
Abstract
Paroxysmal nocturnal hemoglobinuria (PNH), a disease characterized by intravascular hemolysis, thrombosis, and bone marrow failure, is associated with mutations in the PIG-A gene, resulting in a deficiency of glycosylphosphatidylinositol-anchored proteins. Many hypotheses have been posed as to whether PNH and PIG-A mutations result in an intrinsic survival benefit of CD55−/CD59− cells or an extrinsic permissive environment that allows for their clonal expansion within the bone marrow compartment. Recent data have identified the concurrence of PIG-A mutations with additional genetic mutations associated with myeloproliferative disorders, suggesting that some presentations of PNH are the result of a stepwise progression of genetic mutations similar to other myelodysplastic or myeloproliferative syndromes. We report for the first time in the literature the development of clinically significant PNH in a patient with JAK2V617F-negative, CALR-positive essential thrombocythemia, providing further support to the hypothesis that the development of PNH is associated with the accumulation of multiple genetic mutations that create an intrinsic survival benefit for clonal expansion. This case study additionally highlights the utility of genomic testing in diagnosis and the understanding of disease progression in the clinical setting.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a disease characterized by intra-vascular hemolysis, thrombosis, and bone marrow failure. The disease is associated with mutations in the PIG-A gene in hematopoietic stem cells, resulting in a deficiency of glycosylphosphatidylinositol (GPI)-anchored proteins.Citation1 This deficiency results in loss of CD55 and CD59, which are believed to be the main GPI-anchored proteins that serve to protect red blood cells (RBCs) from complement-mediated destruction.Citation2
This rare disease, estimated at two to five new cases per million US inhabitants, has led to the development of multiple hypotheses that seek to explain the role of PIG-A gene mutations and the survival, and clonal expansion, of CD55−/CD59− cells (PNH cells).Citation1 These hypotheses seek to understand whether there is either an extrinsic permissive environment or an intrinsic survival benefit to PNH cells that allows for the clonal expansion within the bone marrow compartment.Citation1,Citation3,Citation4 Additionally, the identification of CD55−/CD59− cells is not pathognomonic for clinical pathology, as PNH cells have been identified in normal individuals, thus further complicating the understanding of PNH and the role of PIG-A.Citation5,Citation6
While early hypotheses of PNH pathophysiology considered the entity as part of a myelofibrosis (MF)/myelodysplastic syndrome, these conceptualizations largely fell out of favor until recently with the advent of new high-throughput genetics and deep sequencing.Citation7 Deep sequencing studies have identified acquired somatic mutations in genes associated with myeloid neoplasms not only in hematologic malignancies but also in aging and in nonmalignant hematologic diseases such as aplastic anemia, suggesting that the development of a malignant process is bridged by the acquisition of multiple genetic mutations.Citation8,Citation9 Recent data have identified the concurrence of PIG-A mutations with genetic mutations associated with myeloproliferative disorders such as JAK2, HMGA2, and BCR-ABL, thus further supporting the hypothesis that in some occurrences of PNH, the development of clinically significant PNH is the result of a stepwise progression of multiple genetic mutations similar to other myelodysplastic or myeloproliferative syndromes.Citation10–Citation12
Myeloproliferative disease evolves to PNH
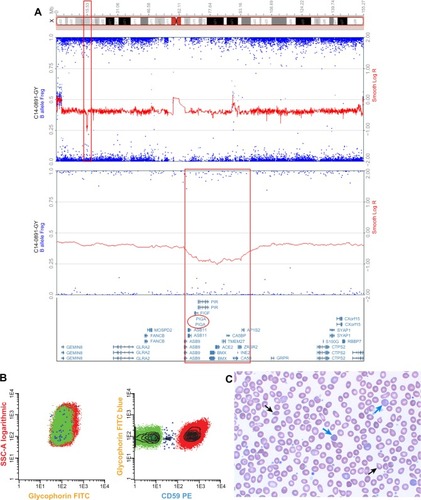
Here, we report for the first time in the literature the development of clinically significant PNH in a patient with JAK2V617F-negative, CALR-positive essential thrombocythemia (ET). The patient was initially diagnosed with ET in 2005 following the incidental finding of elevated platelets. The patient’s initial bone marrow examination was consistent with a myeloproliferative neoplasm (MPN). In 2011, the patient was noted to have ongoing thrombocytosis (platelets 795 K/mm3) but developed anemia (Hgb 9.0 g/dL), elevated lactate dehydrogenase (1,440 U/L), and marked reticulocytosis (absolute reticulocyte count 197.3 K/mm3) (). A repeat bone marrow biopsy showed megakaryocyte hyperplasia with myelofibrosis, consistent with post-ET myelofibrosis. The patient had a normal karyotype and a negative JAK2V617F mutation but was found to have CALR p.K385fs*47, an acquired somatic mutation strongly associated with the development of MPN.Citation13 His hemoglobin improved following iron supplementation, but hemolysis persisted with ongoing elevated lactate dehydrogenase, reduced haptoglobin, and reticulocytosis. Clinically, the patient complained of dyspnea on exertion and tea-colored urine. Urinalysis, which had previously been normal, revealed hemoglobinuria, 56 RBC/μL, and 4+ hemosiderin. High-resolution karyotype with a single-nucleotide polymorphism array revealed hemizygous loss of Xp22.2, an area of the genome that contains the PIG-A gene (). This microdeletion, which has been previously described, is a genetic aberration associated with the development of PNH.Citation14 Peripheral blood samples were used for multiparametric flow cytometry analysis based on fluorescent inactive aerolysin and the GPI-anchored proteins CD59 on RBCs and CD14 on monocytes and granulocytes. Flow cytometry identified loss of GPI-anchored proteins (PNH clone) comprising 14% of RBCs, 60% of granulocytes, and 73% of monocytes, thus confirming his diagnosis of PNH ().
Figure 1 Molecular and clinical phenotype of PNH in CALR mutation-positive MPN.
Abbreviations: ET, essential thrombocythemia; FITC, fluorescein isothiocyanate; MF, myelofibrosis; MPN, myeloproliferative neoplasm; PNH, paroxysmal nocturnal hemoglobinuria; SNP array, single-nucleotide polymorphism array; SSC, side scatter; PE, phycoerythrin.
Conclusion: insights into pathogenesis of PNH and the utility of genomic testing in the clinic
Though the development of PNH has been documented to be associated with a number of genetic aberrations associated with MPN and myelodyplasia, this is the first description of the development of PNH in a patient with a CALR mutation.Citation14 The progressive quality of his disease, initially from ET to post-ET myelofibrosis with eventual evolution of clinically significant PNH driven by microdeletion of Xp22.2, which encompassed the PIG-A gene, illustrates the evolving nature of myelodysplastic/myeloproliferative conditions and supports the hypothesis that multiple genomic “hits” may occur in order to develop clinically significant PNH. While not fully elucidated, the CALR mutation has been shown to lead to excessive cell proliferation.Citation15 We hypothesize that the CALR mutation conferred the survival benefit needed for clonal expansion and survival benefit within the bone marrow stem cell compartment of PIG-A mutant cells, thus cooperating to generate the PNH phenotype.
This case also importantly illustrates the utility of genomic testing outside the research setting and its utility in diagnosis when embedded within the clinical milieu. This patient’s normal karyotype coupled with his CALR mutation status allowed for appropriate identification of disease driving lesions and prognostication in ET. As his disease continued to evolve, single-nucleotide polymorphism microarray appropriately identified the etiologic cause of his worsening anemia and changing clinical presentation, thus demonstrating how the clinical availability of sensitive genetic testing leads to more accurate diagnosis, pathogenic understanding of disease process, and the development of more targeted and personalized treatments.Citation16
Disclosure
The authors report no conflicts of interest in this work.
References
- BrodskyRAParoxysmal nocturnal hemoglobinuria: stem cells and clonalityHematology Am Soc Hematol Educ Program20082008111111519074067
- BrodskyRAParoxysmal nocturnal hemoglobinuriaBlood2014124182804281125237200
- TiuRGondekLO’KeefeCMaciejewskiJPClonality of the stem cell compartment during evolution of myelodysplastic syndromes and other bone marrow failure syndromesLeukemia20072181648165717554386
- MaciejewskiJPSloandEMSatoTAndersonSYoungNSImpaired hematopoiesis in paroxysmal nocturnal hemoglobinuria/aplastic anemia is not associated with a selective proliferative defect in the glycosylphosphatidylinositol-anchored protein-deficient cloneBlood1997894117311819028939
- AratenDJNafaKPakdeesuwanKLuzzattoLClonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individualsProc Natl Acad Sci U S A19999695209521410220445
- MeletisJTerposESamarkosMDetection of CD55 and/or CD59 deficient red cell populations in patients with aplastic anaemia, myelo-dysplastic syndromes and myeloproliferative disordersHaematologia (Budap)200131171611345408
- HansenNEKillmannSAParoxysmal nocturnal hemoglobinuria in myelofibrosisBlood19703644284315455268
- YoshizatoTDumitriuBHosokawaKSomatic mutations and clonal hematopoiesis in aplastic anemiaN Engl J Med20153731354726132940
- XieMLuCWangJAge-related mutations associated with clonal hematopoietic expansion and malignanciesNat Med201420121472147825326804
- ShenWClementeMJHosonoNDeep sequencing reveals stepwise mutation acquisition in paroxysmal nocturnal hemoglobinuriaJ Clin Invest2014124104529453825244093
- TominagaRKatagiriTKataokaKParoxysmal nocturnal hemoglobinuria induced by the occurrence of BCR-ABL in a PIGA mutant hematopoietic progenitor cellLeukemia Epub2015106
- SugimoriCPadronECaceresGParoxysmal nocturnal hemoglobinuria and concurrent JAK2(V617F) mutationBlood Cancer J Epub2012323
- KlampflTGisslingerHHarutyunyanASSomatic mutations of calreticulin in myeloproliferative neoplasmsN Engl J Med2013369252379239024325356
- O’KeefeCLSugimoriCAfableMDeletions of Xp22.2 including PIG-A locus lead to paroxysmal nocturnal hemoglobinuriaLeukemia20112537938221116280
- SunCZhangSLiJCalreticulin gene mutations in myeloproliferative neoplasms without Janus kinase 2 mutationsLeuk Lymphoma20155661593159825115511
- FraimanYSMoliternoARHigh-density genomic analysis reveals basis of spherocytosis in myelodysplastic syndromeBlood201512522351726217825