Abstract
Background and methods
Problems with the clinical management of prostate cancer include the lack of both specific detection and efficient therapeutic intervention. We report the encapsulation of superparamagnetic iron platinum nanoparticles (SIPPs) and paclitaxel in a mixture of polyethyleneglycolated, fluorescent, and biotin-functionalized phospholipids to create multifunctional SIPP-PTX micelles (SPMs) that were conjugated to an antibody against prostate-specific membrane antigen (PSMA) for the specific targeting, magnetic resonance imaging (MRI), and treatment of human prostate cancer xenografts in mice.
Results
SPMs were 45.4 ± 24.9 nm in diameter and composed of 160.7 ± 22.9 μg/mL iron, 247.0 ± 33.4 μg/mL platinum, and 702.6 ± 206.0 μg/mL paclitaxel. Drug release measurements showed that, at 37°C, half of the paclitaxel was released in 30.2 hours in serum and two times faster in saline. Binding assays suggested that PSMA-targeted SPMs specifically bound to C4-2 human prostate cancer cells in vitro and released paclitaxel into the cells. In vitro, paclitaxel was 2.2 and 1.6 times more cytotoxic than SPMs to C4-2 cells at 24 and 48 hours of incubation, respectively. After 72 hours of incubation, paclitaxel and SPMs were equally cytotoxic. SPMs had MRI transverse relaxivities of 389 ± 15.5 Hz/mM iron, and SIPP micelles with and without drug caused MRI contrast enhancement in vivo.
Conclusion
Only PSMA-targeted SPMs and paclitaxel significantly prevented growth of C4-2 prostate cancer xenografts in nude mice. Furthermore, mice injected with PSMA-targeted SPMs showed significantly more paclitaxel and platinum in tumors, compared with nontargeted SPM-injected and paclitaxel-injected mice.
Introduction
The continued prevalence and resistance to treatment of prostate cancer in the United States suggests that detection and therapeutic methods must be improved in order to combat this disease, especially in the deadly, advanced hormone-refractory stage. As of 2011, prostate cancer remained the most commonly detected male cancer in the United States and the second most common reason for cancer death in men. With over 200,000 newly diagnosed cases, and in excess of 30,000 mortalities, prostate cancer continues to be a major burden on the health and financial security of countless men and families.Citation1,Citation2 After a rapid increase in diagnosed cases in the 1990s, mostly due to prostate-specific antigen testing, the number of newly diagnosed cases has reached a plateau over the past few years. Numerous new therapies have entered clinical trials in recent years, without a meaningful decline in mortality rate,Citation1–Citation6 so innovative therapies continue to be required.
Over the past decade, significant progress has been made with respect to the development of novel nanoparticles designed for the detection or treatment of cancers.Citation7–Citation13 The most common types of nanoparticles used for the detection of cancer were fluorescent, radioactive, or superparamagnetic core nanoparticles which were rendered biocompatible by encapsulation with polymers or phospholipids.Citation7,Citation11,Citation14,Citation15 Similarly, chemotherapeutic drugs were added to the encapsulants with the expectation that more efficacious therapies, with reduced global toxicity, would result.Citation16–Citation18 These two types of nanoparticles are typically targeted to primary and/or metastatic tumors either passively by the enhanced permeability and retention effect, which relies on the leaky vasculature often found in tumors, or through active targeting of specifically overexpressed or highly expressed membrane antigens on the tumor cells or neovasculature. For this latter purpose, antibodies or peptides are often conjugated to the surface of the particles.Citation19–Citation21 The development of nanoparticles that combine these two functions, while highly desirable, has not received as much attention as that directed towards the synthesis of particles with separate functions.
Previously, superparamagnetic iron oxide nanoparticles (SPIONs) have been used as experimental magnetic resonance imaging (MRI) contrast agents to detect cancers.Citation22–Citation25 In fact, two SPION contrast agents, Resovist® and Feridex®, have been approved for use by the Food and Drug Administration.Citation25,Citation26 Although SPIONs produce contrast enhancement in MRI, the signal enhancement is often weaker than that found in radioisotopic imaging modes, such as positron emission tomography.Citation27 Therefore, better MRI contrast agents are urgently needed in order to increase signal enhancement for the noninvasive detection of cancer. We have previously reported that superparamagnetic iron platinum nanoparticles (SIPPs) are superior MRI contrast agents compared with the more commonly used SPIONs.Citation28,Citation29 For this reason, we chose to use SIPPs as our core magnetic particle for MRI. Additionally, we previously showed that these SIPP particles could be encapsulated into micelles using a mixture of PEGylated phospholipids and that these particles could be specifically targeted to prostate cancer cell lines, in vitro, using an anti-prostate-specific membrane antigen (PSMA) antibody.Citation29
Here, we report the synthesis, characterization, and application of specifically-targeted, multifunctional SIPPsCitation28 encapsulated in a mixture of phospholipids and combined with the drug, paclitaxel. These SIPP-PTX micelles (SPMs) were subsequently conjugated to a humanized monoclonal antibody (J591) against PSMA to produce targeted SPMs for the combined MRI and therapy of prostate cancer. We measured the physical, magnetic, binding, and cytotoxic properties of the particles in vitro, and the MRI contrast enhancement, biodistribution, and efficacy in vivo were compared with controls.
Materials and methods
Materials
Iron nitrate nonahydrate (Fe(NO3)3·9H2O), platinum (II) acetylacetonate (Pt(Acac)2), and 1-octadecylamine were purchased from Fisher Scientific (Pittsburgh, PA). The phospholipids:1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino-(polyethyleneglycol)-2000] (DSPE-PEG), DSPE-PEG with biotin conjugated to the head group (biotin-DSPE-PEG), and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[lissamine rhodamine B sulfonyl] (Liss-Rhod) were purchased from Avanti Polar Lipids (Alabaster, AL). The C4-2 human prostate cancer cell line was a generous gift from Marco Bisoffi (UNM Cancer Center, University of New Mexico). All other chemicals and supplies were purchased from common manufacturers.
Synthesis of SIPP cores
One mmol of Fe(NO3)3·9H2O and 1.0 mmol of Pt(Acac)2 were added to 12.5 mmol of 1-octadecylamine in a 25 mL, three-neck, round-bottomed flask fitted with a reflux condenser. The mixture was heated to 330°C (at a rate of 200°C per hour) with 10°C recirculated cooling in the reflux condenser. Refluxing was continued for an additional 45 minutes, at which point the reaction was allowed to cool to room temperature. The resulting black particles were collected in hexane and subjected to repeated washing before resuspending in hexane.
Encapsulation of SIPP cores and paclitaxel
Phospholipid-encapsulated SIPP cores with and without paclitaxel (SPMs and SMs, respectively) were prepared using a thin film method. Then, 0.5 mL of SIPP cores (1.4% solids) in hexane was added to a 20.0 mL glass scintillation vial. A chloroform mixture of (56:1:1 mole ratio) DSPE-PEG, biotin-DSPE-PEG, and Liss-Rhod was then added to the SIPP cores. In the SPM preparations, 0.4 mL of 8 mg/mL paclitaxel in chloroform was also added to the reaction. In the SPM preparations used for confocal binding experiments, 1.0 μg of Oregon Green® 488-paclitaxel conjugate (Invitrogen, Eugene, OR) was added to the mixture. The mixture was further diluted in 0.5 mL of methanol and vortexed. The mixture was evaporated under a nitrogen stream to produce the thin film. Next, 5.0 mL of double-distilled water was heated to 90°C and added to the thin film. Hydration was immediate upon vortexing. The hydrated particles were then extruded at 67°C through an 80 nm Nucleopore track-etch membrane filter using a mini-extruder (Avanti Polar Lipids) to produce micelles of approximately 45 nm. The SPMs and SMs were then purified from SIPP-free micelles, excess phospholipids, and drug by collecting the particles using an LS magnetic column placed in a VarioMACS™ magnetic separator (Miltenyi Biotec, Carlsbad, CA). After the non-magnetic material had passed through the column, 8.0 mL of double-distilled water was added to wash the particles. The column was removed from the magnet and 1.0 mL of sterile saline was used to elute the purified SPMs and SMs. For SPMs and SMs for injections into mice, the eluted particles were applied to smaller (0.5 mL) microcolumns (Miltenyi Biotec) and eluted with 200–300 μL of sterile saline.
Physical characterization of SPMs
Transmission electron microscopy (TEM) was used to determine the size and polydispersity of the particle populations. For SIPPs, a 10 μL drop of particles was applied to a carbon-coated grid and allowed to dry. For SPMs and SMs, a 10 μL drop of the aqueous suspension was applied to a carbon-coated grid and allowed to dry for 10 minutes. Adding 5 μL of 2% uranyl acetate negatively stained the grid. The samples were imaged using Hitachi 7500 TEM with an acceleration voltage of 80 kV. Particle diameters were calculated using ImageJ Software.Citation30 At least 100 particles were counted and the mean Feret’s diameters and standard deviations were calculated. Diameters of the SPMs were additionally measured using dynamic light scattering with a Microtrac Nanotrac™ Ultra device (Microtrac, Largo, FL). Inductively coupled plasma-optical emission spectroscopy (ICP) was used to measure the metal content of each synthesis. Prior to analysis, aliquots of the particles were digested at 180°C with nitric acid and hydrochloric acid in a PDS-6 pressure digestion system (Loftfields Analytical Solutions, Neu Eichenberg, Germany). After cooling, the samples were made up to a known volume, mixed, and centrifuged. Samples were then analyzed using a PerkinElmer Optima 5300DV ICP. The recommended wavelengths for each of the analytes were used, and a blank set of calibration standards were used to establish a three-point calibration curve. Calibration and instrument verification samples were analyzed before and after analyzing the samples, as well as periodically throughout the measurements. Analyte peaks were examined and peak identification and background points were adjusted for optimum recoveries.
Magnetic resonance relaxometry
Increasing concentrations of SPMs (0–400 μM iron) were added to 1% agarose in 2.0 mL self-standing microcentrifuge tubes (Corning, Corning, NY). Samples were imaged on a 4.7 Tesla Bruker Biospin (Billerica, MA) MRI system with Paravision 4.0 software. Samples were imaged with a 512 × 256 matrix, a variable echo time, and repetition time of 10 seconds. T1 measurements were acquired by inversion recovery with 15 interpulse delays. Spin-echo and gradient-echo sequences were used to measure T2 and T2*, respectively. The MRI samples were then analyzed as above to determine the iron concentration using ICP. The relaxation rates, Rn = 1/Tn, were calculated and plotted versus the ICP-determined iron concentration of each sample. The relaxivity (rn) of each sample is given as the slope of the linear regression line in units of s−1 mM−1 (Hz/mM) of iron.
Drug-loading capacity and drug release rates
The amount of paclitaxel loaded into the particles was quantitated using a competitive enzyme-linked immunosorbent assay (Cardax Pharmaceuticals, Aiea, HI), according to the manufacturer’s instructions. Briefly, a 100 μL aliquot of SPMs was added to 200 μL of a 1:1 (vol/vol) acetonitrile:methanol mixture. The mixture was incubated with occasional vortexing. After 30 minutes, the solution was centrifuged at 14,000 rpm for 5 minutes. Ten-fold dilutions of the supernatant were prepared in phosphate-buffered saline containing methanol. Next, 50 μL of each dilution was used in the enzyme-linked immunosorbent assay, in triplicate, to determine the amount of paclitaxel loaded into the particles. For the drug release rate experiments, a 100 μL aliquot of freshly prepared particles (approximately 1.0 mL) was collected as the zero-hour time point. At various subsequent time points, the particles were collected on a magnetic column placed in a VarioMACS™ magnetic separator. The nonmagnetic material flow through (released paclitaxel) was collected for enzyme-linked immunosorbent assay quantitation, and the collected particles were then eluted in the original volume of serum or saline; an aliquot of particles was then taken for the enzyme-linked immunosorbent assay, and the collected particles were incubated at 4°C, 20°C, or 37°C until the next time point. This routine was repeated until the completion of the experiment. As above, particles were added to an equal volume of acetonitrile:methanol prior to the enzyme-linked immunosorbent assay. The amount of paclitaxel in each well was measured based on a baccatin III-protein standard curve.
Antibody conjugation, cell culture, and confocal binding assay
J591 (purchased from Neal Bander, Cornell College of Medicine) and polyclonal goat antirabbit IgG (Sigma, St Louis, MO) were conjugated to streptavidin using a Lightning-Link™ streptavidin conjugation kit (Innova Biosciences, Cambridge, UK). Concentrations of streptavidin, antibodies, and streptavidin-antibody conjugates were quantitated using a NanoDrop™ 2000 spectrophotometer (Wilmington, DE). Briefly, the antibody alone (unconjugated) was measured at 280 nm using the nanoDrop spectrophotometer. After conjugating the streptavidin to the antibody, the conjugated antibody was again measured at 280 nm. Using the molecular weights of antibody and streptavidin and the increase in protein concentration measured at 280 after conjugation, we then calculated the amount of streptavidin conjugated to the antibody. Streptavidin-conjugated antibodies (about 1.0 μg) were then incubated with SPMs or SMs (about 150 μg iron) at 4°C to conjugate the antibodies to the SIPP micelles through the biotin groups of the biotin-DSPE-PEG. Briefly, after conjugating the antibodies to the iron platinum micelles, the magnetic nanoparticles were collected (purified away from unbound antibody) using a magnetic column. The flow through (containing unbound antibody) was then collected. The particles were washed three times with water and then eluted from the column with sterile saline. The amount of unbound antibody in the flow through was quantified using a micro BCA™ protein assay (Thermo Scientific, Rockford, IL) and a BioSpec-mini spectrophotometer (Shimadzu, Columbia, MD) at a wavelength of 562 nm. The difference in the amount of antibody added to the micelles minus the amount of antibody in the flow through was then determined to be the amount of antibody conjugated to the micelles. For binding experiments utilizing confocal microscopy, 20,000 C4-2 human prostate cancer cells in RPMI 1640 medium containing 10% fetal bovine serum and 100 U/mL penicillin-streptomycin solution were seeded onto polylysine-coated cover slips in 6-well polystyrene plates (Corning) and incubated at 37°C with 5% CO2 for 24 hours. The medium was then exchanged with medium containing J591-SPMs (about 4.0 μg iron), IgG-SPMs (about 4.0 μg iron), or phosphate-buffered saline (40 μL). The SPMs for the binding experiments were prepared by doping with green fluorescent paclitaxel (Invitrogen) prior to encapsulation. The cells were incubated with the particles for 10 minutes at 4°C, the media were aspirated off, and 5.0 mL phosphate-buffered saline was added to wash the unbound particles away from the cells. Washing was repeated three times. Cover slips were mounted on slides containing a drop of ProLong® Gold Antifade Reagent with DAPI (Invitrogen). Confocal images were acquired using an Olympus DSU spinning disk confocal microscope in the University of New Mexico and Cancer Center Fluorescence Microscopy Shared Resource.
Cytotoxicity
Five thousand C4–2 cells in 100 μL of RPMI 1640 medium were cultured in 96-well plates. The following day, media were exchanged with 100 μL of media containing the treatment or controls in increasing concentrations of paclitaxel and platinum, determined by measuring the paclitaxel and platinum content of the preparations using the paclitaxel enzyme-linked immunosorbent assay and ICP, respectively (see earlier). A WST-1 cytotoxicity kit was used to quantitate the number of metabolically active cells at 24, 48, or 72 hours. The absorbencies of the samples were normalized to the no-treatment control. Dose-response curves were generated as a function of increasing concentration of treatments/controls. The dose to inhibit the metabolic activity of 50% of the cells was then determined.
Animal experiments
The University of New Mexico International Animal Care and Use Committee approved all experiments involving animals. Athymic nude mice had 3 × 106 C4-2 human prostate cancer cells in 50% (vol/vol) Matrigel® (BD Bioscience, Bedford, MA) subcutaneously injected into the right flank. The mice were monitored, and the length, width, and height of the tumors were measured using a digital caliper. The volumes of the xenografts were determined using the equation V = (4/3)abc, where V is the tumor volume and a, b, and c are half the length, width, and height, respectively. Once xenografts had reached volumes of about 75 mm3, the mice were subjected to MRI and injections of either treatments or controls.
In vivo MRI and injections
Once the xenografts had reached the appropriate volume, the mice were anesthetized using a nose cone that delivered an isoflurane and oxygen mixture and imaged on a 4.7 Tesla Bruker Biospin MRI system with Paravision 4.0 software. Mice were imaged with a 256 × 256 pixel matrix with 156 μ m pixels and a 40 mm field of view. T1 measurements were acquired with a echo time of 14 msec and a rapid acquisition with relaxation enhancement with variable repetition time sequence. T2 measurements were acquired with an echo time of 12 msec and a multi-slice multi-echo sequence. After preinjection imaging, the mice were injected retro-orbitally with 150 μL of of either treatments or controls. Injections contained approximately 200 μg of iron (SPMs and SMs) and 702.6 ± 206 μg of paclitaxel (SPMs). The paclitaxel-only mice were injected with 400 μg of paclitaxel in a total volume of 150 μL of castor oil and saline. Additionally, some mice were not injected as a “no-injection” control group. After the preinjection imaging and subsequent injections, the mice were imaged at various time points ranging from 15 minutes to 24 hours following injection. The T1 and T2 images were analyzed using ImageJ.Citation30 Regions of interest in the tumor and muscle were selected and the mean pixel intensity was recorded. Contrast was then calculated as C = (It − Im/Im), where C is the contrast and It and Im are the pixel intensity in the tumor or muscle, respectively. The contrast was normalized to the preinjection images to produce the contrast (%), calculated as C% = (Ct/Co) × 100, where Ct and Co are the contrast of the tumor at the time point and initial contrast of the tumor in the preinjection image, respectively. Contrast (%) was then plotted versus time after injection.
Biodistribution and therapeutic efficacy
Mice were monitored for 20 days following injection with either treatments or controls. The tumor volumes were measured weekly and mice were monitored for adverse reactions. On day 20 following injection, the mice were euthanized using asphyxiation with carbon dioxide, and the tumor and organs were collected and weighed. Portions of the tumor and organs were then sectioned and again weighed for ICP and analysis of platinum and paclitaxel content, respectively. The amounts of platinum and paclitaxel were calculated as percent of the platinum or paclitaxel in the original injection. The average and standard deviation of platinum and paclitaxel in each group of mice was then calculated and plotted for each tissue or xenograft to determine the biodistribution and percent targeting. Tumor volumes were compared between each of the groups of mice. Efficacy was measured by decreases in tumor volume in the treatment versus control groups.
Results
Size and composition of SPMs
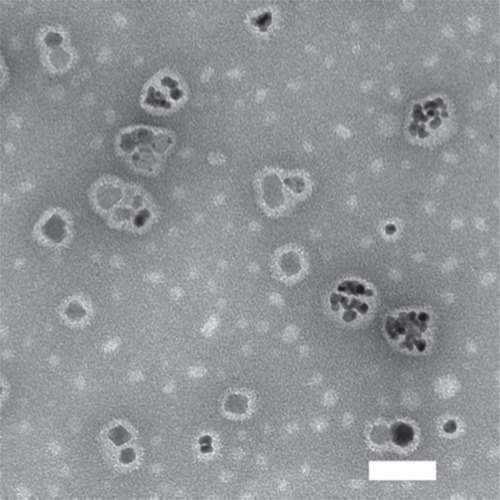
shows a TEM image of the SPMs, which had diameters of 45 ± 25 nm as determined using dynamic light scattering. This large standard deviation was representative of the polydispersity that can be seen in the TEM image (). The SPMs appeared to fall into two morphological groups. One group had multiple, approximately 9 nm diameter SIPPs (in agreement with our earlier dataCitation23,Citation30) encapsulated in the core and were larger in overall diameter (about 50 nm), whereas the other group of particles had smaller diameters of 29 ± 2 nm, and appeared to contain only a single 17 ± 2 nm SIPP core encapsulated in the center. It is important to note that all particles were first purified with a magnetic column; this fact implies that all of the particles in the TEM image possessed a magnetic SIPP core. It is possible that the smaller micelles resulted from a reaction between the FePt alloy and paclitaxel, which generated a crystalline complex between the drug and the alloy. The metal content of the SPMs was determined using ICP. We compared seven separate preparations of SPMs and found that they contained 161 ± 23 μg/mL of iron, 247 ± 33 μg/mL of platinum, and an iron to platinum stoichiometry of 2.3 ± 0.4, suggesting that our method of making SPMs provided good reproducibility.
Figure 1 Transmission electron microscopic image of SPMs.
Notes: SPMs were applied to a carbon-coated grid and allowed to dry. Adding a drop of 2% uranyl acetate negatively stained the grid. The samples were imaged on an Hitachi 7500 transmission electron microscope with an acceleration voltage of 80 kV. The scale bar is 50 nm.
Abbreviation: SPMs, superparamagnetic iron platinum nanoparticles and paclitaxel in a mixture of PEGylated and biotin-functionalized phospholipids.
Magnetic relaxivities of micelles
We next compared the relaxivities of micelles with and without drug using magnetic resonance relaxometry. As expected from our previous characterizations of SIPP cores,Citation28,Citation29 SIPP micelles without paclitaxel (SMs) and SPMs had high transverse relaxivities of r2 = 300 ± 12 and 389 ± 16 Hz/mM iron, respectively, making them superior contrast agents for T2-weighted MRI, compared with SPIONs that generally have transverse relaxivities between 30 and 180 Hz/mM iron.Citation31–Citation34
Paclitaxel loading of micelles
The amount of paclitaxel encapsulated in the SPM preparations (drug-loading capacity) was determined using a paclitaxel enzyme-linked immunosorbent assay. The average drug-loading capacity for seven preparations of particles was 703 ± 206 μg/mL paclitaxel. The high standard deviation suggests that the amount of paclitaxel incorporated into the micelles was subject to some unknown variation, perhaps due to phase fractionation, and that other methods of incorporating the drug into the particles, such as anchoring the drug to the micelles by conjugating a lipid chain to the drug, may be useful in the future.
Paclitaxel release from micelles
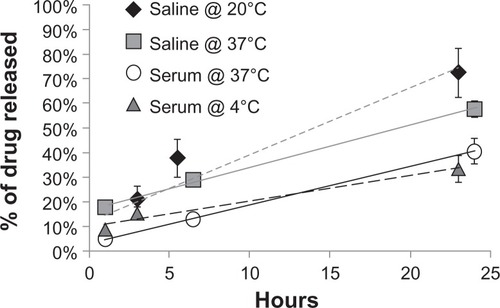
We next aimed to measure how fast the drug was released from the micelles (drug release rate) in different solutions and at different temperatures. shows the drug release rates for the particles in serum and saline at 4°C and 37°C. The time at which half of the paclitaxel had been released from the particles (R0.5) under the different conditions was 19.4 and 14.0 hours for SPMs in saline at 20°C and 37°C, respectively. In contrast, the SPMs in serum at 4°C and 37°C had R0.5 times of 38.9 and 30.2 hours, respectively. It was clear that the drug was released half as fast in serum (R0.5 about 30 hours) than in saline (R0.5 about 14 hours) at 37°C. It has previously been suggested that drugs were released at different rates from nanoparticles in the presence of different serum proteinsCitation35–Citation37 and, therefore, it is possible that serum proteins may have been binding to our SPMs to some extent, causing the drug to be released more slowly due to an increase in the hydrodynamic diameter of the micelles when protein-bound. An interesting future study would be to determine which proteins and to what extent these proteins bind to our PEGylated micelles.
Figure 2 Temperature dependence of drug release rates for SPMs in serum and saline.
Notes: A 100 μL aliquot of freshly prepared particles was collected as the zero-hour time point. At subsequent time points, the particles were magnetically retained on a column and the amount of paclitaxel in the nonmagnetic flow-through and in the magnetic particles was measured using an enzyme-linked immunosorbent assay. The amount of drug release is shown as the percentage of drug released compared with the initial amount of drug loaded into the particles, as measured with enzyme-linked immunosorbent assay immediately after encapsulation. SPMs were incubated in saline at 20°C (diamonds), saline at 37°C (squares), serum at 37°C (circles), or serum at 4°C (triangles).
Abbreviation: SPMs, superparamagnetic iron platinum nanoparticles and paclitaxel in a mixture of PEgylated and biotin-functionalized phospholipids.
Specific binding of antibody-conjugated micelles to prostate tumor cells
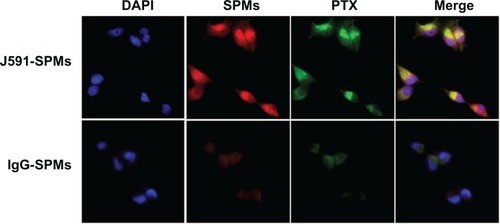
We conjugated J591, raised against the extracellular portion of PSMA, to our SPMs and measured the specific binding of the J591-SPMs to C4-2 human prostate cancer cells that express over one million PSMA receptors on the cell surface.Citation38 shows that, compared with C4-2 cells incubated with nontargeted IgG-SPM micelles, PSMA-targeted J591-SPMs bound specifically to C4-2 cells. Also of importance is that fluorescent paclitaxel can also be seen inside C4-2 cells that were specifically targeted and not in the cells that were incubated with IgG-SPMs, suggesting not only specific delivery of the contrast agent, but also specific intracellular delivery of the drug.
Figure 3 Specific binding of J591-SPMs to C4-2 prostate cancer cells.
Notes: Confocal images of PSMA-targeted, rhodamine red-containing SPMs containing fluorescent paclitaxel (green, top row) and control IgG-SPMs (bottom row) incubated with C4-2 human prostate cancer cells and stained with DAPI. The last column on the right shows the summed images, which display all three colors for the J591-SPMs, and only shows DAPI staining for the Igg-SPMs.
Abbreviations: PSMA, prostate-specific membrane antigen; SPMs, superparamagnetic iron platinum nanoparticles and paclitaxel in a mixture of PEGylated and biotin-functionalized phospholipids.
Cytotoxicity comparison of SPMs versus paclitaxel
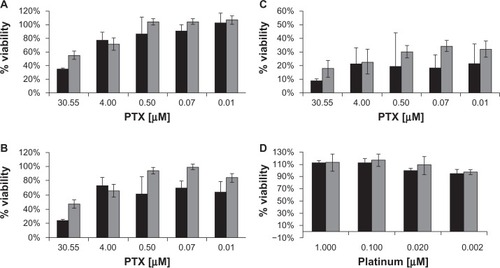
Because the above results showed that SPMs specifically bound to and were taken up by prostate cancer cells, it was of interest to determine if this intracellular delivery of paclitaxel was cytotoxic to C4-2 cells and to compare the cytotoxicity of the SPMs with that of the SMs. shows the cytotoxicity measurements for our SIPP micelles with and without drug over the course of 72 hours. The intracellular delivery of paclitaxel by the SPMs was equally cytotoxic to the cells compared with paclitaxel alone after 72 hours of incubation. However, paclitaxel alone was somewhat more cytotoxic at 24 and 48 hours compared with SPMs; the drug concentration needed to inhibit 50% of the metabolic activity of the cells (IC50) at 24 hours was 22 μM and 50 μM for paclitaxel alone and SPMs, respectively. At 48 hours, the IC50s for paclitaxel and SPMs were 17 μM and 28 μM drug. In comparison, by 72 hours, both paclitaxel and SPMs had the same IC50 of 0.1 μM drug. Thus, in vitro, paclitaxel was 2.2 and 1.6 times more cytotoxic to C4-2 cells at 24 and 48 hours, respectively, but were equally cytotoxic at 72 hours. This difference is most likely due to the fact that it takes about 30 hours for the drug encapsulated in the SPMs to escape (). The SIPP micelles in the absence of drug were not found to be cytotoxic to the C4-2 cell line () using platinum concentrations that were in the same range as those used in the SPM cytotoxicity experiments.
Figure 4 Cytotoxicity measurements of paclitaxel, SPMs, and SMs in C4-2 prostate cancer cells. The graphs show C4-2 cell viability measured with a WST-1 assay after incubation with paclitaxel or SPMs for (A) 24hours, (B) 48hours, and (C) 72hours. Viability after incubation with SPMs (gray bars) and paclitaxel (black bars) is shown as the percentage of viable cells compared with control samples not incubated with particles or drug. (D) Image showing the lack of cytotoxicity when C4-2 human prostate cancer cells are incubated with SIPP micelles without drug (SMs) for 24hours (black bars) and 48hours (gray bars).
Abbreviations: SIPP, superparamagnetic iron platinum nanoparticle; SMs, superparamagnetic iron platinum nanoparticle micelles without drug; SPMs, superparamagnetic iron platinum nanoparticles and paclitaxel in a mixture of PEgylated and biotin-functionalized phospholipids.
SPMs and SIPPs as MRI contrast agents
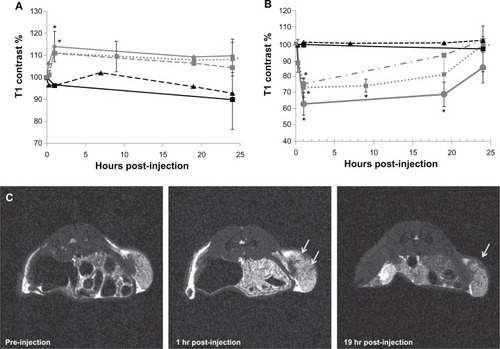
We next produced C4-2 human prostate cancer xenografts in nude mice and performed MR imaging of the tumor both before and after injection of the treatments or controls. shows the T1- and T2-contrast enhancements measured in the tumors as a function of time, with representative images of an SPM-injected tumor. The J591-SPMs and IgG-SPMs, as well as J591-SMs, all showed contrast enhancement in T1-weighted and T2-weighted MR images of the tumors after injection. The T2-contrast was more pronounced and retained in tumors targeted with J591-SPMs and J591-SMs, whereas contrast enhancement in the tumors of mice injected with nontargeted IgG-SPMs was lost more quickly. Significant T2-contrast at 19 hours following injection was only measured with J591-targeted micelles.
Figure 5 In vivo magnetic resonance imaging and contrast measurements of a mouse bearing a C4-2 xenograft. Longitudinal (A) and transverse (B) contrast percentage measured for mice bearing C4-2 xenografts and injected with either J591-SPMs (diamond, solid gray line), J591-SMs (square, dotted gray line), IgG-SPMs (square, dashed gray line), paclitaxel only (triangle, dashed black line), or nothing (square, solid black line). Representative T2-weighted magnetic resonance images of a mouse injected with J591-SPMs are shown in (C).
Notes: The arrows point to areas in the C4-2 xenograft that showed dark contrast enhancement at one hour following injection in the middle frame and an area that still showed contrast enhancement 19hours after injection in the far right frame. *P < 0.05.
Abbreviations: SMs, superparamagnetic iron platinum nanoparticle micelles without drug; SPMs, superparamagnetic iron platinum nanoparticles and paclitaxel in a mixture of PEgylated and biotin-functionalized phospholipids.
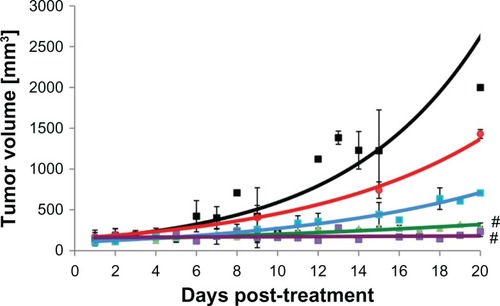
Xenograft growth inhibition by SPMs
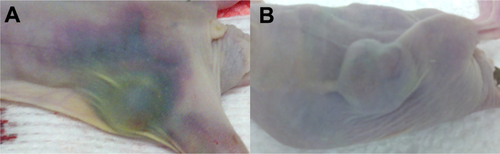
The tumor volumes of the mice were measured over time. The data in show that only the J591-SPMs and paclitaxel were able to prevent growth of tumors in the mice significantly. The nontargeted IgG-SPMs did not significantly reduce the tumor burden in the animals, nor did the J591-SMs. None of the mice in the treatment or control groups showed significant differences in tissue weight or overall body weight over the course of the experiment (Supplemental Figures 1 and 2). In contrast with the J591-SPM injected mice that showed no adverse effects of the treatment, paclitaxel-injected mice suffered from severe hemorrhaging around the tumor (Supplemental Figure 3), two went blind, and one mouse was euthanized early due to neurological impairment.
Figure 6 Tumor volume growth curves for nude mice bearing human C4-2 prostate cancer xenografts treated with various treatments or controls. (A) Black squares, no treatment controls. (B) Red squares, targeted SIPPs without drug, showing no effect on tumor growth. (C) Blue squares, SIPPs containing paclitaxel conjugated to a control Igg antibody, showing no effect on tumor growth. (D) Green triangles, paclitaxel alone, without SIPPs, showing the efficacy of this chemotherapeutic drug by itself. (E) Purple squares, SIPPs containing paclitaxel targeted to PSMA showing that targeting specifically brings the drug to the tumors and prevents tumor growth.
Note: #P < 0.05.
Abbreviations: SIPPs, superparamagnetic iron platinum nanoparticles; PSMA, prostate-specific membrane antigen.
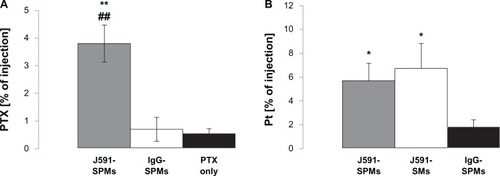
Twenty days after injection of either treatments or controls, the mice were euthanized and the tumors and tissues were collected to quantify the paclitaxel and platinum in their tissues. From the data shown in , it is evident that significantly more paclitaxel and platinum were found in the tumors of mice injected with J591-SPMs compared with mice injected with paclitaxel alone or IgG-SPMs.
Figure 7 Tumor biodistribution of paclitaxel and platinum in mice bearing C4-2 xenografts and injected with treatments or controls. Mice were injected with paclitaxel alone (black bars), J591-SPMs (gray bars), or IgG-SPMs (open bars) and 20days after injection their tumors were collected and paclitaxel was measured as a percentage of the injected dose. Significantly more paclitaxel was measured in the tumors of mice injected with J591-SPMs, compared with nontargeted SPMs and paclitaxel alone (A). Likewise, the biodistribution of platinum from the SIPPs in the mice injected with J591-SPMs (gray bar), J591-SMs (open bar), or IgG-SPMs (black bar) was measured, using ICP, as percent of the injected dose. Again, significantly more platinum was measured in the tumors of mice injected with J591-SPMs, compared with nontargeted SPMs and paclitaxel alone (B).
Notes: *,#P < 0.05 compared with paclitaxel alone or Igg-SPMs, respectively. **,##P < 0.07 compared with paclitaxel alone or Igg-SPMs, respectively.
Abbreviations: SMs, superparamagnetic iron platinum nanoparticle micelles without drug; SPMs, superparamagnetic iron platinum nanoparticles and paclitaxel in a mixture of PEgylated and biotin-functionalized phospholipids.
Discussion
Although SPIONs are the most common type of contrast agents used in contrast agent-enhanced MRI, nanoparticles with increased MRI relaxivities are needed in order to increase the contrast enhancement used for various medical conditions, including cancer. We have previously reported that SIPPs are superior contrast agents for MRI.Citation28,Citation29 The next important step was to determine if SIPPs could be beneficial as in vivo imaging agents and to measure any cytotoxicity of the particles. We have shown both in vitro () and in vivo (Supplemental Figures 1–3) that encapsulated iron platinum particles do not have any significant toxic effects on cells (over 48 hours) or mice (over 20 days). Platinum salts are known to be toxic,Citation39–Citation41 but the platinum in our particles was metallic and contained in a crystal structure alloyed with iron. Upon dissociation from the crystal, the platinum would still be metallic and not in the form of a salt. Platinum metal is chemically inert.
The biodistribution data (Supplemental Figure 4) for our SIPPs suggested that, in addition to being in the tumors, the platinum was primarily retained in the kidney, spleen, liver, and lungs. An important point when considering clinical application is that both platinum and paclitaxel were found in the feces of the mice 20 days following injection. This showed that the platinum and paclitaxel were excreted from the mice in their feces and, although a large percentage of both metal and drug was retained in the body 20 days after injection, the platinum and paclitaxel may eventually be eliminated. It would be of interest to perform biodistribution studies for at least a year to follow complete excretion.
Interestingly, both the J591-targeted micelles and the non-targeted IgG-micelle controls caused contrast enhancement in the tumors (). Nonetheless, it was clear that the tumors of mice injected with J591-SPMs contained a higher concentration of both paclitaxel and platinum (). This, in addition to the fact that the contrast enhancement in tumors of IgG-SPM injected mice was lost more quickly in comparison with mice treated with J591-SPM (), suggests that the enhanced permeability and retention effect is adequate for bringing the contrast agent into the tumor for MR imaging. In contrast, the data in show that mice injected with IgG-SPM did not have a therapeutic response to nontargeted treatment. This suggests that although the concentration of micelles in the tumors was high enough to generate MRI contrast enhancement due to the enhanced permeability and retention effect, it did not lead to accumulation of enough micelles to prevent tumor growth over time. It is also possible that the IgG-SPMs were not endocytosed and, therefore, the drug did not cause cell death because it did not reach the cytoplasm. Paclitaxel is a microtubule stabilizer that inhibits cell division, leading to cell death.Citation42 The confocal images in show that only the J591-SPMs were internalized into the C4-2 cells in vitro, whereas the IgG-SPMs were not. This strongly suggests that, in vivo, only the J591-targeted micelles were able to enter into the cells, where the drug was released.
In order to achieve MRI contrast, the particles must only be attached to the vasculature and/or cells of the tumor, but in order to cause cell death, the particles must actually have been taken up by the cells. This could explain some of the discrepancies seen in the literature relating to the question of whether or not active targeting works.Citation43 Our data suggest that an important distinction should be made between whether the nanoparticles generated tumor contrast or were actually therapeutic. Importantly, growth of the human prostate cancer xenografts in the nude mice was only inhibited in mice treated with paclitaxel or J591-SPMs. In contrast with the mice injected with paclitaxel (Supplemental Figures 1–3), the mice injected with J591-SPMs did not suffer from any noticeable side effects or complications over the 20 days that the mice were followed after injection.
J591, a monoclonal antibody raised against PSMA, has been used in numerous clinical trials for prostate cancer.Citation44–Citation49 Our data show that J591 conjugated to paclitaxel-loaded micelles specifically target the micelles to human prostate cancer cells and prevent tumor growth in a PSMA-dependent manner. PSMA is highly expressed in almost all primary and metastatic prostate cancer tumors.Citation50 Our data suggest that drug-loaded micelles targeted to PSMA could successfully treat prostate cancer tumors while possibly reducing the side effects commonly seen when using chemotherapeutics alone. Moreover, PSMA has been shown to be expressed in the neovasculature of almost all solid tumors, but not in healthy vasculature,Citation50 supporting the idea that PSMA-targeting may be beneficial not only as a prostate cancer targeting motif but also as a general cancer target. In addition to the specific, targeted killing of PSMA-positive prostate cancer cells, ours is also the first report of encapsulating SIPPs with a chemotherapeutic drug in a mixture of functionalized and fluorescent phospholipids to produce multifunctional, iron platinum, stealth immunomicelles for specific MRI and treatment of cancer.
Acknowledgements
This research was supported in part by funding from the National Institutes of Health (5RO1CA123194 to LOS) and the NCI New Mexico Cancer Nanotechnology Training Center (NIH R25CA153825 supporting RMT). Portions of this work were performed at the Center for Integrated Nano-technologies, a US Department of Energy, Office of Basic Energy Sciences, user facility. Sandia National Laboratories is a multiprogram laboratory operated by The Sandia Corporation, a Lockheed-Martin Company, for the US Department of Energy under Contract No DE-AC04-94AL85000. MRI was performed at the Brain Imaging Center, Albuquerque, NM. TEM images were generated at the UNM Electron Microscopy Facility. Some experiments used the facilities provided by the Keck-UNM Genomics Resource, a facility supported by grants from the WM Keck Foundation, the State of New Mexico, and the UNM Cancer Research and Treatment Center. We gratefully acknowledge the assistance of Medhi Ali of the Earth and Planetary Sciences Department at UNM, for the ICP-OES analyses, of Stephen Jett from the UNM Electron Microscopy Facility, and of Dale Huber from the Center for Integrated Nanotechnologies.
Disclosure
The authors report no conflicts of interest in this work.
Supplementary figures
Figure S1 Body weights of mice bearing C4-2 xenografts that were injected with treatments or controls.
Notes: Mice bearing C4-2 human prostate cancer cell xenografts were injected with the treatments or controls and weighed on the day of injection and on the day of euthanasia. None of the mice showed significant differences in loss of body weight over the 20 days.
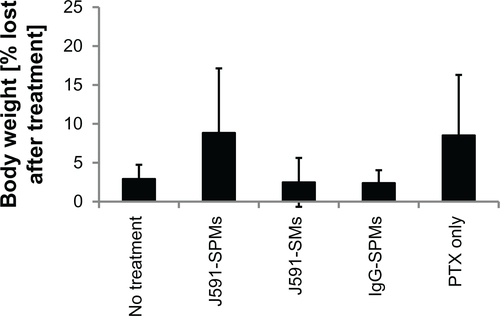
Figure S2 Tissue weights of mice bearing C4-2 xenografts that were injected with treatments or controls.
Notes: Mice bearing C4-2 xenografts were injected with J591-SPMs (white bars), J591-SMs (gray bars), IgG-SPMs (blue bars), PTX alone (beige bars), or nothing (black bars) and 20 days later their tissues and tumors were collected and weighed. None of the mice showed significant differences in tissue weights post-mortem, although the mice injected with PTX alone, J591-SPMs, and IgG-SPMs showed decreased tumor mass compared to mice injected with SIPP-micelles without drug and mice that were not injected. *corresponds to significance of P < 0.05.
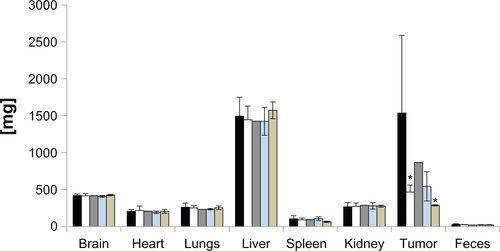
Figure S3 Hematoma around the tumors of mice injected with PTX alone. Representative images are shown for mice injected with PTX alone (A) and J591-SPMs (B) taken post-mortem. The mice injected with PTX alone clearly had extensive hematoma around the tumors and all along the right flanks, whereas the mice injected with J591-SPMs did not have this side effect, suggesting that in addition to targeting the drug, encapsulation of the drug also reduced side effects.
References
- SiegelRNaishadhamDJemalACancer statistics, 2012CA Cancer J Clin2012621102922237781
- MoulJWDawsonNQuality of life associated with treatment of castration-resistant prostate cancer: a review of the literatureCancer Invest201230111222236184
- BeltranHBeerTMCarducciMANew therapies for castration-resistant prostate cancer: efficacy and safetyEur Urol201160227929021592649
- DayyaniFGallickGELogothetisCJCornPGNovel therapies for metastatic castrate-resistant prostate cancerJ Natl Cancer Inst2011103221665167521917607
- GomellaLGGelpiFKellyWKNew treatment options for castrate-resistant prostate cancer: a urology perspectiveCan J Urol20111845767577721854708
- PetrylakDPCurrent clinical trials in castrate-resistant prostate cancerCurr Urol Rep201112317317921533748
- AbeylathSCGantaSIyerAKAmijiMCombinatorial-designed multifunctional polymeric nanosystems for tumor-targeted therapeutic deliveryAcc Chem Res201144101009101721761902
- HoDSunXSunSMonodisperse magnetic nanoparticles for theranostic applicationsAcc Chem Res2011441087588221661754
- KievitFMZhangMSurface engineering of iron oxide nanoparticles for targeted cancer therapyAcc Chem Res2011441085386221528865
- KooHHuhMSSunICIn vivo targeted delivery of nanoparticles for theranosisAcc Chem Res201144101018102821851104
- NamikiYFuchigamiTTadaNNanomedicine for cancer: lipid-based nanostructures for drug delivery and monitoringAcc Chem Res201144101080109321786832
- ShiJXiaoZKamalyNFarokhzadOCSelf-assembled targeted nanoparticles: evolution of technologies and bench to bedside translationAcc Chem Res201144101123113421692448
- YooDLeeJHShinTHCheonJTheranostic magnetic nanoparticlesAcc Chem Res2011441086387421823593
- PuriABlumenthalRPolymeric lipid assemblies as novel theranostic toolsAcc Chem Res201144101071107921919465
- SawantRRTorchilinVPMultifunctionality of lipid-core micelles for drug delivery and tumour targetingMol Membr Biol201027723224620929339
- AriasJLClaresBMoralesMEGallardoVRuizMALipid-based drug delivery systems for cancer treatmentCurr Drug Targets20111281151116521443475
- Estella-Hermoso de MendozaACampaneroMAMollinedoFBlanco-PrietoMJLipid nanomedicines for anticancer drug therapyJ Biomed Nanotechnol20095432334320055079
- FlorenceATJaniPUNovel oral drug formulations. Their potential in modulating adverse effectsDrug Saf19941032332668043223
- PengXHQianXMaoHTargeted magnetic iron oxide nanoparticles for tumor imaging and therapyInt J Nanomedicine20083331132118990940
- AkhterSAhmadZSinghACancer targeted metallic nanoparticle: targeting overview, recent advancement and toxicity concernCurr Pharm Des201117181834185021568874
- HirsjarviSPassiraniCBenoitJPPassive and active tumour targeting with nanocarriersCurr Drug Discov Technol20118318819621513482
- ZhangCLiuTGaoJSuYShiCRecent development and application of magnetic nanoparticles for cell labeling and imagingMini Rev Med Chem201010319320220408802
- LinMMKim doKEl HajAJDobsonJDevelopment of superparamagnetic iron oxide nanoparticles (SPIONS) for translation to clinical applicationsIEEE Trans Nanobioscience20087429830519203873
- ShubayevVIPisanicTR2ndJinSMagnetic nanoparticles for theragnosticsAdv Drug Deliv Rev200961646747719389434
- MailanderVLandfesterKInteraction of nanoparticles with cellsBiomacromolecules20091092379240019637907
- GandhiSNBrownMAWongJGAguirreDASirlinCBMR contrast agents for liver imaging: what, when, howRadiographics20062661621163617102040
- TsukadaHApplication of pre-clinical PET imaging for drug developmentNihon Shinkei Seishin Yakurigaku Zasshi2011315–6231237 Japanese.22256612
- TaylorRMHuberDLMonsonTCEschVSillerudLOStructural and magnetic characterization of superparamagnetic iron platinum nanoparticle contrast agents for magnetic resonance imagingJ Vac Sci Technol B2012302
- TaylorRMHuberDLMonsonTCAliAMBisoffiMSillerudLOMultifunctional iron platinum stealth immunomicelles: targeted detection of human prostate cancer cells using both fluorescence and magnetic resonance imagingJ Nanopart Res201113104717472922121333
- RasbandWSImageJ. US National Institutes of Health, Bethesda, Maryland, USA, 1997–2009 Available from: http//rsbinfo/nih/gov/ij/. Accessed July 4, 2012.
- GossuinYGillisPHocqAVuongQLRochAMagnetic resonance relaxation properties of superparamagnetic particlesWiley Interdiscip Rev Nanomed Nanobiotechnol20091329931020049798
- HuangJZhongXWangLYangLMaoHImproving the magnetic resonance imaging contrast and detection methods with engineered magnetic nanoparticlesTheranostics2012218610222272222
- LeeJHHuhYMJunYWArtificially engineered magnetic nanoparticles for ultra-sensitive molecular imagingNat Med2007131959917187073
- CarrollMRHuffstetlerPPMilesWCThe effect of polymer coatings on proton transverse relaxivities of aqueous suspensions of magnetic nanoparticlesNanotechnology2011223232570221772073
- DieziTABaeYKwonGSEnhanced stability of PEG-block-poly(N-hexyl stearate l-aspartamide) micelles in the presence of serum proteinsMol Pharm2010741355136020575526
- KastantinMMissirlisDBlackMAnanthanarayananBPetersDTirrellMThermodynamic and kinetic stability of DSPE-PEG(2000) micelles in the presence of bovine serum albuminJ Phys Chem B201011439126321264020828210
- LuJOwenSCShoichetMSStability of self-assembled polymeric micelles in serumMacromolecules201144156002600821818161
- TaylorRMSevernsVBrownDCBisoffiMSillerudLOProstate cancer targeting motifs: Expression of alpha(nu) beta(3), neurotensin receptor 1, prostate specific membrane antigen, and prostate stem cell antigen in human prostate cancer cell lines and xenograftsProstate201272552353221748756
- CouraudSPlanusCRioufolCMornexFPlatinum salts hypersensitivityRev Pneumol Clin20086412026 French.18603175
- CristaudoASeraFSeverinoVDe RoccoMDi LellaEPicardoMOccupational hypersensitivity to metal salts, including platinum, in the secondary industryAllergy200560215916415647035
- TouraineFSainte LaudyJBoumedieneAInvestigation of allergic reactions to platinum saltsRev Mal Respir2006235 Pt 1458462 French.17314746
- ZhaoJKimJEReedELiQQMolecular mechanism of antitumor activity of taxanes in lung cancerInt J Oncol200527124725615942666
- NieSUnderstanding and overcoming major barriers in cancer nanomedicineNanomedicine (Lond)20105452352820528447
- AkhtarNHPailOSaranATyrellLTagawaSTProstate-specific membrane antigen-based therapeuticsAdv Urol2012201297382021811498
- BanderNHTrabulsiEJKostakogluLTargeting metastatic prostate cancer with radiolabeled monoclonal antibody J591 to the extracellular domain of prostate specific membrane antigenJ Urol200317051717172114532761
- BayesMRabassedaXProusJRGateways to clinical trialsMethods Find Exp Clin Pharmacol200729642743717922073
- NanusDMMilowskyMIKostakogluLClinical use of monoclonal antibody HuJ591 therapy: targeting prostate specific membrane antigenJ Urol.20031706 Pt 2S84S8814610416
- RiceSLRoneyCADaumarPLewisJSThe next generation of positron emission tomography radiopharmaceuticals in oncologySemin Nucl Med201141426528221624561
- TagawaSTBeltranHVallabhajosulaSAnti-prostate-specific membrane antigen-based radioimmunotherapy for prostate cancerCancer20101164 Suppl1075108320127956
- ChangSSO’KeefeDSBacichDJReuterVEHestonWDGaudinPBProstate-specific membrane antigen is produced in tumor-associated neovasculatureClin Cancer Res19995102674268110537328