Abstract
Boron nitride nanotubes (BNNTs) have generated considerable interest within the scientific community by virtue of their unique physical properties, which can be exploited in the biomedical field. In the present in vitro study, we investigated the interactions of poly-l-lysine-coated BNNTs with C2C12 cells, as a model of muscle cells, in terms of cytocompatibility and BNNT internalization. The latter was performed using both confocal and transmission electron microscopy. Finally, we investigated myoblast differentiation in the presence of BNNTs, evaluating the protein synthesis of differentiating cells, myotube formation, and expression of some constitutive myoblastic markers, such as MyoD and Cx43, by reverse transcription – polymerase chain reaction and Western blot analysis. We demonstrated that BNNTs are highly internalized by C2C12 cells, with neither adversely affecting C2C12 myoblast viability nor significantly interfering with myotube formation.
Introduction
Boron nitride nanotubes (BNNTs)Citation1,Citation2 are of significant interest to the scientific community. As with carbon nanotubes (CNTs), they have attracted wide attention because of their unique and important properties for structural and electronic applications.Citation3,Citation4 Despite their structural similarity, carbon and boron nitride nanotubes have many different properties. While both BNNTs and CNTs possess a very high Young’s modulus, BNNTs exhibit chemical and thermal stability superior to CNTs.Citation5–Citation7 They also have a constant band gap (ie, an energy difference between the top of the valence band and the bottom of the conduction band) of about 5.5 eV:Citation8,Citation9 in contrast, CNTs vary from semi-conducting to conducting behavior, depending on their chirality and diameter.
Recent investigations have confirmed that BNNTs have excellent piezoelectric properties. Ab initio calculations of the spontaneous polarization and piezoelectric properties of BNNTs have demonstrated that they function as excellent piezoelectric systems with response values larger than those of piezoelectric polymers, and comparable to those exhibited by wurtzite semiconductors.Citation10 Moreover, Bai and colleagues have recently experimentally verified a deformation-driven electrical transport and the first signs of piezoelectric behavior in multiwalled BNNTs.Citation11
These properties make BNNTs potentially attractive candidates for a wide range of applications in the nano domain.Citation12 In recent years, several applications of CNTs in the field of biotechnology have been proposed,Citation13 but the biomedical applications of BNNTs remain largely unexplored.Citation14 Zhi and colleagues investigated the interaction between BNNTs and various protein speciesCitation15 and between BNNTs and DNA,Citation16 but the first studies of the interactions between BNNTs and living cells were performed by the authors.Citation17–Citation19 Despite these preliminary and interesting observations, it is mandatory to extend biological investigations to many different cell types and, eventually, in vivo, in order to highlight different behaviors following the treatment with BNNTs. As a matter of fact, in our experience, distinct cell types treated in vitro with BNNTs may respond with different dose sensitivity. Nanovector platforms are particularly complex systems that can lead to opposite phenomena up to the analyzed in vitro model.
In the present report, we focused our study on up-take, cytocompatibility, and differentiation of C2C12 cells, as a model of muscle cells, in presence of BNNTs. To perform such investigations, stable dispersions of BNNTs were prepared using a positively charged protein (poly-l-lysine [PLL]) as dispersion agent. PLL-coated BNNTs were also conjugated with fluorescent molecules (quantum dots) to enable their tracking in living cells.
Due to the piezoelectricity owned by BNNTs, a positive interaction between muscle cells and BNNTs would potentially allow their employment in the future, as intracellular nanotransducers conveying mechanoelectric stimulation to sensitive cells, such as myoblasts. This report lays the basic foundations for this challenge. BNNT cytocompatibility was investigated in order to identify the highest working concentration potentially usable in vitro without affecting cell viability; cellular up-take of BNNTs was then studied; finally, myogenic differentiative capability retained by C2C12 cells internalizing BNNTs was analyzed.
Materials and methods
BNNT dispersion and characterization
BNNTs (provided by the Australian National University, Canberra, Australia) were produced by using a ball milling and annealing method.Citation20 Details of sample purity and composition, as provided by the supplier, include: yield >80%, boron nitride >97 wt%, metal catalysts (Fe and Cr derived from the milling process) ∼1.5 wt% and absorbed O2 ∼ 1.5 wt%.
PLL (81339, MW 70,000–150,000; Fluka, St. Louis, MO) was used for the dispersion and stabilization of BNNTs. Dispersions were prepared with phosphate-buffered solution (PBS). BNNTs (5 mg) were mixed with 10 mL of a 0.1% PLL solution in a polystyrene tube. The samples were sonicated for 12 h (by a Bransonic sonicator 2510; Bransonic, Danbury, CT) using an output power of 20 W for all the experiments. After sonication, they were centrifuged at 1,100 g for 10 min to remove nondispersed residuals and impurities. Excess PLL was removed by ultracentrifugation three times at 30,000 g for 30 min at 4°C (Allegra 64R; Beckman Coulter, Fullerton, CA) resulting in a stable PLL-BNNT dispersion by the noncovalent coating of the nanotube walls with PLL. The concentration of BNNTs was quantified by spectrophotometric analysis, using a LIBRA S12 Spectrophotometer UV/Vis/NIR (Biochrom, Cambridge, UK) as previously reported,Citation17 while the residual concentration of PLL in the dispersions was assessed using the bicinchoninic acid (BCA) method (for details see below: Quantitative studies on differentiated C2C12 cells: double stranded (ds)-DNA and protein quantification). Microphotographs of the final dispersion of BNNTs were obtained with a Zeiss 902 transmission electron microscope (Carl Zeiss, Oberkochen, Germany), dropping a small quantity of aqueous suspension on a copper grid.
PLL-BNNTs were covalently labeled with carboxyl derivatized quantum dots for cellular tracking studies. Carboxyl quantum dots were supplied by Invitrogen (Qdot® 605 ITK™; Invitrogen, Carlsbad, CA). The conjugation reaction between the amino-groups of PLL and carboxylic-groups of quantum dots was carried out as specified by the supplier. Briefly, 4 mL of PLL-BNNT (100 μg/mL) were mixed with 8 μL of Qdots (8 μM) and 150 μL of 1-ethyl-3-(3-dimethylamino-propyl) carbodiimide (10 mg/mL, EDC, 03450; Fluka) as activator. The solution was gently stirred for 90 min at room temperature for optimal conjugation and finally centrifuged (10 min at 1,100 g) to remove large aggregates. Finally, ultracentrifugation (twice at 30,000 g for 30 min at 4°C) was performed to remove unbound quantum dots from the labeled BNNT dispersions. Qdot labeled BNNTs were exploited only for confocal investigation of the up-take: all other tests (transmission electron microscopy [TEM] analysis, cytocompatibility, differentiation) were performed on PLL-BNNTs in order to avoid any misinterpretation of the data due to the presence of Qdots on the nanotubes.
Sample size distribution was analyzed with an N4plus submicron particle size analyzer (Beckman Coulter); Z-potential measurement was performed with a Nano Z-Sizer (Malvern Instruments, Westborough, MA). Each acquisition was performed in triplicate on aqueous nanotube suspensions at a concentration of 1 μg/mL.
Cell culture and BNNT tracking
C2C12 mouse myoblasts (ATCC CRL-1772) were cultured in expansion medium, constituted by Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 IU/mL penicillin, 100 μg/mL streptomycin and 2 mM l-glutamine. Cells were maintained in normal culture conditions (37°C, saturated humidity atmosphere at 95% air/5% CO2). This cell line, widely used as muscle cell model,Citation21 can differentiate rapidly, in appropriate culture conditions, also forming contractile myotubes that produce characteristic muscle proteins.Citation22 In our studies, differentiation of C2C12 myoblasts in myotubes was induced by switching, in cell specimens at 80%–90% confluence, the culture medium from expansion to differentiation medium, composed of DMEM supplemented with 2 mM l-glutamine, 100 IU/mL penicillin, 100 μg/mL streptomycin, 1% Insulin-Transferrin-Selenium ([ITS] I3146; Sigma, St. Louis, MO), and 1% FBS.
Cell internalization studies were performed by both confocal and transmission electron microscopy.
To pursue confocal analysis, 5,000 cells were seeded on coverglasses (of 1.2 cm diameter), and cultured for 3 days. Qdot labeled-BNNTs were then added to the culture medium with a final concentration of 5 μg/mL of PLL-BNNTs and incubated with the cells for 12 h. Calcein AM (2 μM; Molecular Probes, Eugene, OR) was used for cell cytoplasm labeling (green fluorescence), while 5 μg/mL of Hoechst 33342 (H1399; Invitrogen) was used for nucleus staining (blue). Confocal analysis was finally carried out using a Nikon Inverted Microscope TiE equipped with a Nikon Confocal Laser System A1Rsi (software for image analysis was NIS Elements C 3.06; Nikon Instruments, Melville, NY).
Tests using sodium azide ([NaN3] 478484; Carlo Erba, Milan, Italy), a well known endocytosis inhibitor, enabled investigation of the mechanism responsible for internalization of the BNNTs.Citation23 NaN3 acts by blocking production of adenosine triphosphate (ATP) in cells and thus the energy required for the endocytotic pathway. The cell culture was treated for 30 min with 10 mM NaN3-modified medium and then incubated for 3 h in culture medium containing 5 μg/mL of Qdot labeled BNNTs. Confocal analysis followed as previously described.
Internalization was also investigated by TEM. C2C12 cells were seeded at 106 cells/T25 flask. After attachment, cells were incubated in a PLL-BNNT modified medium (at a final concentration of 5 μg/mL). After 12 h of incubation, samples were washed in PBS 0.1 M and thereafter fixed when still adherent to flasks with 1% w/v glutaraldehyde–4% w/v paraformaldehyde in PBS 0.1 M pH 7.2 for 2 h at 4°C. After washing, the cells were detached by scraping, post-fixed in 1% w/v OsO4 PBS 0.1 M pH 7.2 for 1 h, washed and dehydrated with acidified aceton-dimethylacetal (Fluka) for 10 min. Thus, the samples were mixed in Epon-Durcupan resin in BEEM capsules #00 (Structure Probe, West Chester, PA) overnight at room temperature and finally embedded in resin at 56°C for 48 h. Ultra-thin sections (20–30 nm thick) were obtained with an Ultrotome Nova ultramicrotome (LKB, Bromma, Sweden) equipped with a diamond knife (Diatome, Biel/Bienne, Switzerland).
The sections were placed on 200 square mesh nickel grids, counterstained with saturated aqueous uranyl acetate and lead citrate solutions and observed in a Zeiss 902 transmission electron microscope (Carl Zeiss).
Cytocompatibility assays
Both quantitative (Trypan Blue, MTT) and qualitative (LIVE/DEAD®, annexin V-FITC) assays were performed in order to assess cytocompatibility of BNNTs with C2C12 cells and to establish the maximum working concentration to use.
Cells (250,000 in T25 flasks) were incubated for 24, 48, and 72 h in standard culture conditions to determine their viability following incubation with PLL-BNNT modified media, at various concentrations: 0 (k-, as control), 5, 10, and 15 μg/mL of BNNTs. A further control test using 3 μg/mL of PLL was performed, corresponding to the concentration detected in the dispersion of 15 μg/mL of PLL-BNNTs. The number of viable cells was estimated with Trypan Blue exclusion (T0887; Sigma). Briefly, cell monolayers were rinsed twice with PBS and re-suspended with trypsin and EDTA. An aliquot of the cells was immediately stained with 0.4% Trypan Blue, and the number of viable cells was determined using a hemocytometer and light microscopy.
For metabolic activity testing, MTT (3-(4.5-dimethylthiazole-2-yl)-2.5-diphenyl tetrazolium bromide, M2128; Sigma) assay was carried out after 24, 48, and 72 h of incubation with PLL-BNNT modified media. After Trypan Blue counting, 30,000 viable cells were seeded in 96-well plate chambers (n = 6). Once complete cell adhesion has been verified (about 6 h after seeding), cells were incubated with MTT 0.5 mg/mL for 4 h. The supernatant was then removed and the cell samples were treated with 100 μL of dimethyl sulfoxide ([DMSO], D8418; Sigma). The absorbance at 570 nm was measured with a VERSAMax microplate reader (Molecular Devices). A reference control test (k–) was included.
Viability was further investigated with the LIVE/DEAD® viability/cytotoxicity Kit (Molecular Probes). The kit contains Calcein AM (4 mM in anhydrous DMSO) and ethidium homodimer-1 [EthD-1, 2 mM in DMSO/H2O 1:4 (v/v)]. After incubation for 24, 48, and 72 h at different PLL-BNNT concentrations (0, 5, 10, and 15 μg/mL), cells (20,000 in 24-well plate chambers, n = 3) were rinsed with PBS and treated for 10 min at 37°C with 2 μM calcein AM and 4 μM EthD-1 in PBS. A further control with 3 μg/mL of PLL alone was performed. Cells were finally observed with an inverted fluorescence microscope (TE2000U; Nikon Instruments) equipped with a cooled CCD camera (DS-5MC USB2l; Nikon Instruments) and with NIS Elements imaging software (Nikon Instruments) equipped with the appropriate filters.
For early apoptosis detection, 20,000 cells were seeded in 24-well plate chambers (n = 3) and treated with 0, 5, 10, and 15 μg/mL of PLL-BNNTs for 24 h. A further control with 3 μg/mL of PLL alone was performed. ApoAlert Kit (Clontech Laboratories, Mountain View, CA) was used to evaluate differences between normal and apoptotic cells after treatments. The kit contains annexin V-FITC (20 μg/mL in Tris-NaCl), 1× binding buffer and propidium iodide (50 μg/mL in 1× binding buffer). Cells were rinsed with 1× binding buffer and then incubated with 200 μL of 1× binding buffer containing 5 μL of the annexin V solution and 10 μL of the propidium iodide solution. After incubation in the dark at room temperature for 20 min, cells were examined by fluorescence microscopy with the appropriate filters.
Quantitative studies on differentiated C2C12 cells: Double stranded (ds)-DNA and protein quantification
To perform value normalization, both ds-DNA and total protein content assays were carried out in cascade on the same samples after cell lysis. Proteic production was here investigated as an index of cell synthesis compatible, although not specific, with the differentiation process.
For ds-DNA and protein quantifications on differentiated C2C12, 150,000 cells were seeded in 6-well plate chambers and when 90% of confluence was reached, the growth medium was changed to the differentiating medium. All assays (n = 3) were performed following 3 days of incubation with differentiating medium loaded with 0 (control cultures), 5 and 10 μg/mL of PLL-BNNTs. Moreover, individual samples were run in triplicate to minimize operator error.
At the endpoint, the culture medium was removed from the cell samples and double distilled (dd)-H2O was added; the samples were frozen at −20°C and stored for subsequent assays. Cell lysates were thus obtained by two freeze/thaw cycles of the samples: overnight freezing at −20°C, 10 min thawing at 37°C in a water bath with stirring for 15 s to enable both the ds-DNA and the proteins to go into solution.
Ds-DNA content in cell lysates was measured using the PicoGreen kit (Molecular ProbesCitation24). The PicoGreen dye binds to ds-DNA and the resulting fluorescence intensity is directly proportional to the ds-DNA concentration in solution. Standard solutions of DNA in ddH2O at concentrations ranging from 0–6 μg/mL were prepared and 50 μL of standard or sample was loaded for quantification in a 96-well black microplate. Working buffer and PicoGreen dye solution were prepared according to the manufacturer’s instructions and 100 and 150 μL/well added, respectively. After a 10 min incubation in the dark at room temperature, fluorescence intensity was measured on a plate reader (Victor3; PerkinElmer, Waltham, MA) using an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
Total protein concentration was determined by the BCA method (Pierce Biotechnology, Rockford, ILCitation25) following the microplate procedure. Cell lysates and working reagent were loaded inside a 96-well microplate at 25 and 200 μL/well, respectively. The microplate was gently shaken and then incubated at 37°C for 30 min. Samples were cooled down to room temperature and absorbance was read at 562 nm on a plate reader (BioRad Laboratories, Hercules, CA). Protein concentration of cellular specimens was then obtained by reference to bovine serum albumin (BSA) standards. Finally, total protein content was normalized by DNA content as determined by the PicoGreen assay.
Additionally, cell morphology observation by inverted light microscopy on hematoxylin–eosin (H&E) stained specimens was carried out on cell samples analogous to the ones previously described. Briefly, samples were fixed with 1% buffered formalin at 4°C for 10 min, stained with for 5 s with hematoxylin, rinsed in tap water for 5 s, stained with eosin for 1 s, washed in tap and distilled water, and finally dried at room temperature.
Formation of multinucleated myoutubes was assessed with Hoechst staining and observation with fluorescence microscope. Grade of differentiation was quantitatively evaluated calculating the fusion index of the cultures, determined by dividing the total number of nuclei in myotubes (≥2 nuclei) by the total number of nuclei counted.Citation26 More than 30 myotubes were analyzed for each assay (experiments performed in triplicate).
Reverse transcription-polymerase chain reaction (RT-PCR) for detection of MyoD and connexin 43 (Cx43) mRNA in differentiated C2C12 cells
C2C12 cells were cultured as described in the previous section. Assays were performed after 3 days of incubation with standard differentiating medium (control cultures) followed by 3 days of differentiating medium, containing either 0, 5, or 10 μg/mL of PLL-BNNTs.
Total RNA was isolated from cell cultures using High Pure RNA Isolation kit (Roche, Basel, Switzerland) according to the manufacturer’s protocol. RNA concentration was measured in a spectrophotometer at 260 nm. Identical amounts of RNA were reverse transcribed into cDNA using the Transcriptor First Strand cDNA Synthesis kit (Roche). Treatment with DNase I (Invitrogen) was performed following the manufacturer’s indications. Subsequently, cDNA was amplified by PCR with the specific primers and procedures listed in . The housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), was used as internal standard. Each RT-PCR was performed at least twice. The PCR products were loaded on a 2.5% agarose gel and stained with ethidium bromide.
Table 1 Primer sequences and conditions for RT-PCR analysis
Gel electrophoresis and Western blot for MyoD and Cx43 detection in differentiated C2C12 cells and immunocytochemistry for MyoD
C2C12 pellets obtained from control cultures and cultures treated with 0, 5 and 10 μg/ml of PLL-BNNTs (see previous Section for details) were washed in Dulbecco’s modified phosphate saline buffer (D-PBS; Sigma Aldrich), resuspended in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 μg/mL Aprotinin, 2 μg/mL Pepstatin, 10 μM Leupeptin, 200 μM NaVO3 and 200 μM NaF) and incubated for 30 min in an ice bath, vortexing every 10 min. Cell lysates were then centrifuged at 15,000 rpm for 20 min at 4°C, supernatants were recovered and protein concentration was determined for all the samples using the BCA method.
Proteins (30 μg) from the supernatant were separated on a 10% polyacrylamide gel under reducing conditions, transferred to a polyvinyilidene fluoride (PVDF) membrane (Immobilon-P; Millipore, Bedford, MA) and probed with either mouse monoclonal anti-MyoD antibody (Santa Cruz, Santa Cruz, CA), or with mouse monoclonal anti-connexin 43 (Santa Cruz), or with mouse monoclonal anti-β-actin (Sigma Aldrich) as internal control of the total protein content. SNAP i.d. system (Millipore) was used to probe PVDF membranes. Briefly, PVDF membranes were placed in the device holders, saturated in 0.1% Tween Tris-buffered saline (T-TBS) containing 0.25% dry fat-free milk (immediately eliminated by vacuum application) and incubated for 10 min with either anti-MyoD (1:100 in T-TBS), or anti-Cx43 (1:10,000 in T-TBS), or anti-β-actin (1:2,000 in T-TBS) antibodies, respectively. After three washings in T-TBS under vacuum application, the membranes were incubated with goat anti-mouse immunoglobulin-horseradish peroxidase conjugate (KPL, 1:2,000 in T-TBS). Five washings with T-TBS were performed (under vacuum application) and the immunocomplexes were then detected on PVDF membranes by chemiluminescence (ECL Turbo; Euroclone, Milan, Italy) according to the manufacturer’s instructions.
For immunocytochemical analysis of MyoD, C2C12 were seeded at a cell density of 104 cells/well onto chamber slides and cultured either with or without BNNT (5 μg/mL). Thereafter, the cells were fixed with 1% buffered formalin in PBS for 10 minutes at 4°C and then washed in PBS. MyoD antigen retrieval was performed in citrate buffer pH 6.0 (Diapath, Bergamo, Italy) with microwaves (6 × 5 min at 600 W). Aspecific antibody binding sites were saturated by normal goat serum (Vector Laboratories, Burlingame, CA) 1:20 in PBS (for 20 min at 37°C); the cells were then incubated overnight at 4°C either with the specific anti-MyoD antibody (1:50 PBS containing 0.1% BSA) or without primary antibody, as negative control.
MyoD antibody binding was revealed using the ABC kit (Vector Laboratories) and diaminobenzidine (DAB; GeneTex, San Antonio, TX) as chromogen substrate. Moreover, the cells were counterstained by hematoxylin and subsequently dehydrated in a graded series of ethanol, clarified with xylene and mounted with DPX balsam. The samples were observed by an optical microscope.
Statistical analysis
Analysis of the data was performed by analysis of variance (ANOVA) followed by Student’s t-test to test for significance, which was set at 5%. MTT tests were performed in exaplicate; all the other assays in triplicate. In all cases, three independent experiments were carried out. Results are presented as mean value ± standard error of the mean (SEM).
Results and discussion
Interactions between C2C12 cells and BNNTs
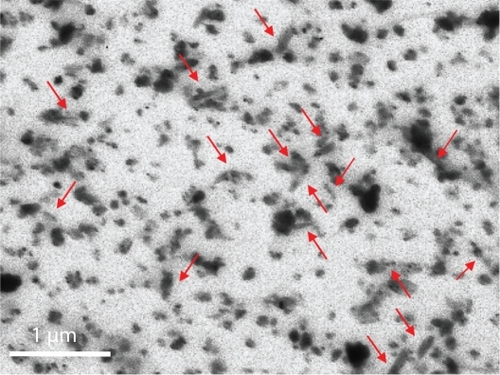
Stable dispersions of BNNTs were obtained by means of PLL as a dispersion agent. PLL is a cytocompatible polymer with positive amino-terminal groups. shows a TEM image of a 200 μg/mL dispersion after preparation: both single nanotubes (red arrows) and small aggregates of nanotubes are visible, with a size congruent to the results achieved with the dynamic light scattering analysis, that yielded an average particle value of 242 ± 92 nm. Moreover, Z-potential analysis revealed a strong positive Z-potential (43.40 ± 3.26 mV), highlighting the high stability of the dispersions. After the preparation procedure, the residual PLL concentration, evaluated with the BCA method, resulted to be about 1:5 w/w with respect to that of the BNNTs present in the dispersion (ie, for a dispersion containing 15 μg/mL of BNNTs, residual PLL concentration was about 3 μg/mL).
Figure 1 TEM image of PLL-BNNT dispersion: both single nanotubes (red arrows) and small aggregates of nanotubes are visible.
shows the results of the internalization assays performed with confocal microscopy. depicts consecutive slides of cells (magnification 60X, z-stack step 500 nm) following incubation with BNNTs, showing Qdot-labelled BNNTs (red) strongly internalized in the cell cytoplasm (green). No evidence of BNNTs in the nuclei (blue) of cells was observed.
Figure 2 Confocal analysis of BNNT internalization a) and investigation of its mechanism: BNNT uptake b) and its inhibition after treatment with sodium azide c).
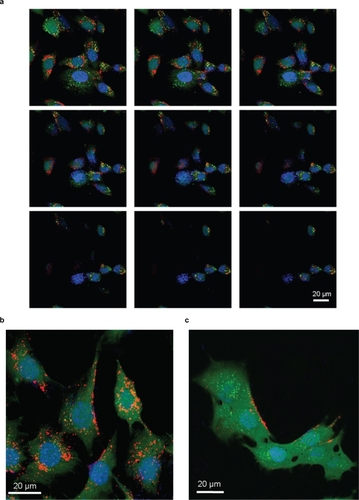
These observations demonstrated that PLL-BNNTs were internalized by the cells, but provided no information on the nature of the mechanism responsible for this up-take. Sodium azide was exploited to investigate the energy-dependence of the up-take mechanism of PLL-BNNTs. The chosen incubation time was 30 min, as optimal compromise between effective ATP blocking by sodium azide, and reduced cytotoxic effects resulting from a prolonged energy deficiency. shows a confocal image of cells incubated for 1 h in a medium containing 5 μg/mL of Qdot-labelled BNNTs: internalization occurred as shown in the previous tests. An image of C2C12 cells analogously incubated but following pre-treatment with sodium azide is shown in . Here a clear inhibition of cell up-take was evidenced by a negligible amount of internalized BNNTs and a massive BNNT accumulation on the cell membrane. This result demonstrates that the internalization of PLL-BNNTs by C2C12 is energy-dependent, as already noticed for PEI-coated BNNTs in other cell lines.Citation18
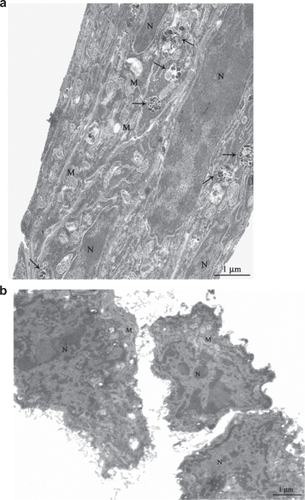
TEM analysis reported in confirmed that PLL-BNNTs are abundantly internalized by C2C12 cells, showing the presence of noncellular electron-dense material, compatible for appearance and dimensions with BNNTs (see ), in cytoplasmic vacuoles. Arrows are used to indicate the vacuoles/vesicles in the intracellular environment. , as control, shows C2C12 cells cultured in the same conditions but without BNNT treatment: absence of BNNT-like material in the cytoplasmic compartments is evident. Such observations are consistent with those of Yehia and colleagues,Citation27 Raffa and colleagues,Citation28 and Tutak and colleaguesCitation29 performed on carbon nanotubes: in these studies different kinds of mammalian cells were exposed to CNTs dispersed in cell growth medium that presented high-resolution TEM images of dense dark material within intracellular vacuoles.
Figure 3 TEM imaging of C2C12 cells incubated for 12 h with (a) and without (b) a 10 μg/mL BNNT modified medium. The TEM micrograph shows C2C12 with vesicles containing BNNT (arrows) in the treated sample. Magnification 7000×.
Abbreviations: BNNT, boron nitride nanotubes; M, mitochondria; N, nuclei; TEM, transmission electron microscopy.
The data concerning the effects of BNNT up-take are reported in .
Figure 4 MTT assay results after 24, 48, and 72 h of incubation of C2C12 cells with 0, 5, 10, and 15 μg/mL of PLL-BNNTs, and with 3 μg/mL of PLL (n = 6).
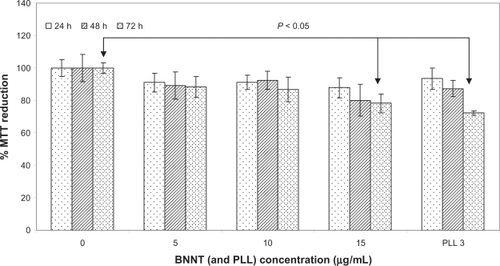
Viability assays show that after 24, 48, and 72 h of incubation at different PLL-BNNT concentrations (5, 10, and 15 μg/mL), cell densities did not differ from their respective controls. In all cases, preliminary cell viability assessed with Trypan Blue was greater than 90%. The data at 72 h confirmed that BNNTs did not affect cellular replication: the cells reached confluence in each test, undergoing almost three complete replication cycles. The same considerations are valid for controls performed with 3 μg/mL of PLL, ie, the amount of PLL present in the highest concentrated BNNT sample.
shows MTT results for each test. C2C12 exhibited a slight decrease in metabolic activity following incubation with PLL-BNNTs. Cells incubated with 5 μg/mL of nanotubes sustained a decrease of about 10% after 24, 48, and 72 h of incubation, but all these results were not statistically different from the respective controls (in all cases, P > 0.05). Similar results were obtained by culturing C2C12 with 10 μg/mL of PLL-BNNTs. Incubation with 15 μg/mL of nanotubes provided a nonsignificant MTT reduction after 24 and 48 h (about 15%; P > 0.05), but the decrease in metabolic activity became significant (about 20%; P < 0.05) by the third day. The same trend was followed by cells incubated with 3 μg/mL of PLL alone (statistical significance after 3 days of incubation), thus demonstrating that, at these concentrations, the slight cytotoxicity is mainly due to the surfactant rather than to a negative effect of the nanotubes.
On the basis of these results, we decided to use 5 and 10 μg/mL as the working concentrations for the subsequent studies.
Optimal cell viability up to 10 μg/mL of BNNTs was confirmed by the LIVE/DEAD® viability/cytotoxicity assay. Live cells are distinguished by the presence of ubiquitous intracellular esterase activity, determined by the enzymatic conversion of the nonfluorescent cell-permeant agent, calcein AM, to an intensely fluorescent molecule, calcein. Calcein is well retained within live cells, producing an intense uniform green fluorescence. Conversely, EthD-1 enters cells with damaged membranes and undergoes a 40-fold enhancement of fluorescence upon binding to nucleic acids, thereby producing a bright red fluorescence in dead cells. EthD-1 is excluded by the intact plasma membrane of live cells.
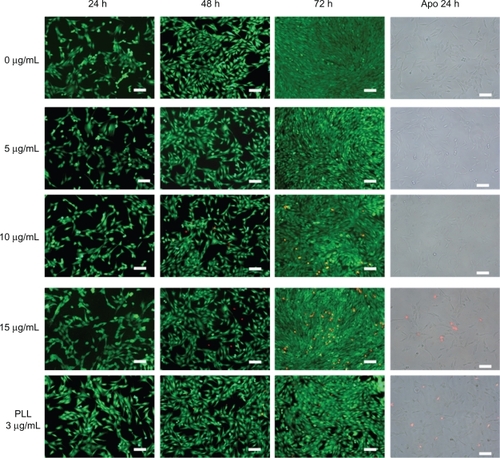
shows the results of the viability/cytotoxicity assay, performed after 24, 48, and 72 h of incubation with 0, 5, 10, and 15 μg/mL of BNNTs. A further test with 3 μg/mL of PLL alone was also carried out. No evidence of cell membrane damage was present following treatment of C2C12 cells up to 10 μg/mL of PLL-BNNTs, clearly demonstrating optimal cell viability, completely comparable to that of the control cells (0 μg/mL). A nonnegligible amount of red stained cells was present in the samples treated with either 15 μg/mL of PLL-BNNTs or 3 μg/mL of PLL, thus confirming the MTT assay quantitative results.
Figure 5 LIVE/DEAD® viability/cytotoxicity assay and early apoptotic detection (annexin V-FITC/PI assay) performed for different incubation times and concentrations.
Note: Scale bar: 100 μm.
Early apoptotic phenomena were investigated as described in Cytocompatibility assays. One of the methods employed to study apoptosis detects changes in the position of phosphatidylserine (PS) in the cell membrane. In nonapoptotic cells, most PS molecules are localized on the inner layer of the plasma membrane, but soon after the initiation of apoptosis, PS is redistributed to the outer layer of the membrane, and becomes exposed to the extracellular environment.Citation30 Exposed PS can be easily detected with annexin V, a 35.8 kDa protein with strong affinity for PS.
Propidium iodide (PI) is a DNA red fluorescent intercalating agent; it is membrane impermeant and generally excluded from viable cells. Therefore, it can be used jointly with annexin V-FITC to differentiate necrotic (red stained), early apoptotic (green stained), apoptotic (green and red stained) and normal cells (unstained).
also shows the results of apoptotic assay carried out on C2C12 cells incubated for 24 h with 0, 5, 10, and 15 μg/mL of PLL-BNNTs and a control culture incubated with 3 μg/mL of PLL. No evidence of apoptosis was observed in all the treatments, with a few dead cells in the sample treated with 15 μg/mL of PLL-BNNTs or 3 μg/mL of PLL. Such observations are again in agreement with the results obtained with the LIVE/DEAD® assay and the Trypan Blue test.
Taken together, these findings clearly demonstrate the absence of significant negative effects of PLL-BNNTs on C2C12 myoblasts up to a concentration of 10 μg/mL.
C2C12 differentiation
Differentiation of C2C12 myoblasts (80%–90% confluent) in myotubes was obtained after a 3-day culture of these samples under differentiating conditions (1% FBS, 1% ITS). Cultures supplemented with PLL-BNNTs were monitored and compared to control cultures as previously reported, in order to verify whether BNNTs may contrast the differentiation process. Proliferation and protein synthesis of differentiating myoblasts were assessed after 3 days of incubation in differentiating medium supplemented with 0, 5 and 10 μg/ml of PLL-BNNTs by measuring ds-DNA and total protein contents.
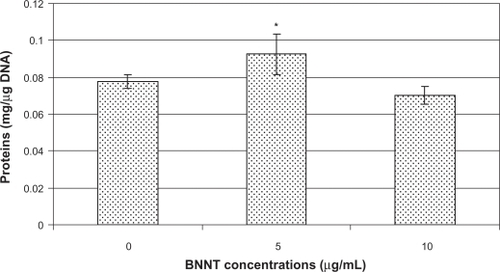
Cultures treated with 5 μg/ml of PLL-BNNTs versus controls exhibited a nonsignificant drop in ds-DNA amount (<2%), but corresponding to a more sustained increase in protein content (about 15%). Consequently, when normalized by ds-DNA (and therefore by cell number), the total proteins resulted in a significant rise (about 20%), which was statistically different with respect to the control (P < 0.05). Quantitative results showed that normalized protein content is about 92 μg/μg-DNA in BNNT-treated samples, whereas it is about 78 μg/μg-DNA in controls. Cultures treated with 10 μg/mL of PLL-BNNTs versus controls exhibited DNA increment (about 10%) and slight protein decrement (about 3%), resulting in a normalized protein content of about 70 μg/μg-DNA in BNNT-treated samples, nonsignificantly different from the control.
These findings, summarized in , confirmed that in presence of BNNTs C2C12 cells retained a good protein synthesis capacity under differentiating conditions.
Figure 6 Total protein content normalized by ds-DNA content in C2C12 cell differentiated in presence of 0, 5, and 10 μg/mL of PLL-BNNTs (n = 3, *P < 0.05).
The morphology of differentiating cells was assessed by phase contrast microscopy and H&E staining after 3 days of culture. shows differentiated C2C12 cultured with 0 (a), 5 (b), and 10 (c) μg/mL of PLL-BNNTs, demonstrating that treated cells developed myotubes without qualitative differences from the control cultures.
Figure 7 Light microscopy on H&E-stained cell samples showing myotube formation: differentiated C2C12 cultured with 0 (a), 5 (b), and 10 (c) μg/mL of PLL-BNNTs; fusion index evaluated for the three cultures (d).
Abbreviations: BNNTs, boron nitride nanotubes; H&E, hematoxylin–eosin; PLL, poly-l-lysine.
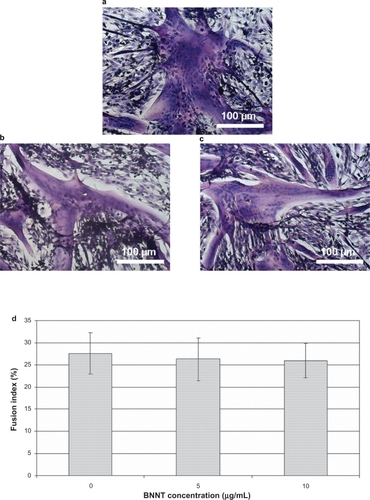
Quantitative assessment of myotube formation was performed evaluating the fusion index of the three different cultures (). Also in this case, no statistically significant differences were observed between samples treated with 0, 5 and 10 μg/mL of BNNTs: in all experiments the calculated fusion index was higher than 25% (P > 0.5), indicating well sustained differentiative processes.
We can therefore conclude that BNNTs showed a negligible effect on the proliferation of differentiating C2C12 myoblasts; moreover, they were found to affect neither the cellular protein synthesis nor the qualitative/quantitative development of myotubes in vitro.
C2C12 cell differentiation was also investigated throughout the expression of some constituent myoblastic markers, such as MyoD and Cx43, both at the gene and protein level.
Differentiation of myogenic cells is regulated by multiple factors. Among these MyoD is known to play a key role in regulating myoblast differentiation. MyoD belongs to a family of proteins known as myogenic regulatory factors (MRFs).Citation31 Some of these bHLH (basic helix loop helix) transcription factors act synergistically, others sequentially in the myogenic differentiation cascade. MRF family members include MyoD, Myf5, myogenin, and MRF4 (Myf6). MyoD is one of the earliest markers of myogenic commitment, being expressed in activated satellite cells and in their progenitors.Citation32
Connexins, such as Cx43, are proteins present in the myoblasts involved in the formation of gap junctions, composed of connexin-based connexons. Such junctions regulate the intercellular passage of small molecules, including inorganic ions, small metabolites and second messengers, thus achieving electrical, as well as metabolic, coupling of the cells.Citation33
shows MyoD and Cx43 mRNA levels in differentiated C2C12 cells treated with 0, 5 and 10 μg/mL of PLL-BNNTs, as obtained by RT-PCR. These results highlighted that the expression of both Cx43 and MyoD genes was not drastically affected by the administration of BNNTs at a concentration lower than 10 μg/mL: Cx43 and MyoD mRNAs were found to be well expressed in all the tested samples.
Figure 8 RT-PCR results for MyoD and Cx43 expression in C2C12 cells differentiated in presence of 0, 5, and 10 μg/ml of PLL-BNNTs; GAPDH was used as internal standard (a) Western blot results for MyoD and Cx43 production in the same cultures; β-actin was used as internal standard (b) Immunocytochemical analysis for MyoD expression on BNNT-treated cell samples (c) and controls without BNNTs (d) “Bn” indicates visible dotting in the cytoplasm due to BNNTs, the arrows indicate MyoD positive cells at nuclear level, while the arrowhead indicates MyoD positive cells at cytoplasm level.
Abbreviations: BNNTs, boron nitride nanotubes; H&E, hematoxylin–eosin; PLL, poly-l-lysine; RT-PCR, reverse transcription – polymerase chain reaction.
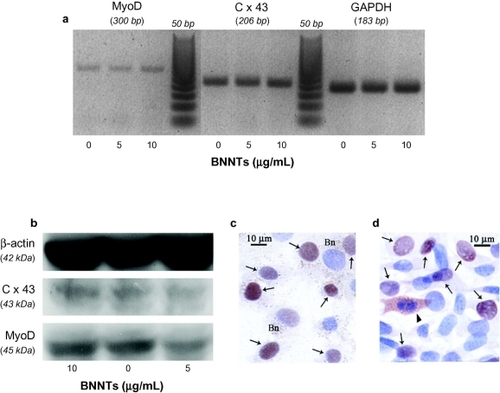
Western blot analysis fully agrees with the RT-PCR results: shows a good synthesis level of both Cx43 and MyoD proteins for all the tested concentrations. Finally, immunocytochemistry highlighted the main nuclear localization of MyoD, with no appreciable difference between BNNT-treated and untreated cells. Cytoplasmatic detection of MyoD was only visible in mitotic cells.
All together, these results corroborated the findings obtained using the fusion index evaluation and the qualitative analysis of the myotube morphology reported in . Therefore, myotube formation resulted to be neither inhibited nor delayed by BNNT up-take, indicating that BNNTs in the tested range can be compatible with C2C12 differentiation.
Conclusions
In sharp contrast to the applications of CNTs, that have been proposed in biotechnology in recent years, the biomedical applications of BNNTs have received scant attention despite their unique and impressive chemical and physical properties. As regards CNTs, a huge amount of data can be found in the literature about their biocompatibility,Citation34 often with contrasting results, and the possibility of demonstrating the safety of these nanomaterials is quite remote. On the contrary, a few studies are available for BNNTs, hence the importance to pursue rational and systematic investigations of the interactions between BNNTs and living matter.
In this paper, we reported on the results of the interaction between PLL-coated BNNTs and C2C12 myoblasts, as model of skeletal muscle cells. After obtaining a stable dispersion of BNNTs based on PLL wrapping, we used confocal microscopy and TEM analysis to demonstrate that a high internalization of PLL-BNNTs, following an energy-dependent mechanism, occurred in C2C12 cells. Moreover, cytocompatibility assays performed on C2C12 myoblasts denoted excellent proliferation and metabolic activity up to a concentration of 10 μg/mL of PLL-BNNTs and up to a 72 h culture time. At these concentrations, apoptotic, necrotic and membrane permeabilisation phenomena were completely absent, as demonstrated by both the annexin V-FITC/PI assay and the LIVE/DEAD® test. Differentiation of C2C12 cells and formation of myotubes did not prove to be significantly affected by incubation with BNNT-modified differentiating medium for 72 h. The protein content of the differentiating cells was comparable or even superior in treated with respect to control cultures; mRNA of constitutive myoblastic markers, such as MyoD and Cx43, resulted to be still expressed in cells treated up to 10 μg/ml of BNNTs. Moreover, Western blot analysis showed a similar MyoD and Cx43 synthesis in all the treated samples, thus confirming no severe adverse effects on C2C12 differentiation.
Collectively, these experimental findings confirm that BNNTs are suitable for the development of novel nanovectors for biomedical applications. The exploitation of their peculiar physical properties, especially piezoelectricity, could in fact provide the basis for clinical electrostimulation therapies for various cardiac, skeletal and visceral muscular, and neurogenic disorders.
Acknowledgements
The work described in this paper was partially supported by the IIT (Italian Institute of Technology) Network. The authors gratefully acknowledge the Australian National University of Canberra, and in particular Dr Jun Yu and Prof Ying Chen from the Department of Electronic Materials and Engineering, Research School of Physical Sciences and Engineering for providing the BNNT sample tested in this work. Moreover, the authors wish to thank Dr Matilde Masini (Department of Experimental Pathology BMIE, University of Pisa, Pisa, Italy) for TEM technical support. The authors report no conflicts of interest in this work.
References
- ChopraNGLuykenRJCherreyKBoron-nitride nanotubesScience1995269522696696717807732
- GolbergDBandoYTangCZhiCBoron nitride nanotubesAdv Mater2007191824132432
- TerronesMRomo-HerreraJMCruz-SilvaEPure and doped boron nitride nanotubesMater Today20071053038
- ZhiCBandoYTangCHondaSKuwaharaHGolbergDBoron nitride nanotubes/polystyrene compositesJ Mater Res2006211127942800
- SongJHuangYJiangHHwangKCYuMFDeformation and bifurcation analysis of boron-nitride nanotubesInt J Mech Sci2006481111971207
- SuryavanshiAPYuMFWenJTangCBandoYElastic modulus and resonance behavior of boron nitride nanotubesApp Phys Lett2004841425272529
- ChenYZouJCampbellSJLe CaerGBoron nitride nanotubes: pronounced resistance to oxidationAppl Phys Lett2004841324302432
- OkuTKoiNSuganumaKElectronic and optical properties of boron nitride nanotubesJ Phys Chem Solids20086912281231
- FarmanzadehDGhazanfarySFirst principle electric field response of single-walled boron nitride nanotube: a case study of zigzag (4,0) modelStruct Chem2009204709717
- DaiYGuoWZhangZZhouBTangCElectric-field-induced deformation in boron nitride nanotubesJ Phys D Appl Phys2009428085403
- BaiXDGolbergDBandoYZhiCYTangCMitomeMDeformation-driven electrical transport of individual boron nitride nanotubesNano Lett20077363263717288485
- GolbergDBaiXMitomeMTangCZhiCBandoYStructural peculiarities of in-situ deformation of an individual boron nitride nanotube inside a high-resolution transmission electron microscopeActa Mater200755412931298
- LacerdaLBiancoAPratoMKostarelosKCarbon nanotubes as nanomedicines: from toxicology to pharmacologyAdv Drug Deliv Rev200658141460147017113677
- CiofaniGRaffaVMenciassiACuschieriABoron nitride nanotubes: an innovative tool for nanomedicineNano Today200941810
- ZhiCBandoYTangCGolbergDImmobilization of proteins on boron nitride nanotubesJ Am Chem Soc200512749159961599716287265
- ZhiCBandoYWangWTangCKuwaharaHGolbergDDNA-mediated assembly of boron nitride nanotubesChem Asian J20072121581158518041790
- CiofaniGRaffaVMenciassiADarioPPreparation of boron nitride nanotubes aqueous dispersions for biological applicationsJ Nanosci Nanotechnol20088126223623119205187
- CiofaniGRaffaVMenciassiACuschieriACytocompatibility, interactions and uptake of polyethyleneimine-coated boron nitride nanotubes by living cells: confirmation of their potential for biomedical applicationsBiotechnol Bioeng2008101485085818512259
- CiofaniGRaffaVMenciassiACuschieriAFolate functionalised boron nitride nanotubes and their selective uptake by glioblastoma multiforme cells: implications for their use as boron carriers in clinical boron neutron capture therapyNanoscale Res Lett200942113121
- YuJChenYWuhrerRLiuZRingerSPIn situ formation of BN nanotubes during nitriding reactionsChem Mater20051751725176
- NedachiTFujitaHKanzakiMContractile C2C12 myotube model for studying exercise-inducible responses in skeletal muscleAm J Physiol Endocrinol Metab20082955E1191E120418780777
- LeeJHTachibanaHMorinagaYFujimuraYYamadaKModulation of proliferation and differentiation of C2C12 skeletal muscle cells by fatty acidsLife Sci2009841314415420
- Shi KamNWLiuZDaiHCarbon nanotubes as intracellular transporters for proteins and dna: an investigation of the uptake mechanism and pathwayAngew Chem Int Ed2006454577581
- SingerVLJonesLJYueSTHauglandRPCharacterization of PicoGreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitationAnal Biochem199724922282389212875
- StoscheckCMQuantitation of proteinMethod Enzymol19901825069
- CharrasseSComunaleFFortierMPortales-CasamarEDebantAGauthier-RouviereCM-cadherin activated Rac1 GTPase through the Rho-GEF trio during myoblast fusionMol Biol Cell2007181734174317332503
- YehiaHNDraperRKMikoryakCSingle-walled carbon nanotube interactions with HeLa cellsJ Nanobiotechnology20075817956629
- RaffaVCiofaniGNitodasSCan the properties of carbon nanotubes influence their internalization by living cells?Carbon20084616001610
- TutakWParkKHVasilovAToxicity induced enhanced extracellular matrix production in osteoblastic cells cultured on single-walled carbon nanotube networksNanotechnology2009202525510119487801
- MartinSJReutelingspergerCPMcGahonAJEarly redistribution of plasma membrane phophatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and AblJ Exp Med1995182154515567595224
- NodaTFujinoTMieMKobatakeETransduction of MyoD protein into myoblasts induces myogenic differentiation without addition of protein transduction domainBiochem Bioph Res Commun20093822473477
- KanisicakOMendezJJYamamotoSYamamotoMGoldhamerDJProgenitors of skeletal muscle satellite cells express the muscle determination gene, MyoDDev Biol2009332113114119464281
- El OakleyRYacoubMHSuzukiKOverexpression of connexin 43 in skeletal myoblasts: Relevance to cell transplantation to the heartJ Thorac Cardiovasc Surg2001122475976611581610
- SmartSKCassadyAILuGQMartinDJThe biocompatibility of carbon nanotubesCarbon200644610341047