
Abstract
Aphallia (absence of penis) is an extremely rare abnormality which has rarely been described in medical literature and can be part of the urorectal septum malformation sequence (URSMS). URSMS has hardly been reported in medical literature and includes the absence of perineal and anal openings in association with ambiguous genitalia and urogenital, colonic, and lumbosacral anomalies. This case report tells the importance of detailed examination of infants that are diagnosed with aphallia. We report a case of a newborn who was diagnosed as aphallia with the URSMS syndrome after birth. The neonate had an endocardial cushion defect (atrial septal defect and ventricular septal defect) and bilateral agenesis of the kidney. The neonate succumbed to death secondary to hypoplastic lung leading to respiratory failure.
Introduction
The urorectal septum malformation sequence (URSMS) was first described as a separate entity by Escobar et al in 1987. The various abnormalities associated with URSMS include the presence of ambiguous genitalia, absence of urethral and vaginal openings, imperforate anus, hypoplastic bladder/rectum/urethra/uterus, urorectal-vaginal fistulae, mullerian duct defects, and renal defects like renal agenesis. URSMS has been described as a sporadic condition. Current embryological understanding of the pathology seen in hindgut development and URSMS is not fully understood, but appears to involve a deficiency of caudal mesoderm.Citation1 We report a rare case of a newborn who was diagnosed as a case of aphallia with the URSMS syndrome after birth.
Case
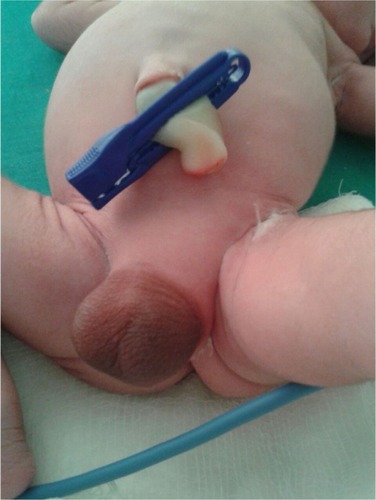
A term male infant with a birth weight of 3 kg was referred to us within hours of life in view of aphallia. The infant was born of a non-consanguineous marriage to a primi mother with an uncomplicated antenatal period. The baby had Apgar score of 8/8/9, respectively at 1, 5, and 10 minutes of life. There was no family history of any congenital anomalies. Examination revealed an infant with respiratory distress and also with absent penis (). The scrotum was well developed with good amount of rugae and bilateral testes were palpable. The mother was unsupervised and had only one antenatal visit during the pregnancy. There was only one antenatal scan available that showed amniotic fluid index of 2, suggestive of severe oligohydramnios. On detailed physical examination, the infant was noted to have imperforate anus (high anal opening) and single umbilical artery. Other systemic examination showed normal facies except the depressed nasal bridge. There was no cleft lip, cleft palate, cataract or ophthalmological malformation, and low-set ears. On cardiovascular system examination, a pan systolic murmur was heard. The infant was evaluated with echocardiography which showed endocardial cushion defect (atrial septal defect and ventricular septal defect). Ultrasound of abdomen showed bilateral agenesis of kidney with no other abnormality. Chest radiograph was suggestive of low-volume lungs (mild pulmonary hypoplasia) with five intercostal spaces. In the gastrointestinal system, there was no tracheoesophageal fistula and the whole body radiograph was not suggestive of any vertebral abnormalities. There was no neural tube defect like myelomeningocele or meningocele. Cranial ultrasound was suggestive of prominent ventricles with bulky choroid plexus with normal corpus callosum. There were no digital abnormalities with normal upper-segment and lower-segment ratio. Serum electrolytes of the neonate at the time of birth was suggestive of serum sodium of 146 meq/dL, serum potassium of 5.6 meq/dL, serum calcium of 8.3 mg/dL, and sugar value of 72 mg/dL. Renal function of the neonate showed serum creatinine of 1.8 mg/dL, blood urea of 128 mg/dL and blood urea nitrogen of 62 mg/dL. On the basis of physical examination and associated malformations, the infant was diagnosed as a case of URSMS. Parents were counseled about the poor prognosis in view of multiple malformations and the baby succumbed to respiratory failure next day. No active intervention was done, as parents were not willing for any aggressive management and opted for minimal supportive care. The postmortem examination of the infant was deferred, as the parents were not willing for postmortem. Informed parental consent was obtained for case report. Institutional Review Board approval was not sought as per guidelines on retrospective reports.
Figure 1 Shows the absence of penis with well developed scrotum with good rugosity.
Discussion
Aphallia or absence of penis is a very rare congenital anomaly and has rarely been reported in literature. It is estimated to have an incidence of around in 1 in 30 million live births.Citation1 A total of nearly approximately 80 cases have been reported in the medical literature till now.Citation2 Aphallia develops as a result of failure of development of the genital tubercle into phallus. This leads to the total absence of all three components of penile shaft ie, both corpora cavernosa and spongiosum.Citation3 This agenesis takes place at 5th to 7th week of embryonic development which explains its association with other associated system malformations like renal seen in the index case.Citation4 Aphallia may also be seen as a component of a more severe malformation complex syndrome that includes the perineal area, such as sirenomelia, cloacal exstrophy, or the URSMS which tells that this combined malformation may be the effect of defective blastogenesis, which is usually seen around 5th to 7th week of gestation.
The URSMS is characterized by an absence of perineal openings, ambiguous genitalia, and renal agenesis. The URSMS has been postulated to be due to defect in cloacal differentiation during early embryogenesis. It results because of defective fusion of the urorectal septum membrane with the cloacal membrane or because of absence of caudal mesoderm.Citation5 URSMS is classified in two division namely as “partial URSMS” that is characterized by a single perineal or anal opening draining a common cloaca along with an absent (imperforate) anus or “full URSMS” which is characterized by the absence of both perineal and anal openings, and the two together are called as “URSM spectrum”.Citation6 In the index case, we thought of URSMS as the neonate had absence of perineal opening, ambiguous genitalia in the form of aphallia and also bilateral renal agenesis. In our case, complete urogenital agenesis was present, with neither urethral nor anal sphincters, compatible with a male full URSMS. In our index case, severe oligohydramnios was the result of bilateral renal agenesis leading to decreased urine output and amniotic fluid index of only 2. This oligohydramnios was responsible for secondary pulmonary hypoplasia which was seen as low-volume lung on X-ray and was the cause of death. The infant did not have typical Potter’s facies in spite of severe oligohydramnios which is difficult to explain. The presence of heart defect and renal agenesis can also be explained by the defect occurring in first trimester leading to multiple malformations. The full URSMS embryologically results due to a failure of the breakdown of the cloacal membrane combined with a defective urorectal septal development. Due to lack of mesodermal cells in the caudal region, the development of the hindgut and surrounding tissues is severely impaired whereas in the partial URSMS, there is a partial breakdown of the cloacal membrane, resulting in a single perineal/anal orifice. The incomplete defect or descent of the urorectal septum causes specific internal genital anomalies such as septate or bifid vagina and uterus. There has been case reports that have shown overlap between URSMS and limb-body-wall-complex or exstrophy of the cloaca, which have led clinicians to think that they represent a continuous spectrum of abnormalities in the lower mesoderm organization.Citation7 Karyotype analysis of the patient with aphallia is typically XY. The scrotum is usually normally developed and contains normal testis, as in the index case. The testes produce normal testosterone production in response to human chorionic gonadotropin stimulation. The testosterone is the most important hormone that plays significant role in the development of male genitals like testes and prostate and also in the development of secondary sexual characteristics.Citation8 URSMS has also been classified as malformative (non-endocrine) disorders of sex development in which a genetic etiology has been identified by few authors.Citation9 Urethral opening can be located near anal verge (postsphincteric) under a skin tag or rarely into the rectum (presphincteric).Citation10
Other system anomalies usually associated with URSMS are renal system malformations (unilateral renal agenesis, bilateral renal agenesis and bilateral renal dysplasia, unilateral renal agenesis with contralateral kidney showing features of cystic dysplastic kidney and renal hypoplasia, agenesis/hypoplasia of the bladder, vesico-rectal fistula), genital system (cryptorchidism, uterus, and vagina anomalies like hemi-uterus with right vestigial fallopian tube, bicornuate uterus),Citation11 abnormalities of the gastrointestinal tract (intestinal malrotation, anal atresia, rectovaginal fistula, tracheoesophageal fistula, Meckel diverticulum),Citation6 nervous system (dysgenesis of the lumbar spine and sacrum, arachnoid cyst, frontal pachygyria, myelomeningocele, sacral abnormalities, tethered cord)Citation12 musculoskeletal (flexion contractures of the hips and knees, tibial bowing, bilateral equinovarus of the feet, hypoplastic legs),Citation11 and cardiopulmonary anomalies (pulmonary hypoplasia, agenesis of right lung, atrioventricular canal defect, tetralogy of Fallot, truncus arteriosus with atrial septal defect, ventricular septum defect, and right-sided descending aorta, preductal coarctation of the aorta and right ventricular myocardial hyperplasia),Citation11 therefore all such patients must be evaluated for all systems.Citation13 Aphallia has been reported very rarely as a part of URSMS. There has been also speculation of association between VACTERL and URSMS but the final relationship still remains too explained.Citation14 In the first case report of its type Aggarwal and Phadke reported two male siblings, affected with a spectrum of anomalies simulating URSM and suggested the existence of hitherto unknown genetic mechanisms for this pattern of malformation.Citation15
Gérard-Blanluet et alCitation5 reported a male fetus, diagnosed antenatally to have severe bilateral renal dysplasia with no residual renal function. At birth the examination showed aphallia with normal scrotum, imperforate anus, bilateral renal dysplasia, bilateral club foot, and complete right lung agenesis. The fetus had Potter sequence with dysmorphic features; large, rotated ears, a short compressed nose, and deep subocular grooves.Citation6
The recent few case reports in medical history have raised doubt regarding the existence of URSMS and tried to show it as a variant of VACTERL association. Patra and Purkait reported a newborn that had characteristic features of URSMS and also had features of VACTERL association in the form of congenital varus deformity of leg, polydactyly, tracheoesophageal fistula, cardiac defect, anal atresia, and hydronephrosis.Citation14 In another case report, authors tried to highlight the association between URSMS and VACTERL association. But the authors finally concluded that URSMS and VACTERL association should be viewed as separate entities until their respective etiologies have been clarified.Citation7 There has been few case reports of aphallia from India.Citation16–Citation18 In a recent report published by Bahe et al showed the long-term complication of aphallia.Citation18
The treatment needs an individualized approach and parents should be explained about all treatment options available with their advantages and disadvantages. The male sex assignment consists of penile reconstruction (phalloplasty) using various techniques.Citation19,Citation20 The female sex assignment is a multistaged approach and consists of assignment of the female sex to the infant. Bilateral orchidectomy, urethral perineal transposition, and feminizing genitoplasty is done in newborn period prior to postnatal testosterone surge.Citation2,Citation21 Vaginoplasty is done in the latter part of life. Hormonal therapy with estrogens forms an important part of management and is started at the time of puberty to have secondary sexual characteristics and continued lifelong.Citation6 The female sex reassignment is easier and the child can have a normal sexual life whereas male assignment has a high rate of failure because of erectile dysfunctions in later life.Citation22,Citation23 Kanamori et al reported survival in a case with complete URSMS who had moderate pulmonary hypoplasia and preserved left renal function. The infant was managed with vesico-amniotic shunts that prevented severe pulmonary hypoplasia caused by oligohydramnios.Citation24 The infant with URSM syndrome have guarded prognosis and parents should be counseled about the condition.
URSM is a very rare sequence seen in perinatal medicine. The treating neonatologist should keep this rare anomaly in mind whenever dealing with aphallia. Quick referral should be made as these infants need an early surgical intervention. Infants should be examined thoroughly for associated malformations of all systems as the prognosis of infants also depends on them as in our case the infant had bilateral renal agenesis. Parents should be counseled in details about the need for sex assignment but their decision should be respected while planning the treatment of their child.
Disclosure
The authors report no conflicts of interest in this work.
References
- EscobarLFWeaverDDBixlerDHodesMEMitchellMUrorectal septum malformation sequence. Report of six cases and embryological analysisAm J Dis Child19871419102110243618561
- HendrenWHThe genetic male with absent penis and urethrorectal communication: experience with 5 patientsJ Urol19971574146914749120985
- ElderJSWalshPCRetikABVaughanEDJrWeinAJCampbell’s Urology8th edSaunders: Elsevier Science200223432345
- GuptaAGuptaMAphallia. A rare congenital anomalyJK Science200810142143
- Gérard-BlanluetMLambertVKhung-SavatovskySPerrin-SabourinLAphallia, lung agenesis and multiple defects of blastogenesisFetal Pediatr Pathol2011301222621204662
- WheelerPGWeaverDDPartial urorectal septum malformation sequence: a report of 25 casesAm J Med Genet200110329910511568914
- ChienJ-CChenS-JTiuC-MChenY-JHwangBNiuD-MIs urorectal septum malformation sequence a variant of the vertebral defects, anal atresia, tracheo-oesophageal fistula, renal defects and radial dysplasia association? Report of a case and a review of the literatureEur J Pediatr2005164635035415729561
- GautierTSalientJPenaSImperto-McGinleyJPetersonRETesticular function in 2 cases of penile agenesisJ Urol198112645565577288953
- GrinsponRPReyRAWhen hormone defects cannot explain it: malformative disorders of sex developmentBirth Defects Res Part C Embryo Today Rev20141024359373
- EvansJAErdileLBGreenbergCRChudleyAEAgenesis of the penis: patterns of associated malformationsAm J Med Genet1999841475510213046
- GrippKWBarrMAnadiotisGMcDonald-McGinnDMZdericSAZackaiEHAphallia as part of urorectal septum malformation sequence in an infant of a diabetic motherAm J Med Genet199982536336710069704
- WheelerPGWeaverDDObeimeMOVanceGHBullMJEscobarLFUrorectal septum malformation sequence: report of thirteen additional cases and review of the literatureAm J Med Genet19977344564629415474
- SkoogSJBelmanABAphallia: its classification and managementJ Urol198914135895922918598
- PatraSPurkaitRUrorectal septum malformation sequence in a newborn with VACTERL associationJ Coll Physicians Surg Pak201222211611722313652
- AggarwalSPhadkeSRRecurrence of urorectal septum malformation sequence spectrum anomalies in siblings: time to explore the geneticsAm J Med Genet A2013161A71718172123686839
- TalwarSKapoorRAphalliaIndian Pediatr19882565795813235197
- KothareSVMarfatiaNSVidwansASShahMDCongenital penile agenesisIndian Pediatr19882565725763235196
- BahePSharmaDBaheAPanditaAAn infant with aphallia and its associated complication: a rare case report and review of literatureTrop Doct Epub2015317
- GoyalABianchiAThe parascrotal flap phallo-urethroplasty for aphallia reconstruction in childhood: report of a new techniqueJ Pediatr Urol201410476977224553074
- BajpaiMScrotal phalloplasty: A novel surgical technique for aphallia during infancy and childhood by pre-anal anterior coronal approachJ Indian Assoc Pediatr Surg201217416216423243368
- StolarCJWienerESHensleTWSilenMLSukarochanaKSieberWKReconstruction of penile agenesis by a posterior sagittal approachJ Pediatr Surg19872212107610803440890
- SarinYKSinhaAAphalliaIndian Pediatr200340436736812736414
- RattanKNKajalPPathakMKadianYSGuptaRAphallia: experience with 3 casesJ Pediatr Surg2010451E13E1620105566
- KanamoriYIwanakaTNakaharaSSurvival in a neonate with complete urorectal septum malformation sequence after fetal vesico-amniotic shunting for a prominently dilated cloacaFetal Diagn Ther200824445846119039229