Abstract
Background
Dapsone is described as being active against Mycobacterium leprae, hence its role in the treatment of leprosy and related pathologies. Despite its therapeutic potential, the low solubility of dapsone in water results in low bioavailability and high microbial resistance. Nanoemulsions are pharmaceutical delivery systems derived from micellar solutions with a good capacity for improving absorption. The aim of this work was to develop and compare the permeability of a series of dapsone nanoemulsions in Caco-2 cell culture against that of effective permeability in the human body simulated using Gastroplus™ software.
Methods and results
The release profiles of the dapsone nanoemulsions using different combinations of surfactants and cosolvent showed a higher dissolution rate in simulated gastric and enteric fluid than did the dispersed dapsone powder. The drug release kinetics were consistent with a Higuchi model.
Conclusion
This comparison of dapsone permeability in Caco-2 cells with effective permeability in the human body simulated by Gastroplus showed a good correlation and indicates potential improvement in the biodisponibility of dapsone using this new system.
Introduction
Leprosy, also known as Hansen’s disease, is a worldwide health problem, varying from tuberculoid to lepromatous leprosy (ie, paucibacillary to multibacillary disease) according to the host immune response. It is caused by Mycobacterium leprae, and affects the skin, eyes, and nerves, leading to skin lesions, eye pain, loss of vision, weakness, and numbness.Citation1 The final diagnosis is based on a combination of findings on skin biopsy, smear, and physical examination. The treatment options differ according to the clinical manifestations.Citation1 Leprosy can be associated with type 1 (reversal) and type 2 (erythema nodosum leprosum) immunologic reactions that may occur at any time before, during, or after the start of treatment.Citation1 During this process, this disease can worsen dramatically, and antibacterial therapy should continue during these periods.Citation1,Citation2
Since 1947, dapsone (4,4-diaminodiphenyl sulfone) has been known to be a useful drug in the treatment of leprosy.Citation2 Its mechanism of action is via competitive inhibition of M. leprae dihydropteroate synthase, thus inhibiting the synthesis of folic acid.Citation3,Citation4 Dapsone () is classified as a class II drug according to the Biopharmaceutics Classification System, and has high permeability and low solubility in water (log P = 0.97).Citation5 Thus, despite its therapeutic potential, the low solubility of dapsone in water results in low bioavailability and microbial resistance.Citation5 A viable alternative for dose reduction and therefore limitation of side effects, never described before in the literature for dapsone, is the use of an intestinal permeation enhancer system capable of maintaining the drug in a soluble state after administration.
Figure 1 Structure of dapsone.
Nanoemulsions are homogenous systems with high solubilization capacity, thermodynamic stability over a wide range of pH and ionic environments, low viscosity, and can be formed spontaneously by simple mixture of various components.Citation6,Citation7 A nanoemulsion formulation of dapsone could improve its bioavailability and stability, and reduce the dosage necessary and the side effects of this drug, thereby representing a novel alternative in the treatment of leprosy. The aim of this study was to develop and evaluate the potential for better bioavailability of dapsone when the drug is incorporated into a nanoemulsion system using in vitro and in silico approaches.
Materials and methods
Materials
Dapsone from G Amphray Laboratories (Mumbai, India) was used in the production of all formulations, and dapsone USP standard lot 02/572 A (Rockville, MD) was used in all quantitative analyses. The isopropyl myristate, propylene glycol, ethanol, polyoxyethylene sorbitan monooleate-Tween® 80, 40 and 20, and sorbitan monooleate-Span® 80 and 20 (Sigma Chemical Company, St Louis, MO) used in preparation of the formulation were all of pharmaceutical grade. Methanol and hydrochloric acid 37% were of chromatography grade (Tedia Company Inc, Fairfield, OH). Potassium phosphate monobasic, sodium phosphate dihydrate, and sodium hydroxide (Vetec Chemicals, Rio de Janeiro, Brazil), and fluorescein (LGC Biotechnology, Sao Paolo, Brazil) of analytical grade were also used. Glucose, sodium bicarbonate, fetal bovine serum, nonessential amino acids, penicillin, streptomycin, and L-glutamine were purchased from Sigma Chemical Company. Water was obtained from a Milli-Q Gradient A-10 water purification system (Millipore, Bedford, MA).
Nanoemulsion preparation
Oral nanoemulsion systems containing dapsone were prepared using isopropyl myristate as oil phase and propylene glycol and ethanol as cosurfactant, as well as the surfactants Span 80 or 20, combined with Tween 80, 40 or 20, and, lastly, water. The formulation was prepared according to the method reported by Nandi et al.Citation8
A pseudoternary phase diagram was constructed to determine the exact proportion of each component needed in the formulation to obtain the best parameters for the ideal nanoemulsion.Citation9 A Tween 80 and Span 80 surfactant mixture was used at a fixed ratio of 1:1 in the first vertex. The other vertex represented the proportion of oil and cosolvent in a 8:1 mass ratio of isopropyl myristate and propylene glycol, respectively, while the third vertex represented water added in 10 to 10 μL using an automatic micropipette in the titration process.Citation8 This process was carried out to evaluate the maximum amount of water that could be incorporated into the system containing the mixture of surfactants, oil, and cosolvent.
A pseudoternary phase diagram was then developed using the following proportions of oil phase + cosolvent and surfactants: 90:10, 80:20, 70:30, 60:40, 50:50, 40:60, 30:70, 20:80, and 10:90. Water was titrated in each of these proportions and, finally, the proportions were recalculated.
We then evaluated the solubility of dapsone in the finished formulation and in the oil phase of the nanoemulsion. Dapsone concentrations of 1.5, 2.0, 2.5, 3.0, 3.5, and 4.0% w/w were tested. The dapsone solubility studies were performed with vigorous stirring for 24 hours followed by filtration through a 0.45 μm filter. Determination of the total amount of solubilized dapsone was done by ultraviolet spectroscopy at 295 nm.
Characterization of dapsone nanoemulsion
The oral dapsone nanoemulsion was characterized by droplet size distribution, refractive index, conductivity, and drug content. Determination of the droplet size was performed by dynamic light scattering using a Horiba LB-550 DLS analyzer (Kyoto, Japan), with a detection angle of 90°, 100 scans over two minutes for each sample, a refractive index adjusted to 1.330, and a temperature of 22°C.Citation10 The refractive index was determined using an Abbe refractometer (Model AR-001, AFAB Enterprises, Eustis, FL), calibrated with distilled water at 25°C. This parameter is important for assessment of the stability of nanoemulsions, because it is related to the optical clarity of these systems and helps to determine the type of nanoemulsion as w/o or o/w.Citation11
The conductivity of the oral dapsone nanoemulsion was measured using a FE30 FiveEasy Mettler Toledo conductivity meter (Bedfordshire, UK) calibrated with a NaCl solution at 5.0 mg/L.
A stress stability study of the oral dapsone nanoemulsion was conducted in a climatic chamber (Nova Ética 420/ CLDTS 300 model, São Paulo, Brazil) at 40°C ± 2°C and 75% ± 5% relative humidity over 3 months.Citation12 Samples were collected on days 15, 30, 60, and 90. At each interval, the formulation was characterized for droplet size distribution, refractive index, drug content, and conductivity. Precipitation was monitored by visual inspection. A sample of each formulation was stored under shelf conditions, ie, 25°C ± 2°C and 75% ± 5% humidity, for comparison with samples stored under stress temperature and humidity conditions.
High-performance liquid chromatography analysis
Quantification of the amount of dapsone in the formulations was carried out using high-performance liquid chromatography (HPLC) coupled with a photodiode array detector (Elite Labchrom model, Merck-Hitachi, Darmstadt, Germany), according to a method described elsewhere,Citation13 with some modifications. The dapsone assay was carried out on a Kromasil C18 column (150 mm × 4.6 mm, 5 μm). The mobile phase consisted of a mixture of methanol and 30 mM phosphate buffer pH 5.9 (30:70, v/v), and was isocratically delivered at 1.0 mL per minute. The column temperature was maintained at 25°C and the injection volume was 100 μL. Quantification of dapsone in the samples was based on the ultraviolet absorption at 295 nm using a six-level standard curve prepared from the stock solution in simulated gastric fluid and simulated intestinal fluid, with diluents at concentrations of 2.5, 5, 10, 15, 20, and 25 μg/mL for both conditions. Stock dapsone solutions were prepared by transferring 25 mg of dapsone USP standard to a 100 mL volumetric flask. The required volume was completed using methanol, reaching a final concentration of 250 μg/mL.
Dissolution testing of dapsone capsules
Dissolution tests were carried out with the nanoemulsion in capsules in order to standardize the dose and facilitate the assay. The nanoemulsion was placed in hard gelatin capsules (number 00), corresponding to a total volume of 0.95 mL, thereby simulating a commercial formulation. The maximum quantity of dapsone nanoemulsion incorporated in this capsule was 800 mg, equivalent to 16 mg of the drug considering the formulation containing 2.0% (w/w). The capsules were weighed on an analytical balance before and after being filled with the nanoemulsion. The dissolution tests were performed in USP apparatus I using 900 mL of simulated gastric fluid and simulated intestinal fluid at 100 rpm.Citation14 During the study, the temperature was maintained at 37°C ± 0.5°C. Each test was performed six times. A capsule containing dapsone nanoemulsion was added to each vessel and aliquots of 5 mL were collected at intervals of 15, 30, 45, 60, and 120 minutes, with replacement of the medium on each occasion.
In order to evaluate the effect of the surfactant on the solubility and release of the active pharmaceutical ingredient, dapsone was encapsulated at the same concentration as in formulation II with addition of Tween 80 and Span 80 to the dissolution medium. The dissolution test was performed following the same conditions described for the nanoemulsion, and aliquots were collected at intervals of 15 and 120 minutes. The dissolution aliquots were filtered through a 0.45 μm filter and analyzed by HPLC.
Evaluation of release kinetics
The data obtained in the dissolution test were fitted to four different equations to describe the drug release kinetics, ie, zero order, first order, Higuchi, and Hixon-Crowell.Citation14,Citation15 The mathematical model that best expressed the kinetic release profile was selected based on the highest coefficient of determination (R²), obtained from linear regression analysis using the Statistica 7 software package (Statsoft Company, Tulsa, OK).Citation15
Permeability testing conditions
A Caco-2 human intestinal cell line (HTB-37; American Type Culture Collection, Manassas, VA) was obtained from the cell bank at the Universidade Federal do Rio de Janeiro. The cells were routinely grown in 75 cm2 culture flasks with complete Dulbecco’s modified Eagle’s medium (containing 4.5 g/L glucose, 3.7 g/L NaHCO3, supplemented with 10% fetal bovine serum, 1% nonessential amino acids, penicillin 100 U/mL, streptomycin 100 μg/mL, and glutamine 4 mM). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2. The medium was changed every 2 days until confluence of the culture. At confluence, the cells were washed with phosphate-buffered saline, without Ca2+ and Mg2+, and detached from the flasks by trypsin buffer (0.25% in phosphate-buffered saline containing 0.2% ethylenediamine tetra-acetic acid). The cells were then resuspended in complete Dulbecco’s modified Eagle’s medium, and 5 × 104 cells were seeded on culture inserts fitted into 24-well culture plates (Transwell, Costar, Cambridge, MA). After 14–18 days in culture, the monolayers were used for the permeability assays.Citation16
The integrity of the monolayers was determined using an FEI Morgagni microscope operating at 80 kV (Hillsboro, OR), as well as by measuring transepithelial electrical resistance and fluorescein passage. Transepithelial electrical resistance values, expressed in Ω. cm2, were obtained using Millicell ERS-2 apparatus (Millipore). During the growth and differentiation period, the cell monolayer status was evaluated on days 7 and 14 after seeding. The cell monolayer was considered to be fully formed when stable values were ≥300 Ω · cm2.
During the permeability experiments, transepithelial electrical resistance was measured at different time points, including at the start and end of the assays. Fluorescein permeation was also used to assess the integrity of the monolayer. At the end of the permeability experiments, 200 μL of fluorescein 1 mg/mL in phosphate-buffered saline was added to each apical chamber and 400 μL of phosphate-buffered saline was added to each basolateral chamber.Citation17–Citation19 After one hour of incubation at 37°C, the fluorescein content in the basolateral medium was determined by HPLC, as previously described.
Permeability study of dapsone nanoemulsion
All transport studies were performed at 37°C in an atmosphere of 5% CO2 in phosphate-buffered saline (pH 7.2) as the transport medium. Before the permeability assay, the cell monolayers were washed using the transport medium and preincubated for 30 minutes. Dapsone nanoemulsion or phosphate-buffered saline solution (200 μL, donor solution) at final concentrations of 14 μg/mL in phosphate-buffered saline were added to the apical chamber. The concentration of the dapsone nanoemulsion was defined according to the cytotoxicity tests. The basolateral chamber contained only phosphate-buffered saline as the accept solution. All permeability tests were performed in the apical to basolateral direction. Transport was monitored for a period of 240 minutes, after which time 100–150 μL samples were removed from the basolateral chamber for HPLC analysis and replaced with an equal volume of phosphate-buffered saline.Citation17,Citation20
To calculate the permeability of the dapsone nanoemulsion in Caco-2 cells, the apparent permeability coefficient (Papp) was obtained from the following equation: Papp = (V/A × C) × (dC/dt), where dC/dt is the permeate rate (the amount of drug transported per unit time), V is the volume in the receptor chamber, A is the surface area of the filter, and C is the initial drug concentration in the donor chamber.
In silico studies
The GastroplusTM version 7.0 (Simulations Plus Inc, Lancaster, CA) was used to simulate the effective permeability (Peff) of dapsone and output pharmacokinetic parameters in the human body. The program has three tabs used for data input; the first is a compound tab, followed by a physiology tab, and finally a pharmacokinetic tab. The compound tab inputs the basic physicochemical data for dapsone, such as solubility, pKa, and dose, as derived from the literature. The input parameter required for Peff simulation was experimentally determined based on permeability studies using the Caco-2 cell line.Citation21 In the physiology tab, the default values for transit time were selected for each compartment, with the fasted option checked. Pharmacokinetic parameters, such as clearance and distribution volume, were used in the pharmacokinetic tab for simulation by Gastroplus. These values were taken from the literature.Citation22,Citation23
Statistical analysis
Statistical analysis was performed with the one-way analysis of variance test (Tukey’s multiple comparisons) using Graph- Pad Prism® 5.0 software (La Jolla, CA). Linear regression and others statistics were conducted using Statistica 7.
Results and discussion
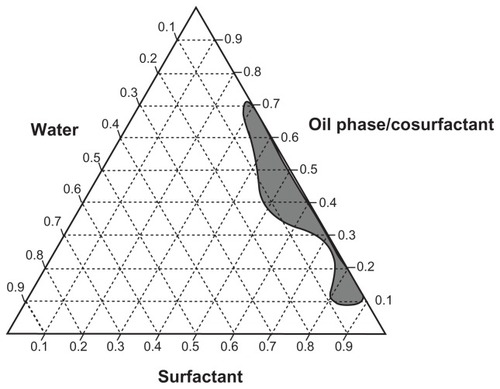
Different combinations of surfactants and cosurfactants were evaluated to identify a stable w/o nanoemulsion. These nanoemulsions can improve the biodisponibility of dapsone and the in vivo stability of this drug by in vivo phase inversion to an o/w nanoemulsion of dapsone due to the aqueous environment of the gastrointestinal tract.Citation24,Citation25 It was not possible to obtain stable nanoemulsions when constructing the pseudoternary phase diagrams using combinations of Tween 40/Span 20 and Tween 20/Span 20 as surfactants, ethanol or propylene glycol as the cosurfactant, or isopropyl myristate as oil phase. However, it was possible to establish several stable combinations when Span 80 and Tween 80 were used with propylene glycol as the cosurfactant and isopropyl myristate as oil phase (). Use of propylene glycol as cosurfactant has been shown to be promising in oral pharmaceutical formulations. Yang et al used this excipient in the preparation of a paclitaxel nanoemulsion and Sharma et al used it in oral formulations of insulin.Citation26,Citation27 Thus, based on the pseudoternary phase diagram (), a stable formulation able to incorporate more water was selected for the characterization tests ().Citation9 Dapsone was efficiently incorporated in the nanoemulsion formulation selected () in two ways, ie, dissolved in oil phase (I and III) and in the finished formulation (II and IV) at concentrations of 2.0% (I and II) and 2.5% (III and IV) w/w (). characterizes the nanoemulsions at time zero and after 90 days of storage under stress conditions. The parameters evaluated were droplet size, polydispersity index, refractive index, conductivity, and dapsone content (). The nanometer scale droplets in these formulations were found to have a low polydispersity index. Conductivity was 0 μS/cm for all the formulations studied at time zero and after storage, indicating that all the nanoemulsions produced were of the w/o type. The droplet size below 100 nm and surfactant concentration used in the nanoemulsions produced allowed prediction of in vivo phase inversion and formation of a stable o/w nanoemulsion.Citation24 The results of the stress stability studies showed that the nanoscale dimensions were maintained with uniform emulsion droplets and a narrow size distribution in all the study formulations (I–IV) during the 3 months of testing. Formulations III and IV showed an increase in droplet size, and formulation I showed a slight reduction. Moreover, formulations I to IV in the stress stability studies were considered to be stable, and formulations I and II did not show drug precipitation during the shelf-life studies.
Table 1 Basic composition of the formulations prepared according to the pseudoternary phase diagram
Table 2 Nanoemulsion formulations classified as I to IV, prepared solubilizing dapsone in the oil phase or in the finished formulation
Table 3 Nanoemulsion characterization at time zero and after 90 days of storage under stress conditions
Figure 2 Pseudoternary phase diagram using as components: isopropyl myristate (oil phase), propylene glycol (cosurfactant), Tween® 80 + Span® 80 (surfactants), and water.
Note: The area marked in dark gray represents the area of nanoemulsion formation.
Like dapsone, paclitaxel is a chemotherapeutic agent with high lipophilicity and low water solubility, and has a high potential for bioavailability problems.Citation28 Paclitaxel nanoemulsions are described in the scientific literature as being systems capable of increasing the solubility of the active drug.Citation29,Citation30 Although an o/w type of paclitaxel nanoemulsion is more common, researchers have developed a paclitaxel nanoemulsion of the w/o type, which has been shown to have increased clinical efficacy.Citation30 This is probably due to in vivo phase inversion, as expected for the I and II dapsone nanoemulsions. This hypothesis reinforces the use of a nanoemulsion system to modulate the bioavailability of dapsone.
Formulations I and II showed the best results in the stability studies. However, formulation II was chosen for the permeability and kinetic release studies because of its capacity to incorporate a larger volume of water (), thereby avoiding precipitation of dapsone and in vivo formation of an o/w nanoemulsion.Citation24
The drug release profile from formulation II was studied in the different media. Using simulated gastric fluid, more than 90% of drug release occurred within the first 15 minutes, with 91.5% of the drug similarly released in 15 minutes using simulated intestinal fluid. Dapsone appeared to be maintained in a solubilized form in the dissolution media when the nanoemulsion was tested, and was confirmed by a mean droplet size of 84.5 ± 12 nm, a mean maximum size of 98.0 ± 26 nm measured with phase inversion, and by conductivity measurement (1.12 μS/cm).
In order to evaluate the enhancement of dissolution of dapsone in nanoemulsion by the surfactant effect, a control study based on a dissolution test of the dapsone contained in the capsules was performed. The total amount dissolved was only 76.0% in simulated gastric fluid medium and 78.8% in simulated intestinal fluid at 120 minutes. At 15 minutes, the total amount dissolved was 27.9% in simulated gastric fluid and 5.6% in simulated intestinal fluid. An R2 of 0.95441 for simulated gastric fluid and 0.96026 for simulated intestinal fluid was found for release of the active pharmaceutical ingredient from the dapsone capsules based on the dissolution data fitted to the Higuchi model. This indicates slow diffusion of dapsone and low solubility of the powdered active pharmaceutical ingredient in the dissolution media used in our study.Citation24
Our results for permeation across human Caco-2 cells showed that formulation II had higher permeation than dapsone in phosphate-buffered solution with or without addition of surfactant (). These data indicate that the nanoemulsion was able to enhance the bioavailability of dapsone, and the apparent permeability coefficient (Papp) of the nanoemulsion was increased compared with dapsone solution alone ().
Table 4 Pharmacokinetic parameters and permeability coefficients simulated in Gastroplus™ for dapsone solution and nanoemulsion
Figure 3 Permeation of dapsone nanoemulsion (NE) and solution through Caco-2 cells.
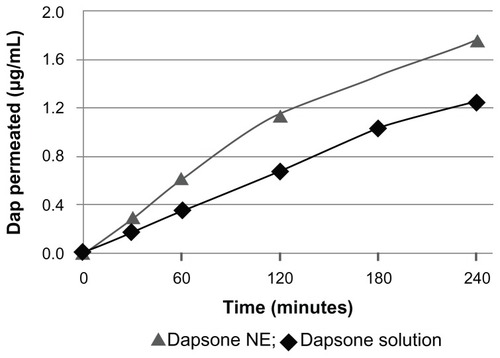
Thus, dapsone, a class II drug, when incorporated in a nanoemulsion, behaves as a class I drug according to the Biopharmaceutics Classification System, due to its higher solubility and higher permeability, with a Papp of 11.04 × 10−6 cm/sec.Citation31,Citation32 Moreover, dapsone formulated as a nanoemulsion showed more mass permeated by area over time than dapsone dissolved in phosphate-buffered saline only, indicating that the presence of pharmaceutical adjuvants helped in the process of permeation. The small droplet size contained in the nanoemulsion enables better adherence to the membrane during transport of the drug and optimizes intestinal absorption and permeation.Citation33 Therefore, both the dose required and the ensuing adverse effects can be reduced, with improved absorption and permeability parameters.
Drugs incorporated into nanoemulsion systems show reproducible bioavailability and a constant plasma concentration profile, which is clinically important for drugs known to have severe adverse effects.Citation33,Citation34 All these characteristics and benefits contributed to a high coefficient of permeability for the dapsone nanoemulsion. Additionally, the presence of pharmaceutical adjuvants has an important role in this context. The surfactants, Span 80 and Tween 80 confer stability to the nanoemulsion, decreasing the surface tension of the two immiscible liquids (water and oil). Moreover, these surfactants are classified as promoters of permeation because they form pores in the cell membrane (enterocytes in this model), which increase drug permeation across the intestinal epithelium after oral administration.Citation35 The dapsone solution containing the Tween 80 and Span 80 surfactants did not produce significantly different permeability results.
The pharmacokinetic parameters simulated by Gastroplus indicate that dapsone nanoemulsion increases the drug absorption rate (time taken to reach peak concentration decreased from 3.0 to 2.3 hours), with a slight increase in peak dapsone concentration (1.12 μm/mL for solution and 1.21 μm/mL for nanoemulsion). The efficiency of dapsone nanoemulsion in promoting drug absorption can be attributed to this drug delivery system ().
The peak plasma dapsone concentration values reported by Mirochnick et alCitation36 using in vivo assays in health volunteers were 1.94 ± 0.31 μm/mL for a propylene glycol solution and 1.86 ± 0.47 μm/mL for a tablet formulation. Both formulations contained 100 mg dapsone and the times taken to reach peak concentration (1.01 and 1.13 hours) were not significantly different. These findings confirm that Gastroplus is a useful tool for prediction of bioavailability in silico and the effect of nanoemulsion systems in increasing the dapsone absorption rate, as well as the robustness of the experimental procedure established in this work.Citation36
Conclusion
We have developed an oral dapsone nanoemulsion system with propylene glycol as the cosolvent. The nanoemulsion contains droplets of a nanoscale size, and its refractive index and conductivity characterizes it as a w/o system with probable in vivo phase inversion. Formulation II enhanced the drug dissolution rate compared with a powdered dapsone dispersion, with kinetic release according to the Higuchi model. The oral dapsone nanoemulsion also showed improved drug permeability. Therefore, the dapsone nanoemulsion system could modulate absorption, bioavailability, and stability parameters, and be an alternative to the class II antibacterial agents structurally related to dapsone, with feasible reduction of adverse effects while delivering a pharmacologically active dose.
Acknowledgments
We are grateful to FAPERJ, CAPES Edital CAPES Nanobiotecnologia 2008, CNPq, and FOPESQ-UFF for financial support and fellowships.
Disclosure
The authors report no personal or financial conflicts of interest in this work.
References
- LegendreDPMuznyCASwiatloEHansen’s disease (leprosy): current and future pharmacotherapy and treatment of disease-related immunologic reactionsPharmacotherapy201232273722392826
- DhopleAMIn vitro activity of epiroprim, a dihydrofolate reductase inhibitor, singly and in combination with brodimoprim and dapsone, against Mycobacterium lepraeInt J Antimicrob Agents19991231932310493608
- ZhuYIStillerMJDapsone and sulfones in dermatology: overview and updateJ Am Acad Dermatol20014542043411511841
- WozelVEInnovative use of dapsoneDermatol Clin20102859961020510768
- LindenbergMKoppSDressmanJBClassification of orally administered drugs on the World Health Organization model list of essential medicines according to the Biopharmaceutics classification systemEur J Pharm Biopharm20045826527815296954
- YuanYLiSMoFZhongDInvestigation of microemulsion system for transdermal delivery of meloxicamInt J Pharm200632111712316876972
- JainJFernandesCPatravaleVFormulation development of parenteral phospholipid-based microemulsion of etoposideAAPS Pharm Sci Tech201011826831
- NandiIBariMJoshiHStudy of isopropyl myristate microemulsion systems containing cyclodextrins to improve the solubility of two model hydrophobic drugsAAPS Pharma Sci Tech20034E10
- NornooAOZhengHLopesLBRestrepoBJKannanKReedROral microemulsions of paclitaxel: in situ and pharmacokinetic studiesEur J Pharm Biopharm20097131031718793723
- MoulikSPPaulBKStructure, dynamics and transport properties of microemulsionsAdv Colloid Interface Sci19987899195
- LawrenceMJReesGDMicroemulsion-based media as novel drug delivery systemsAdv Drug Deliv Rev2000458912111104900
- International Conference on HarmonizationQ1A(R2). Stability testing of new drug substance and productsUS Food and Drug Administration Federal Register2003 Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1A_R2/Step4/Q1A_R2__Guideline.pdfAccessed October 4, 2011
- QueirozRHCDreossiSACarvalhoDJPotentiation the effect methe-moglobinemia dapsone in rats by N-acetylcysteineJ Anal Toxicol1997212032079171203
- US Pharmacopeia NF-28 1092The Dissolution Procedure: Development and Validation33th edRockville, MDUS Pharmacopeia2010
- CostaPLoboJMSModeling and comparison of dissolution profilesEur J Pharm Sci20011212313311297896
- BiganzoliECavenaghiALRossiRBrunatiCMNolliLMUse of a Caco-2 cell culture model for the characterization of intestinal absorption of antibioticsIl Farmaco19995459459910555261
- CalatayudMGimenoJVélezDDevesaVMontoroRCharacterization of the intestinal absorption of arsenate, monomethylarsonic acid, and dimethylarsinic acid using the Caco-2 cell lineChem Res Toxicol20102354755620078116
- SandriGBonferoniMCRossiSFerrariFBoselliCCaramellaCInsulin-loaded nanoparticles based on n-trimethyl chitosan: in vitro (Caco-2 model) and ex vivo (excised rat jejunum, duodenum, and ileum) evaluation of penetration enhancement propertiesAAPS Pharma Sci Tech201011362371
- MarkowskaMOberleRJuzwinSHsuCPGryszkiewiczMStreeterAJOptimizing Caco-2 cell monolayers to increase throughput in drug intestinal absorption analysisJ Pharmacol Toxicol Methods200146515512164260
- RanaldiGIslamKSambuyYEpithelial cells in culture as a model for the intestinal transport of antimicrobial agentsAntimicrob Agents Chemother199236137413811510430
- GrbicSParojcicJIbricSDjuricZIn vitro-in vivo correlation for gliclazide immediate-release tablets based on mechanistic absorption simulationAAPS Pharm Sci Tech201112165171
- OkumuADimasoMLobenbergRComputer simulations using GastroPlus™ to justify a biowaiver for etoricoxib solid oral drug productsEur J Pharm Biopharm200972919819056493
- WeiHLöbenbergRBiorelevant dissolution media as a predictive tool for glyburide a class II drugEur J Pharm Sci200629455216815694
- TangJLSunJHeZGSelf-emulsifying drug delivery systems: strategy for improving oral delivery of poorly soluble drugsCurr Drug Ther200728593
- LuoMShenQChenJTransdermal delivery of paeonol using cubic gel and microemulsion gelInt J Nanomedicine201161603161021904450
- YangSGursoyRNLambertGBenitaSEnhanced oral absorption of paclitaxel in a novel self-microemulsifying drug delivery system with or without concomitant use of P-glycoprotein inhibitorsPharm Res20042126127015032307
- SharmaGWilsonKVan der WalleCFSattarNPetrieJRRavi KumarMNVMicroemulsions for oral delivery of insulin: design, development and evaluation in streptozotocin induced diabetic ratsEur J Pharm Biopharm20107615916920655382
- DateAANagarsenkerMSParenteral microemulsions: an overviewInt J Pharm2008355193018295991
- GursoyNGarrigueJSRazafindratsitaALambertGBenitaSExcipient effects on in vitro cytotoxicity of a novel paclitaxel self-emulsifying drug delivery systemJ Pharm Sci2003922411241814603486
- NornooAOOsborneDWChowDSLCremophor-free intravenous microemulsions for paclitaxel I: formulation, cytotoxicity and hemolysisInt J Pharm200734910811617869459
- LamKWXuJNgKMPharmaceutical salt formation guided by phase diagramsInd Chem Res2010491250312512
- YeeSIn vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man – fact or mythPharm Res1997147637669210194
- TalegaonkarSAzeemAAhmadFJKharRKPathanSAKhanZIMicroemulsion: a novel approach to enhanced drug deliveryRecent Pat Drug Deliv Formul2008223825719075911
- KoganAGartiNMicroemulsions as transdermal delivery vehiclesAdv Colloid Interface Sci2006123–126369385
- PorterCJHTrevaskisNLCharmanWNLipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugsNat Rev Drug Discov2007623124817330072
- MirochnickMClarkeDFMcNamaraERCabralHBioequivalence of a propylene glycol-based liquid dapsone preparation and dapsone tabletsAm J Health Syst Pharm2000571775177711030029