-gene therapy. The products of these genes do not depend on prodrugs and may act by killing both quiescent and rapidly dividing tumor cells, possibly by inducing tumor cell apoptosis. Apoptosis deficiency is a critical factor in colorectal cancer therapy failureCitation13,Citation14 and the development of gene therapy strategies that improve apoptosis may therefore provide a complementary strategy for the treatment of colon cancer.
Toxic genes from noneukaryotic organisms such us bacteria, plants, viruses, and bacteriophages are being widely used in cancer gene therapy.Citation15–Citation17 The lysis gene from phage ϕX174 (gene E) has recently shown antiproliferative activity in cancer cells.Citation18 The E protein (91 aa) has no detectable cell-wall degrading activity, and its primary structure does not suggest any important enzymatic activity. However, electron microscopy images of cells undergoing E-mediated lysis have shown discrete 50–200 nm holes in the cell membrane. This observation has led to a proposed model in which the E protein oligomerizes to form a transmembrane tunnel that spans the entire cell envelope, thereby releasing the cytoplasmic contents.Citation19
In this study, we investigated a new antitumor strategy for the treatment of colon cancer that combines 5-FU-loaded nanoparticles (NPs) with gene therapy using gene E. We tested this approach using a SW480 human adenocarcinoma colon cancer cell line that is resistant to both apoptosis and chemotherapy.
Materials and methods
Materials
All chemicals used, except for 5-FU, poly(ε-caprolactone) and Pluronic® F-68 (Sigma-Aldrich, Munich, Germany), were of analytical quality and were obtained from Panreac (Barcelona, Spain). The pcDNA3.1/GFP and pcDNA3.1-TOPO plasmids and the DH5α chemically competent Escherichia coli were purchased from Invitrogen (Carlsbad, CA). The pTRE plasmid (Tet-Off gene-expression system) was obtained from Clontech Laboratories, Inc, (Mountain View, CA). Gene E was kindly provided by Dr Ramos (Zaidín Experimental Station, CSIC, Granada, Spain).
Synthesis and characterization of PCL NPs
PCL NPs were prepared using the interfacial polymer disposition method.Citation11,Citation20 Briefly, 200 mg of polymer was dissolved in 10 mL of dichloromethane under mechanical stirring (300 rpm). The resulting organic solution was transferred dropwise into 0.05 L of a 2% (w/v) aqueous solution of Pluronic® F-68 stirred at 1200 rpm. The organic phase was then completely evaporated using a Büchi Rotavapor® (Büchi, Flawil, Switzerland) rotary evaporator to obtain an aqueous suspension of pure PCL NPs. These were then cleaned using repeated cycles of centrifugation (45 minutes at 10,000 rpm, Centrikon T-124 high-speed centrifuge; Kontron, Paris, France) and resuspension in water, until the conductivity of the supernatant was ≤10 μS/cm.
Pure PCL NPs were loaded with 5-FU using an entrapment procedure. The method for drug absorption onto the NPs was similar to that described above except that the aqueous phase contained appropriate amounts of the chemotherapy agent. The influence of the concentration of stabilizing agent and polymer on drug absorption was also studied. Thus, the amount of polymer added to the organic solution was varied from 0.2 to 1 g, and the concentration of stabilizing agent in the aqueous phase was varied between 0 and 2% (w/v). The production performance (yield, %) of all the formulation conditions was also determined:
The mean particle size was determined by photon correlation spectroscopy (PCS; Malvern Autosizer® 4700; Malvern Instruments Ltd, Malvern, UK), at 25.0°C ± 0.5°C. The scattering angle was set at 60°, and after suitable dilution of the NP suspensions (≈0.1%, w/v) measurements were taken in triplicate. The stability of the formulations was evaluated by measuring the size of the NPs after 2 weeks of storage at 4.0°C ± 0.5°C in water. In addition to the mean diameter, the system reported a polydispersity index with a value between 0 and 1. A polydispersity index of 1 indicated great differences in particle size. On the opposite, a reported value of 0 means that size variation is absent.
To corroborate the size determinations and to elucidate the shape of the NPs, nanoformulations were checked through analysis by Zeiss DSM 950 scanning electron microscopy (Carl Zeiss, Oberkochen, Germany), and by STEM CM20 high resolution transmission electron microscopy (Philips, Amsterdam, The Netherlands). Prior to observation, dilute suspensions (≈0.1%, w/v) were sonicated for 5 minutes, and drops were placed on copper grids with Formvar film. The grids were then dried at 25.0°C ± 0.5°C in a convection oven.
The stability of the formulations was evaluated by measuring both the size and 5-FU loading values after 2 weeks of storage at 4.0°C ± 0.5°C in water. To that aim, the International Conference on Harmonisation guidelines were followed.Citation21 The electrokinetic properties of the blank (nonloaded) and drug-loaded NPs (≈0.1%, w/v) were analyzed by electrophoresis measurements in deionized and filtered water (Milli-Q Academic, Millipore, France), using a Malvern Zetasizer 2000 electrophoresis device (Malvern Instruments Ltd,). These measurements were performed after 24 hours of contact of NPs in water under mechanical stirring (50 rpm) at 25.0°C ± 0.5°C. The experimental uncertainty of the measurements was below 5%. The theory of O’Brien and White was used to convert the electrophoretic mobility (ue) into zeta potential (ξ) values.Citation22
Analysis of the ability of PCL NPs to act as 5-FU nanocarriers: drugloading and in vitro release studies
To determine the 5-FU concentration in all the systems investigated, UV absorption measurements were performed at the maximum absorbance wavelength (266 nm) in a UV-Vis Dinko spectrophotometer (Dinko, Barcelona, Spain), with samples contained in quartz cells with 1 cm path lengths. Good linearity was observed at this wavelength, and the method was validated and verified for accuracy, precision and linearity under all conditions tested.
The loading of 5-FU onto the NPs was evaluated in triplicate by spectrophotometric determination of the drug remaining in the aqueous solution obtained upon filtration of the NP suspension through a membrane filter (pore size: 0.1 μm), which was deduced from the total amount of 5-FU in the NP suspension. For the method to be accurate, we took into account the contribution of sources of absorbance other than the drug, for example, the surfactant agent. This contribution was calculated by measuring the absorbance of the supernatant produced under the same conditions but without the chemotherapy agent. The incorporation of 5-FU into PCL NPs was expressed in terms of 5-FU entrapment efficiency (%) (encapsulated drug [mg]/total drug in the NP suspension [mg] × 100), and 5-FU loading (%) (encapsulated drug [mg]/carrier [mg] × 100).
Release of 5-FU from the NPs was studied using formulations prepared under optimal drugloading conditions of antitumor molecules fixed into the aqueous phase at a concentration of 10 mM. 5-FU release in vitro was studied in triplicate following the dialysis bag method, with phosphate-buffered saline (PBS; pH 7.4 ± 0.1) as the release medium. The bags were first soaked in water at 25.0°C ± 0.5°C for 12 h before use. The dialysis bag (cut-off of Spectra/Por® 6, 2 kD dialysis membrane tubing; Spectrum, New Brunswick, NJ) retained the NPs but allowed the free anticancer molecules to diffuse into the dissolution medium. A 2 mL sample of the NP suspension (containing the antitumor drug at 1.3 mg/mL) was placed into the dialysis bag with the two ends fixed by clamps. The bags were then placed in a glass beaker containing 0.2 L of the dissolution medium and stirred at 200 rpm. The temperature was maintained at 37.0°C ± 0.5°C during the drug-release experiments. At preset time intervals (0.25, 0.5, 1, 2, 3, 6, 9, 12, 24, 48, 72, and 96 hours), 1 mL of the medium was withdrawn and its 5-FU content analyzed by UV-Vis spectrophotometry (266 nm). An equal volume of PBS maintained at the same temperature was added after sample withdrawal to ensure sink conditions. The same analytical procedure used to estimate the drug loading was followed in this investigation.
Plasmid construction for E gene expression
The E gene was amplified from pMC22 by polymerase chain reaction (PCR) using primers with EcoRI and NheI sites incorporated (forward, 5′-GAATTCGATGGTACGCTGGACTTTG-3′; reverse, 5′-CCGGGCTAGCCATT ACATCACTCCTTCCGC-3′). Cycling conditions were: 94°C for 1 minute, 30 cycles at 94°C for 1 minute, 65°C for 90 seconds, 72°C for 90 seconds, and 72°C for 10 minutes. The amplified product and pTRE were each digested with EcoRI and NheI and ligated with T4 ligase to obtain pTRE-E. To determine the intracellular localization of the E product, a GFP-E gene fusion was generated. Gene E was engineered to eliminate the stop codon. The amplified product was ligated into pcDNA3.1/GFP (Invitrogen) following the manufacturer’s protocol, to obtain pcDNA3.1/GFP-E. Finally, the E gene was also subcloned into pcDNA3.1-TOPO to obtain pcDNA3.1/E. Subcloning-efficiency DH5α E. coli were transformed with the generated plasmids and their correct sequences were confirmed by DNA sequencing.
Cell culture and drugs
The simple (ie, 5-FU-loaded PCL NPs and gene E therapy administered separately) and combined treatments were tested in the apoptosis- and chemoresistant SW480 human carcinoma cell line (Instrumentation Service Center, Granada University, Granada, Spain).Citation23 Cells were grown in RPMI 1640 medium (Sigma, St Louis, MO), supplemented with 10% fetal bovine serum (FBS), 15 mM HEPES, 14 mM NaHCO3, 2 mM l-glutamine, 40 μg/mL gentamicin, and 500 μg/mL ampicillin (Antibióticos S.A, Madrid, Spain). Cells were maintained in monolayer culture at 37°C in an atmosphere containing 5% CO2.
Production and selection of stable inducible SW480 cell clones
To analyze E gene activity against colon cancer, SW480 cells were transfected with pTRE-E using the Fugene 6 DNA transfection reagent (Roche, Madrid, Spain). Cells were initially transfected with pTet-On and successfully-transfected clones were selected for geneticin (1 mg/mL) resistance and rtTA expression. Clones were then transfected with pTRE-E, a pTRE2hyg vector containing the E cDNA and selected for resistance to geneticin (1 mg/mL) and hygrovodamycin (0.4 mg/mL). Two of the four clones identified in the first transfection round were used in a second round of transfections with the pTRE-E expression plasmid to give a total of 12 derivative clones. All clones were cultured in the presence of doxycycline (Dox; 0.2 mg/mL, 24 hours) to induce E gene expression, which was detected by reverse transcriptase (RT)-PCR. cDNA was generated using the Promega Reverse Transcription System (Promega, Madrid, Spain) using 1 μg of total RNA (Rneasy Mini kit; Qiagen, Hilden, Germany). PCR amplification of the E gene was then performed using specific primers (forward, 5′-GCTTTCCTGCTCCTGTTGAG-3′; reverse, 5′-TTGACGCACGTTTTCTTCTG-3′) under the following conditions: 94°C for 1 minute, 35 cycles at 94°C for 1 minute, 55°C for 30 seconds, 72°C for 30 seconds, and 72°C for 10 minutes. Amplif ication of β-actin (forward, 5′-ATCATGTTTGAGACCTTCAA-3′; reverse, 5′-CATCTCTTGCTCGAAGTCCA-3′) was used to assess RNA integrity. Amplified PCR products were visualized by agarose gel electrophoresis and quantified using a Bio-Rad documentation system (Quantity One Analysis software; Bio-Rad Laboratories, Hercules, CA). Relative E expression was calculated as the ratio of E to β-actin. One transfected clone, SW480/12/E, presented significant gene E expression in the presence of Dox with no background expression, and was used to test the simple and combined treatments.
Proliferation assays
The antitumor effect of the 5-FU-loaded NPs, E gene therapy, and the combined treatment schedule was evaluated using the proliferation assay. Cells were seeded in a 96-well plate (6 × 103 cells per well). At different times, 20 μL of 3-(4,5-di-methylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/mL) was added to each well followed by incubation at 37°C for 4 hours. The medium was removed and 200 μL of dimethyl sulfoxide (DMSO) was added to each well. The optical density (OD) at 570 and 690 nm was determined using a Titertek multiscan colorimeter (Flow, Irvine, CA). To determine the percentage of relative cell viability (%RCV) we used the formula %RCV = (treated cells OD/untreated cells OD) × 100. In addition, the MTT assay was used to determine the cytotoxicity of the 5-FU-loaded NPs at a wide range of concentrations (10–200 mM).
Apoptosis analysis
Cells were harvested, washed two times with sample buffer (100 mg glucose; 100 mL PBS without Ca2+ or Mg2+) and fixed in 70% (v/v) cold ethanol for 1 hour before staining. The cells were pelleted and resuspended in a solution containing 50 μg/mL propidium iodide (PI) and 0.5 mg/mL RNase in sample buffer at pH 7.4, for 30 minutes in the dark. Apoptosis was evaluated using a fluorescence-activated cell-sorter flow cytometer (Becton Dickinson, Franklin Lakes, NJ) and all data were analyzed using the Cellfit program. To determine possible caspase activation during E gene expression, a sample of transfected cells was preincubated in the pan caspase inhibitor zVAD-fmk (BD Pharmingen, San Jose, CA) at 200 mM, 2 hours before the induction of E gene expression.
Fluorescent microscopy analysis of E protein localization
SW480 cells were transfected with pcDNA3.1/GFP-E using Fugene 6 (Roche), as described above, to determine the intracellular localization of the E protein. For mitochondrial staining, the medium was changed to DMEM containing 500 nM MitoFluor Red (MitoTracker; Invitrogen, Barcelona, Spain), incubated for 15 minutes, and then replaced with normal medium. For nuclear staining, 1 mL of DAPI (Invitrogen) at 100 nM was added to fixed cells in a 60 mm dish, and incubated for 20 minutes at room temperature. The cells were then rinsed with PBS and mounted. GFP was excited at 488 nm, DAPI nuclear stain at 364 nm, and MitoFluor Far Red at 588 nm. Fluorescent microscopy analysis was carried out using a Nikon Eclipse Ti (Nikon Instruments Inc, Melville, NY).
Measurement of the mitochondrial membrane potential (ΔΨm) and the caspase activation
To measure levels of ΔΨm disruption, cells were washed twice with cold PBS and incubated with 40 nM DiOC6(3) (Invitrogen) for 15 minutes at 37°C. Cells were then washed with ice-cold PBS and resuspended in 500 μL of PBS. The fluorescence intensities of DiOC6(3) were analyzed using a FACScan flow cytometer with excitation and emission settings of 484 and 500 nm, respectively. Caspase activation was evaluated by western blot analysis using 30 μg of cell protein extract separated by SDS-PAGE (Mini Protean II cell; Bio-Rad Laboratories) and transferred to a nitrocellulose membrane (20 V at room temperature for 30 min). The blots were treated with blocking solution (20 mM Tris, 0.9 NaCl, 10% nonfat milk) for 3 hours and then incubated with primary rabbit polyclonal IgG anti-caspase-3 (1:1000 dilution), anticaspase-8 (1:200 dilution), anti-caspase-9 (1:500 dilution) and anti-β-actin antibodies (1:5000 dilution; Abcam, Cambridge, MA) overnight at 4°C. After addition of the peroxidase-conjugated secondary antibody, the proteins were detected by enhanced chemiluminescence (ECL; Bonus, Amersham, Little Chalfont, UK). A protein extract from transfected cells treated with zVAD-fmk as described above was also included in the study.
Morphological analysis
Cells were analyzed by optical and transmission electronic microscopy (Hitachi H7000 transmission electron microscope; Hitachi, Tokyo, Japan) according to Prados and colleagues.Citation24
Statistical analysis
The SPSS software package (v. 14; SPSS Inc, Chicago, IL) was used for all statistical analyses. Results were compared using Student’s t-test. All data were expressed as means ± standard deviations of sample means. Differences were considered statistically significant at a P value <0.05.
Results and discussion
Particle geometry, surface charge, and stability
The interfacial polymer disposition method allowed the formation of well-stabilized spherical PCL NPs with an average diameter of 140 ± 20 nm and a narrow size distribution (polydispersity index: 0.046) (). No presence of aggregates or bulky sediments was observed. The size of the polymeric particles and the quality of the suspensions did not vary significantly when loaded with different amounts of 5-FU (). This particle size could be considered as particularly suitable for facilitating the cellular uptake of the nanomedicine; it is assumed that NPs with a diameter in the range 100–200 nm can easily be internalized by receptor-mediated endocytosis, whereas larger particles are generally taken up by phagocytosis.Citation25 Given an appropriate cellular uptake of PCL NPs with a mean diameter of 140 nm ± 20 nm, a significant enhancement of the 5-FU concentration in tumor cells might be expected. No antitumor drug release and/or precipitation, NP aggregation or change in the size of particles was detected after two weeks of storage at 4.0°C ± 0.5°C in water. Thus, if the nanoparticulate preparations were not used immediately after formulation, they were kept under the aforementioned storage conditions until use.
Table 1 Influence of 5-FU loading (%) on size of PCL NPs
Figure 1 Scanning electron microscope (A) and high resolution transmission electron microscope (B) images of PCL NPs.
Note: All scale bars shown in the figure are 150 nm.
Abbreviations: NPs, nanoparticles; PCL, poly(ε-caprolactone).

The zeta potential values of the nonloaded and 5-FU-loaded PCL NPs in deionized and filtered water were found to be −18.3 ± 0.4 and −19.1 ± 0.6 mV, respectively. Such data clearly shows how similar are the surface electrical charges of these NPs which points out that the antitumor agent was not adsorbed onto the polymeric surface. Consequently, a very efficient drug entrapment has led to 5-FU-loaded PCL NPs which, from an electrokinetic point of view, are indistinguishable from nonloaded PCL.
Analysis of the capabilities of PCL NPs as 5-FU nanocarriers: drug loading and in vitro release studies
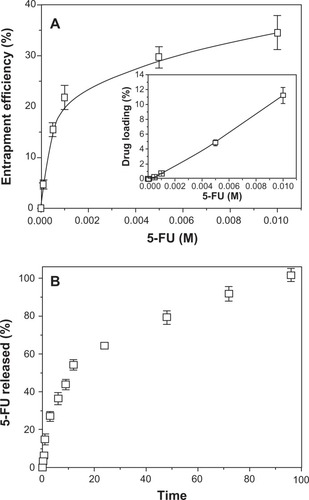
It is possible that 5-FU loading onto PCL NPs benefit from electrostatic attractive forces that may exist between the positively charged drug molecules (generated by protonation of the NH group of the 5-FU chemical structure),Citation26 and the negatively charged polymeric NPs.Citation11 While this might be the case, we attempted to improve drug loading by introducing the 5-FU molecules before the interfacial polymer disposition process to induce mechanical drug trapping. Furthermore, a stabilizing agent (Pluronic® F-68) was added to facilitate the opening of the polymeric chains and to yield a noncompact structure into which 5-FU could be incorporated.Citation11,Citation27 shows the entrapment efficiency (%) and loading (%) of 5-FU onto the PCL NPs as a function of drug concentration. This figure indicates that the 5-FU concentration positively affects the entrapment efficiency. An initial drug concentration of 10−4 M in the entrapment medium gave entrapment efficiency and loading values of 4.7% ± 0.9% and 0.015% ± 0.003%, respectively, while one of 10−2 M gave entrapment efficiency and loading values of 34.5% ± 3.3% and 11.219% ± 1.074%, respectively.
Figure 2 5-FU loading and in vitro release studies. (A) Efficiency of 5-FU entrapment (%) into PCL NPs as a function of antitumor drug concentration (inset: corresponding 5-FU loading values, %). The lines are guides for the eye and have no other significance. (B) 5-FU release (%) from PCL NPs at 37.0°C ± 0.5°C as a function of incubation time in PBS (pH 7.4 ± 0.1).
Abbreviations: 5-FU, 5-fluorouracil; NPs, nanoparticles; PBS, phosphate-buffered saline; PCL, poly(ε-caprolactone).
shows the influence of the Pluronic® F-68 and PCL concentrations used in the formulation of the NPs on 5-FU loading (%). Drug loading was markedly lower in the absence of the surfactant agent, but increased considerably and remained unaltered at and above 0.5% (w/v) Pluronic® F-68 in the concentration range tested. Interestingly, the yield of 5-FU-loaded polymeric NPs was always higher than 94% in all formulations tested with surfactant agent, but decreased to below 9% in the absence of this macromolecule. It can therefore be assumed that smaller polymeric NPs with a narrower size distribution were obtained after addition of the surfactant to the aqueous solution, without influencing the drug incorporation into the polymer. Similarly, the PCL concentration did not significantly influence drug loading onto the NPs.
Table 2 Influence of PCL and Pluronic® F-68 concentration on 5-FU entrapment efficiency (%) and 5-FU loading (%) by entrapment into the polymeric NP network
5-FU release from the polymeric matrix followed a biphasic process typical of PCL at pH 7.4,Citation11,Citation20,Citation28 with an initial fast drug release (up to ≈ 27% in 3 hours) and the remaining 5-FU being released in a sustained manner over a further period of 93 hours (). The initial fast 5-FU release is attributed to the leakage of surface-bound and/or poorly entrapped drug, which rapidly diffused into the incubation medium. It is likely that the sustained release phase corresponds to the 5-FU fraction (ie, ≈ 73%) that was deeply embedded into the polymeric nanomatrix, which must follow a longer diffusion path before leakage. This suggests that the majority of the 5-FU was entrapped in the polymeric network rather than being adsorbed onto the NP surface.
Antitumor activity of 5-FU-loaded PCL NPs on SW480 cells
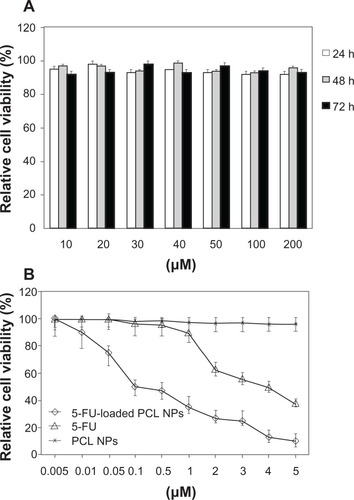
We initiated this investigation with an analysis of the cytotoxic activity of blank (nondrug-loaded) PCL NPs on SW480 human adenocarcinoma colon cancer cells. As shown in , the MTT assay results indicate that there were no significant differences between the absorbance of the negative control and the wells treated with blank NPs after incubation for 72 hours (P > 0.05). Thus, PCL NPs show no cytotoxicity and may be considered to be appropriate for drug-delivery purposes. We then compared the ability of 5-FU and 5-FU-loaded PCL NPs to inhibit the proliferation of SW480 cells. These cells have an inherent resistance to apoptosis that correlates with chemoresistance to 5-FUCitation23 and, along with cell lines SW620, HCT-8 and DLD1/5FU, are amongst the most resistant colon cell lines to this drug.Citation29 This is of interest given the relationship between colon cancer drug resistance and invasive/meta-static ability.Citation30 The nanomedicine inhibited cell proliferation in a dose-dependent manner with a much lower IC50 (concentrations of the drug needed to reduce population growth by 50% in vitro) value (0.1 μM) than that reported previously for SW480 cells (IC50 5-FU: 4 μM),Citation31 showing that the antitumor effect of 5-FU-loaded PCL NPs in SW480 was 40-fold higher than that for the free drug ().
Figure 3 In vitro cytotoxicity of PCL NPs over a range of concentrations (10–200 mM) (A) and cytotoxicity of 5-FU-loaded PCL NPs in comparison to 5-FU (B) in colon cancer SW480 cells after 3 days of incubation. Data are represented as means ± SD of quadruplicate cultures. To calculate the %RCV (see Materials and methods) SW480 cells without treatment were used as control.
Abbreviations: 5-FU, 5-fluorouracil; NPs, nanoparticles; PBS, phosphate-buffered saline; PCL, poly(ε-caprolactone); %RCV, percentage of relative cell viability; SD, standard deviation.
Antitumor activity of gene E on SW480 cells
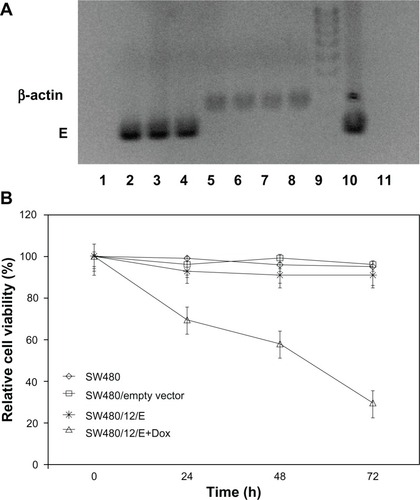
The stable SW480/12/E clone was used to determine the antitumor activity of gene E in apoptosis-resistant colon cancer cells. Cultivation of these cells in the presence of Dox showed effective expression of the E gene compared to the same cells without Dox. Analysis of RT-PCR products amplified from gene E cDNA, normalized to the corresponding β-actin signal, showed a slight time-dependent increase in E expression (2- and 2.7-fold higher at 48 and 72 hours versus cells at 24 hours; ). Cells showed a corresponding significant and time-dependent decrease in viability (30.5%, 41.9%, and 70.4% at 24, 48, and 72 hours, respectively; ).
Figure 4 Growth arrest by E gene expression in SW480 cells. (A) RT-PCR detection of E gene expression in SW480 transfected cells (SW480/12/E) before and after Dox exposure. Amplified RT-PCR products of E and β-actin mRNA at different time periods were separated by 2% agarose gel electrophoresis and visualized with ethidium bromide. RT-PCR of gene E: Lane 1: SW480/12/E cells in the absence of Dox; lanes 2–4: SW480/12/E cells 24, 48, and 72 hours after Dox induction, respectively. RT-PCR of β-actin: Lane 5: SW480/12/E cells in absence of Dox; lanes 6–8: SW480/12/E cells 24, 48. and 72 hours after Dox induction, respectively. Lane 9: DNA ladder; lane 10: pTRE-E (positive control); lane 11: parental SW480 cells (negative control). (B) MTT assay of SW480/12/E cells induced with Dox showed a significantly higher rate of cell death than for SW480/12/E cells in the absence of Dox, parental cells and cells transfected with empty vector (P < 0.05). To calculate the %RCV (see Materials and methods) SW480 cells without transfect reagents were used as control. Data are represented as means ± SD of quadruplicate cultures.
Abbreviations: Dox, doxorubicin; 5-FU, 5-fluorouracil; NPs, nanoparticles; PCL, poly(ε-caprolactone); %RCV, percentage of relative cell viability; SD, standard deviation.
Most colon cancer gene therapy assays investigated to date have used a prodrug therapy system in which the anticancer molecule is activated by exogenous genes that encode enzymes, such as HSV-tk and CD. However, assays in which either of these enzymes were used produced only limited results.Citation32–Citation34 The use of specific promoters, such us the promoter of Cox-2 or uPAR, improves its tissue specificity but not its efficacy.Citation27,Citation35 Recently, Lee and colleaguesCitation36 used a combination of shMDR and HSV1-tk to significantly decrease (63.3%) the volume of tumors generated in mice by HCT-15 colon cancer cells. However, prodrugs continue to be one of the main limiting factors of these strategies.Citation37 In this context, the transfection of cDNA constructs that encode toxins with direct antitumor activities represents a good alternative. These toxin genes have been successfully used in pancreatic cancer (diphtheria toxin gene),Citation15 hepatocellular carcinoma (staphylococcal enterotoxin C2),Citation38 breast and prostate cancer (Clostridium perfringens enterotoxin and Pseudomonas aeruginosa exotoxin A),Citation39 and breast cancer and melanoma (gef gene).Citation40,Citation41 Expressed bacteriophage genes, such as that coding for the lambda-holin protein, have led to a substantial reduction (more than 98%) of cell viability in breast cancer cells.Citation42 We have previously shown that E gene expression efficiently inhibits proliferation of melanoma and colon cancer drug-sensitive cells.Citation18,Citation43 Our present findings show that E gene expression is able to induce a significant antiproliferative effect in a drug-resistant SW480 cell line.
E gene expression induces caspase-3- and caspase-9-mediated apoptosis in SW480 cells
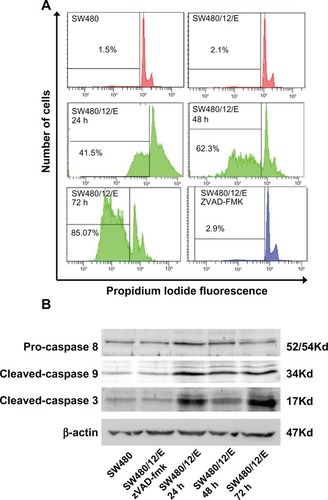
As shown in , SW480/12/E cells at 24 hours after induction with Dox showed an apoptosis fraction of 41.5% ± 0.4%. This was significantly higher than that for the control groups (2.1% ± 0.47% for SW480/12/E and 1.5% ± 0.32% for parental cells). After 48 and 72 hours of transfection, the percentage of apoptotic cells increased to 62.3% ± 0.52% and 85.07% ± 0.9%, respectively. These results show that E gene expression induces an intense apoptosis phenomenon in apoptosis-resistant SW480 colon cancer cells (85.07% at 72 hours). This result is of particular interest as apoptosis resistance is one of the characteristics implicated in colon cancer development and metastasis.Citation30 In addition, defects in apoptotic machinery are a major cause of cytotoxic drug resistance.Citation44 Although the mechanism underlying apoptosis resistance is not clear, colon cancer cells may contain high levels of antiapoptotic proteins (PED, cFLIP, Bcl-xL, and Bcl-2).Citation45 Recently, it was shown that the gene inhibitor of apoptosis 2 (cIAP2) is upregulated in DLD-1/FU-resistant cells (with characteristics similar to SW480, SW620, and HCT-8 cells). Interestingly, downregulation of cIAP2 by siRNA induced an efficient caspase-3-mediated apoptosis.Citation29 Apoptosis induction was efficiently inhibited by zVAD-fmk in transfected SW480 cells (), suggesting the involvement of the mitochondrial apoptotic pathway. Western blot analysis confirmed caspase-9 and capase-3 activation, whereas caspase-8 showed no modulation of expression (). Treatment of SW480/12/E cells with zVAD-fmk efficiently inhibited E-induced caspase-3 and caspase-9 activation, which appeared to be at similar levels to that in SW480/12/E cells without Dox exposure (). E-induced modulation of apoptosis may therefore be a promising new strategy in colon cancer treatment, especially in those that exhibit apoptosis resistance.
Figure 5 Apoptosis signals after E gene expression in SW480 cells. (A) SW480/12/E cells before and after Dox induction at times indicated were analyzed by FACScan to determine apoptotic cell death. The apoptosis was assessed after propidium iodine staining by calculating the percentage of cells in the sub-G1 fraction. SW480/12/E cells treated with zVAD-fmk were also analyzed at 72 hours. SW480 parental cells were used as the control. Data shown are representative results from four independent experiments. (B) Activated (cleaved) caspase-3, caspase-9, and pro-caspase-8 were detected by Western blot analysis using specific antibodies at the indicated time points after Dox treatment. zVAD-fmk was applied to determine whether caspases were involved in the apoptotic process in SW480/12/E cells induced with Dox (72 hours). The filter was probed with a β-actin antibody to determine whether the amounts of protein in each lane were comparable. Immunoblots were visualized using an enhanced chemiluminescence detection system.
Abbreviation: Dox, doxorubicin.
E protein is located in the mitochondria, induces the loss of mitochondrial membrane potential, and changes the cell morphology of SW480 cells
SW480 cells transfected with pcDNA3.1/GFP-E were used to determine the mitochondrial localization of E protein. Immunofluorescence microscopy produced a merged image with yellow regions where green (GFP-E) and red (MitoFluor) fluorescence overlap (). Significant morphological changes were observed in the transfected cells after 48 hours, when cells were swollen, and after 72 hours, when the number of spherical cells increased rapidly (). As shown in , a significant decrease in ΔΨm was detected in SW480/12/E cells induced with Dox at 24, 48, and 72 hours in comparison with SW480/12/E cells not exposed to Dox, or SW480 parental cells (data not shown). This decrease in fluorescence intensity reflects the E gene-induced depolarization of the inner mitochondrial membrane, and defines an early, but already irreversible, stage of apoptosis.
Figure 6 Mitochondrial localization of the GFP/E fusion protein in SW480 cells. Representative fluorescent microscopy images of transfected SW480 cells expressing fusion protein E-GFP at 24 (×10), 48 (×40), and 72 hours (×40). The dotted pattern of GFP-E fluorescence is shown in green. The majority of expressed GFP-E was found to be colocalized with MitoFluor (shown in red color). Colocalization is shown in yellow. Cell nuclei were counterstained with DAPI.
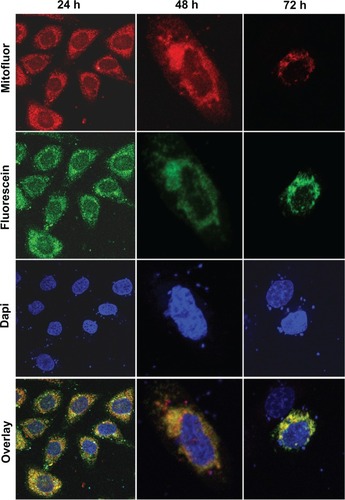
Figure 7 Mitochondrial damage caused by E protein in SW480 cells. (A) SW480 parental cells and SW480/12/E before and 24, 48, and 72 hours after Dox induction were stained with DiOC6, and analyzed by flow cytometry to determine the mitochondrial membrane potential (ΔΨm) disruption caused by E gene expression. Data shown are representative results from four independent experiments. (B) Ultrastructural analysis showed that the morphology of SW480/12/E cells not induced by Dox was similar to that of SW480 parental cells, with a typical presence of a large nucleus and light cytoplasmic complexion containing well-preserved organelles including mitochondria (a, 2000×). In contrast, E gene expression in SW480/12/E after Dox exposure generated a large number of altered mitochondria with disrupted cristae (b, 6000×). These cells eventually presented noticeably dilated mitochondria (c, 12,000×). Furthermore, mitochondrial changes in some cells were accompanied by the presence of dilated smooth endoplasmic reticulum (d, 9000×), and the presence of clusters of intermediate filaments (e, 4000×).
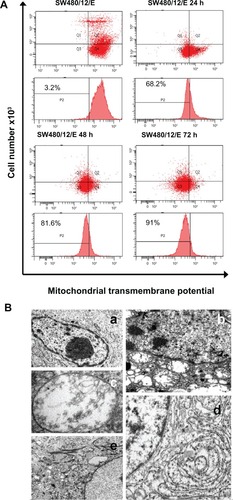
To further investigate the nature of E gene-mediated cytotoxicity, SW480/12/E cells were analyzed by transmission electron microscopy. The most significant morphological change in the SW480/12/E cells induced with Dox was the pronounced swelling of the mitochondria, which showed disrupted cristae after 24 hours of treatment (). The mitochondria in noninduced cells remained unaffected. In some cases, significant accumulations of intermediate filaments were also observed around the nucleus (), along with a dilated endoplasmic reticulum (), although this was not always the case.
Effect of the combined use of 5-FU-loaded PCL NPs and the cytotoxic suicide gene E on SW480 cancer cells
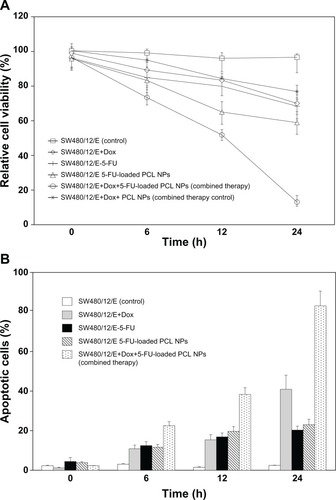
After independently establishing the effect of 5-FU-loaded PCL NPs and E gene on the SW480 colon cancer cells, we investigated the antitumor effect of the combined treatments. The modulation of cell proliferation was determined at 6, 12, and 24 hours. Treatment with both 5-FU-loaded PCL NPs and with Dox (to induce E gene expression) produced a decrease in viability of SW480/12/E cells of 87% ± 3.1%. This is greater than the sum of the effects of separate treatments with 5-FU PCL NPs (40.8% ± 2.6%) and E gene expression (30.5% ± 1.8%), suggesting a synergistic antitumor effect. Treatment with free 5-FU and E gene expression in SW480/12/E cells resulted in a much lower synergistic effect than that of 5-FU-loaded PCL NPs and E gene expression (). Furthermore, this combined therapy (5-FU-loaded PCL NPs and E gene expression) in SW480/12/E cells induced an increase in the percentage of apoptotic cells at 24 hours (83% ± 4.6%) greater than that obtained from the independent use of 5-FU-loaded PCL NPs and E gene expression (22% ± 2.5% and 41% ± 5%, respectively), suggesting the combined use of these treatments has an important enhancement of the apoptotic effect in this resistant tumor cell line ().
Figure 8 Synergistic effect of 5-FU-loaded PCL NPs associated with E gene expression in SW480 cancer cells. The combined therapy showed (A) a synergistic effect on growth arrest in the SW480/12/E colon cancer cells, and (B) a significant increase in apoptosis compared to single treatments. To calculate the %RCV (see Materials and methods) SW480 cells without treatment were used as control. Data are represented as means ± SD of quadruplicate cultures.
Abbreviations: Dox, doxorubicin; 5-FU, 5-fluorouracil; NPs, nanoparticles; PCL, poly(ε-caprolactone); %RCV, percentage of relative cell viability; SD, standard deviation.
Novel advances in the use of combined therapy for the treatment of lung,Citation46 bladder,Citation47 pancreatic,Citation48 and hepatocellular cancer,Citation49 have been reported. In colon cancer, Abaza and colleaguesCitation50 have recently demonstrated that antisense c-myc in combination with cytotoxic drugs (including 5-FU) increased caspase-3 (amongst other apoptotic signals) and exhibited a marked apoptotic effect compared to each component treatment alone. Our objective, to explore the ability of the E gene to enhance the cytotoxic effect of drugs against colon cancer cells, was similar. In light of previous results obtained with 5-FU-loaded PCL NPs against SW480 cells, we assayed a combined therapy involving 5-FU-loaded PCL NPs and cytotoxic E suicide gene expression, and found it to induce a decrease in SW480 cell viability around 20% greater than the sum of the individual treatments. This finding supports the hypothesis that a synergistic effect occurs when both treatments are administered together.
Conclusion
We have described the most advantageous synthetic conditions for producing 5-FU-loaded PCL NPs. 5-FU entrapment into the polymeric NPs has resulted in important drug loading and sustained drug-release properties, thereby opening up opportunities for improved delivery of 5-FU to cancer cells. 5-FU PCL NPs exhibited a 40 times greater in vitro antitumor activity than free 5-FU. Furthermore, the utility of E gene expression for the induction of growth arrest in colon cancer cells with an apoptosis-resistant phenotype was analyzed. E gene expression was found to strongly induce apoptosis mediated by the mitochondrial pathway in these resistant cells, and to significantly enhance the cell growth inhibition induced by 5-FU-loaded PCL NPs. These results suggest that this combined antitumor strategy may be a promising therapy in patients with advanced-stage colon cancer. Future studies will, however, be necessary to determine its in vivo utility.
Acknowledgements
The authors would like to thank Dr B González (Department of Oncology, Virgen de las Nieves Hospital, Granada, Spain) for performing the chemotherapy treatments. The excellent technical assistance of M Tassi and G Ortiz Ferrón is also acknowledged. This study was supported by the Department of Health of the Autonomous Government of Andalusia, Spain (project nos PI-0338 and PE-2008-FQM-3993).
Disclosure
The authors report no conflicts of interest in this work.
References
- ParkinDMBrayFFerlayJPisaniPGlobal cancer statistics, 2002CA Cancer J Clin2005557410815761078
- BerrinoFDe AngelisRSantMSurvival for eight major cancers and all cancers combined for European adults diagnosed in 1995–1999: results of the EUROCARE-4 studyLancet Oncol2007877378317714991
- ZhangNYinYXuSJChenWS5-Fluorouracil: mechanisms of resistance and reversal strategiesMolecules2008131551156918794772
- BertoliniAFiumanoMMuffattiAAcute cardiotoxicity from 5-fluorouracil: an underestimated toxicityMinerva Cardioangiol19994726927310582438
- CookeJWBBrightRColemanMJJenkinsKPProcess research and development of a dihydropyrimidine dehydrogenase inactivator: Large-scale preparation of eniluracil using a Sonogashira couplingOrg Process Res Dev20015383336
- CouvreurPVauthierCNanotechnology: intelligent design to treat complex diseasePharm Res2006231417145016779701
- AriasJLReddyLHCouvreurPPolymeric nanoparticulate system augmented the anticancer therapeutic efficacy of gemcitabineJ Drug Target20091758659819694612
- AriasJLReddyLHCouvreurPSuperior preclinical efficacy of gemcitabine developed as chitosan nanoparticulate systemBiomacromolecules2011129710421117615
- ZhangLYangMWangQ10-Hydroxycamptothecin loaded nanoparticles: preparation and antitumor activity in miceJ Control Release200711915316217400320
- DevalapallyHDuanZSeidenMVAmijiMMModulation of drug resistance in ovarian adenocarcinoma by enhancing intracellular ceramide using tamoxifen-loaded biodegradable polymeric nanoparticlesClin Cancer Res2008143193320318483388
- AriasJLLopez-ViotaMSaez-FernandezERuizMAFormulation and physicochemical characterization of poly(epsilon-caprolactone) nanoparticles loaded with ftorafur and diclofenac sodiumColloids Surf B Biointerfaces20107520420819740632
- DuraiRYangSYSeifalianAMWinsletMCPrinciples and applications of gene therapy in colon cancerJ Gastrointestin Liver Dis200817596718392246
- MillanAHuertaSApoptosis-inducing factor and colon cancerJ Surg Res200915116317018061616
- PrabhudesaiSGRekhrajSRobertsGDarziAWZiprinPApoptosis and chemo-resistance in colorectal cancerJ Surg Oncol200796778817443738
- ShowalterSLHuangYHWitkiewiczANanoparticulate delivery of diphtheria toxin DNA effectively kills Mesothelin expressing pancreatic cancer cellsCancer Biol Ther200871584159019039293
- GedenSEGardnerRAFabbriniMSOhashiMPhanstiel IvOTeterKLipopolyamine treatment increases the efficacy of intoxication with saporin and an anticancer saporin conjugateFEBS J20072744825483617714513
- ZhaoJMWenYJLiQA promising cancer gene therapy agent based on the matrix protein of vesicular stomatitis virusFASEB J2008224272428018716030
- OrtizRPradosJMelguizoCThe cytotoxic activity of the phage E protein suppress the growth of murine B16 melanomas in vitro and in vivoJ Mol Med20098789991119579018
- WitteAWannerGLubitzWHoltjeJVEffect of phi X174 protein E-mediated lysis on murein composition of Escherichia coliFEMS Microbiol Lett19981641491579675861
- SinhaVRBansalKKaushikRKumriaRTrehanAPoly-epsilon-caprolactone microspheres and nanospheres: an overviewInt J Pharm200427812315158945
- Food and Drug Administration, HHSInternational Conference on Harmonisation; Stability Data Package for Registration Applications in Climatic Zones III and IV; Stability Testing of New Drug Substances and Products; availability. NoticeFed Regist200368657176571814631936
- O’BrienRWWhiteLRElectrophoretic mobility of a spherical colloidal particleJ Chem Soc Faraday Trans1978216071626
- ZhangYYuanJZhangHYNatural resistance to apoptosis correlates with resistance to chemotherapy in colorectal cancer cellsClin Exp Med201110.1007/s10238-011-0146-0145
- PradosJMelguizoCOrtizRRegression of established subcutaneous B16-F10 murine melanoma tumors after gef gene therapy associated with the mitochondrial apoptotic pathwayExp Dermatol20101936337119645856
- DecuzziPPasqualiniRArapWFerrariMIntravascular delivery of particulate systems: does geometry really matter?Pharm Res20092623524318712584
- AriasJLRuizMALopez-ViotaMDelgadoAVPoly(alkylcyanoacrylate) colloidal particles as vehicles for antitumour drug delivery: a comparative studyColloids Surf B Biointerfaces200862647017961994
- SoppimathKSAminabhaviTMKulkarniARRudzinskiWEBiodegradable polymeric nanoparticles as drug delivery devicesJ Control Release20017012011166403
- WangZXBianHBYangJSDeWJiXHAdenovirus-mediated suicide gene therapy under the control of Cox-2 promoter for colorectal cancerCancer Biol Ther200981480148819571664
- KarasawaHMiuraKFujibuchiWDown-regulation of cIAP2 enhances 5-FU sensitivity through the apoptotic pathway in human colon cancer cellsCancer Sci200910090391319302291
- LiBHYangXZLiPDIL-4/Stat6 activities correlate with apoptosis and metastasis in colon cancer cellsBiochem Biophys Res Commun200836955456018294957
- KanyamaHTomitaNYamanoTEnhancement of the anti-tumor effect of 5′-deoxy-5-fluorouridine by transfection of thymidine phosphorylase gene into human colon cancer cellsJpn J Cancer Res19999045445910363585
- KerrDJSeymourLWMarutaFGene therapy for colorectal cancerExpert Opin Biol Ther2003377978812880378
- FillatCCarrioMCascanteASangroBSuicide gene therapy mediated by the herpes simplex virus thymidine kinase gene/ganciclovir system: fifteen years of applicationCurr Gene Ther20033132612553532
- ZhangGLiuTChenYHTissue specific cytotoxicity of colon cancer cells mediated by nanoparticle-delivered suicide gene in vitro and in vivoClin Cancer Res20091520120719118047
- Teimoori-ToolabiLAzadmaneshKAmanzadehAZeinaliSSelective suicide gene therapy of colon cancer exploiting the urokinase plasminogen activator receptor promoterBioDrugs20102413114620199127
- LeeSWLeeYLLeeYJEnhanced antitumor effects by combination gene therapy using MDR1 gene shRNA and HSV1-tk in a xenograft mouse modelCancer Lett2010291838919896764
- PortsmouthDHlavatyJRennerMSuicide genes for cancer therapyMol Aspects Med20072844117306358
- ChenLSWangMOuWCEfficient gene transfer using the human JC virus-like particle that inhibits human colon adenocarcinoma growth in a nude mouse modelGene Ther2010171033104120410928
- YaoQCaoSLiCTurn a diarrhoea toxin into a receptor-mediated therapy for a plethora of CLDN-4-overexpressing cancersBiochem Biophys Res Commun201039841341920599713
- BoulaizHPradosJMelguizoCInhibition of growth and induction of apoptosis in human breast cancer by transfection of gef geneBr J Cancer20038919219812838323
- BoulaizHPradosJMelguizoCTumour malignancy loss and cell differentiation are associated with induction of gef gene in human melanoma cellsBr J Dermatol200815937037818565182
- AguCAKleinRSchwabSThe cytotoxic activity of the bacteriophage lambda-holin protein reduces tumour growth rates in mammary cancer cell xenograft modelsJ Gene Med2006822924116170834
- RamaARPradosJMelguizoCE phage gene transfection enhances sensitivity of lung and colon cancer cells to chemotherapeutic agentsInt J Oncol2010371503151421042719
- MeletASongKBucurOJaganiZGrassianARKhosravi-FarRApoptotic pathways in tumor progression and therapyAdv Exp Med Biol2008615477918437891
- WatanabeTKomuroYKiyomatsuTPrediction of sensitivity of rectal cancer cells in response to preoperative radiotherapy by DNA microarray analysis of gene expression profilesCancer Res2006663370337416585155
- PradosJMelguizoCRamaACombined therapy using suicide gef gene and paclitaxel enhances growth inhibition of multicellular tumour spheroids of A-549 human lung cancer cellsInt J Oncol20083312112718575757
- ShiehGSShiauALYoYTLow-dose etoposide enhances telomerase-dependent adenovirus-mediated cytosine deaminase gene therapy through augmentation of adenoviral infection and transgene expression in a syngeneic bladder tumor modelCancer Res2006669957996617047058
- DeharvengtSRejibaSWackSAprahamianMHajriAEfficient electrogene therapy for pancreatic adenocarcinoma treatment using the bacterial purine nucleoside phosphorylase suicide gene with fludarabineInt J Oncol2007301397140617487360
- HagiwaraSKudoMNakataniTCombination therapy with PEG-IFN-alpha and 5-FU inhibits HepG2 tumour cell growth in nude mice by apoptosis of p53Br J Cancer2007971532153717971768
- AbazaMSAl-SaffarAAl-SawanSAl-AttiyahRc-myc antisense oligonucleotides sensitize human colorectal cancer cells to chemotherapeutic drugsTumour Biol20082928730318802399