
Abstract
Background
The mechanisms that regulate fetal hemoglobin (HbF) expression in sickle cell disease (SCD) remain elusive. We previously showed that steady-state SCD patients with high HbF levels due to a γ-globin gene mutation demonstrate strong inverse correlations between HbF levels and leukocyte counts, suggesting that leukocytes play a role in regulating HbF in SCD.
Materials and methods
To further investigate the role of leukocytes in HbF expression in SCD, we examined the presence of HbF silencing factors in the serum of 82 SCD patients who received hydroxyurea (HU) therapy.
Results
HU-mediated HbF induction was associated with elevated total hemoglobin levels and improved red blood cell parameters, but there was no correlation with reticulocyte or platelet counts. Importantly, we again found that HU-induced HbF levels correlated with reductions in both neutrophils and lymphocytes/monocytes, indicating that these cell lineages may have a role in regulating HU-mediated HbF expression. Our in vitro studies using CD34+-derived primary erythroblasts found that patient serum preparations include HbF silencing factors that are distinct from granulocyte-macrophage colony-stimulating factor, and the activity of such factors decreases upon HU therapy.
Conclusion
Together, these results demonstrate the importance of leukocyte numbers in the regulation of HbF levels for SCD patients both in steady state and under HU therapy, and that leukocytes secrete HbF silencing factors that negatively affect HbF expression in erythroid-lineage cells in SCD.
Introduction
The primary genetic defect in sickle cell disease (SCD) is a mutation of the β-globin gene.Citation1 It produces sickle hemoglobin, which polymerizes under low oxygen tension. This formation of sickle hemoglobin polymers is assumed to underlie vaso-occlusive crisis, chronic hemolysis, and ischemia–reperfusion injury associated with inflammation in this disorder.Citation2–Citation4
Despite sharing a common β-globin mutation, the clinical severity among patients with SCD is extremely heterogeneous and the underlying mechanisms remain unknown. Elevated levels of fetal hemoglobin (HbF) expression alleviate the clinical severity of SCD; however, expression levels are also variable.Citation5 HbF expression is regulated by single-nucleotide polymorphisms (SNPs) of multiple genetic loci,Citation6–Citation8 which presumably help to determine the level of HbF production in SCD.Citation9 In addition to HbF, which is otherwise dormant in people without anemia, leukocytosis is frequently observed in untreated SCD patients even in the absence of bacterial infection.Citation10 An elevated baseline leukocyte count is associated with a risk for early death.Citation11 Further, high leukocyte counts in children with SCD predict severe clinical complications later in life.Citation12 These clinical observations suggest that both HbF levels and leukocyte counts are consequential to the clinical severity of SCD.
Although the pharmacological stimulation of HbF expression by hydroxyurea (HU) is an established treatment for patients with clinically severe SCD,Citation13 HbF response to HU varies significantly and a number of SCD patients are resistant to HU therapy. Clarifying the predictors of HbF response to HU will allow clinicians to determine which patients are most likely to respond to HU therapy. This will in turn limit the toxicities associated with the treatment among those who would not benefit from the therapy. A study by Charache et al showed that initial leukocyte count and HbF concentration as well as post-therapy plasma HU levels are predictors of high post-therapy HbF levels.Citation14 Ware et al also demonstrated that greater changes in blood counts in SCD children on HU therapy result in better HbF response to HU.Citation15 However, the mechanisms by which these hematologic parameters predict HbF response remain elusive.
As to the mechanisms responsible for leukocytosis in SCD, we had reported a positive correlation between leukocyte count and plasma granulocyte-macrophage colony-stimulating factor (GM-CSF) levels in SCD patients,Citation16 indicating that plasma GM-CSF levels help to regulate leukocyte count. We subsequently analyzed retrospective data from SCD patients receiving HU therapy and found that the rate of HbF induction associated with HU therapy is proportional to the reduction of peripheral blood leukocyte count.Citation17 We also found that GM-CSF has a negative regulatory effect on HbF expression.Citation17 These results suggest that leukocytes are an important regulator for determining HbF response to HU.
To determine whether leukocytes are also involved in the regulation of HU-mediated HbF expression in SCD, in this study we analyzed retrospectively collected pre-HU and post-HU therapy hematological data. We found that HbF response was again associated with reduced leukocyte count, but not reticulocyte and platelet counts, suggesting that leukocytes likely play a critical role in HbF response to HU. Furthermore, our in vitro studies suggest that patient sera contain as-yet unidentified factors that appear to inhibit HbF expression in CD34+-derived erythroid progenitors cultured with erythropoietin (Epo). Interestingly, the ability to inhibit HbF activity varied among steady-state patients and was significantly decreased upon HU therapy. This study has revealed novel aspects of the molecular mechanisms by which HU regulates HbF expression, which will help us to better understand HU resistance among SCD patients.Citation15,Citation18
Materials and methods
SCD patient blood serum
The study was performed in accordance with the principles of the Declaration of Helsinki and approved by the institutional review board of Augusta University. We collected retrospective data on 337 adult SCD patients who were homozygous for the βS mutation and under the care of the Sickle Cell Center of the Medical College of Georgia at Augusta University; the clinical characteristics of this cohort were reported previously.Citation17 Of these patients, 82 were receiving HU therapy (15–35 mg/kg/day) and had not received transfusions for at least 6 months. Hematologic values were reported as the average of at least 3 months of data. Serum preparations that had no visible hemolysis were obtained from multiple patients under HU therapy, before daily intake of HU to minimize carryover of HU. Written informed consent was obtained from all patients.
In vitro culture of human CD34+-derived erythroid progenitor cells
Human CD34+ cells obtained from the National Heart, Lung and Blood Institute Programs of Excellence in Gene Therapy Hematopoietic Cell Processing Core (Fred Hutchinson Cancer Research Center, Seattle, WA, USA) were cultured by a method described previouslyCitation19 with minor modifications. Briefly, CD34+ cells (1–10 × 104 cells/mL) were cultured in Iscove’s modified Dulbecco’s medium containing 30% fetal bovine serum (FBS) or 30% human AB serum, 3 unit/mL Epo, 20 ng/mL stem cell factor, and 10 ng/mL interleukin 3. Culture media were replaced every 4 days. Anti-human GM-CSF antibody (Thermo Fisher Scientific, Waltham, MA, USA) was added to sera from SCD patients at a dilution ratio of 1:10 followed by a 30-minute incubation at room temperature before adding to culture media. Cells were harvested on day 14. Cell photographs were taken by EVOS FL Cell Imaging System (Advanced Microscopy Group, Bothell, WA, USA).
Flow cytometry
Cells were suspended in FACS buffer (phosphate-buffered saline containing 5% FBS and 0.1% sodium azide). Following incubation with human Fc blocker (Thermo Fisher Scientific) for 10 minutes at room temperature, cells were then washed twice in FACS buffer and stained at 4°C for 30 minutes with a phycoerythrin (PE)-labeled anti-human glycophorin A monoclonal antibody (CD235a PE) (BD Biosciences, San Jose, CA, USA) and fluorescein isothiocyanate-labeled CD71 monoclonal antibody (BD Biosciences). Paraformaldehyde was added in an equal volume to a final concentration of 0.1% to fix cells for 30 minutes at room temperature. The expression of glycophorin A and CD71 on erythroblasts was analyzed in a Becton-Dickinson FACScan using Cell Quest software (Franklin Lakes, NJ, USA).Citation20 Data were represented as percent of positive cells.
Isolation of whole cell extracts from erythroblasts and immunoblotting
Whole cellular extracts were prepared from CD34+-derived erythroblasts as described.Citation21 Briefly, cells (5–10 × 106 cells) were suspended with 1 × RIPA lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA) supplemented with 1 mM phenyl-methyl sulfonyl fluoride, 100 mM sodium orthovanadate, and protease inhibitor cocktail. Whole cellular extracts were obtained by centrifugation at 14,000× g for 15 minutes. Immunoblotting was performed as described previously.Citation22 Approximately 2–3 micrograms of cellular extracts were separated on 12% SDS polyacrylamide gels and transferred to nitrocellulose membranes (Invitrogen, Carlsbad, CA, USA). All antibodies used for immunoblotting analyses were purchased from Santa Cruz Biotechnology unless otherwise stated. Protein bands were visualized by the Phototope HRP Western blot detection system (Cell Signaling Technology, Danvers, MA, USA) according to the protocol provided by the supplier. Immunoblotting for γ-globin expression was performed by using serum preparations with no visible hemolysis that were isolated from multiple SCD patients.
Real-time (RT)-PCR
Expression of γ-globin mRNA in human CD34+-derived erythroid progenitors treated with or without SCD patients’ serum was examined by RT-PCR as described previously.Citation23 To determine expression levels of human γ-globin mRNA in primary erythroblasts, human CD34+ cells were cultured as described above. Total RNA was extracted from primary erythroblasts using RNeasy Mini Kit (Qiagen, Germantown, MD, USA), and cDNA was generated with the SuperScript II Reverse Transcriptase kit (Invitrogen). RT-PCR was carried out with the Mx3000P QPCR System (Agilent Technologies, Santa Clara, CA, USA) using SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. All amplifications were performed in triplicate, and 18S rRNA was used as internal control. Relative expression was quantitated using the standard ΔΔCt method. The primers used were as follows: human γ-globin, forward-5′-TGGATGATCTCAAGGGCAAC-3′; reverse-5′-TCAGTGGTATCTGGACA-3′; human 18S rRNA, forward-5′-TTGGAGGGCAAGTCTGGTG-3′; reverse-5′-CCGCTCCCAAGATCCAACTA-3′. All primers were designed and obtained from Integrated DNA Technologies (Coralville, IA, USA). RT-PCR for γ-globin mRNA expression was carried out by using several serum preparations with no visible hemolysis from multiple SCD patients.
Statistical analysis
The Spearman correlation coefficient (rs) was used for correlations with non-Gaussian distributed data, which included hematologic values (–). Other data were analyzed by Student’s t-test. P-values less than 0.05 were considered statistically significant.
Figure 1 HbF response to HU is associated with hematological improvements in SCD patients.
Abbreviations: HbF, fetal hemoglobin; HU, hydroxyurea; SCD, sickle cell disease.
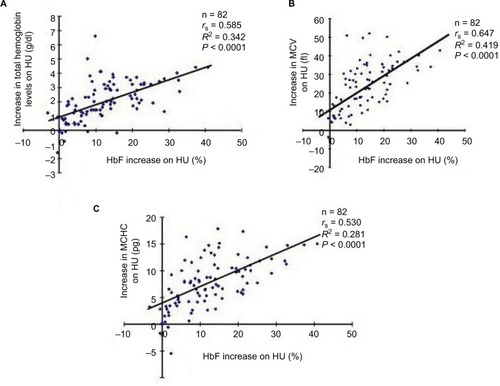
Figure 2 There were no correlations between HU-induced HbF increases and reductions in the number of reticulocytes (A) and platelets (B) among the SCD patients who received HU therapy.
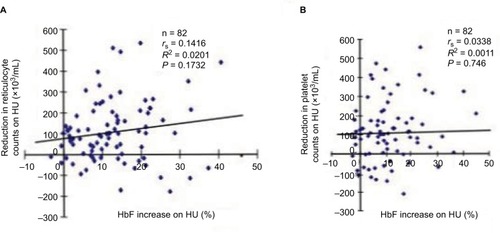
Figure 3 Correlations between the levels of HbF increases upon HU therapy and pre-HU therapy leukocyte counts (A), post-HU therapy leukocyte counts (B), or pre-HU therapy HbF levels (C). Eighty-two SCD patients who received HU therapy were analyzed.
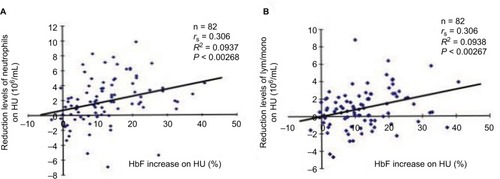
Figure 4 Correlations between neutrophil (A) or lymphocyte/monocyte counts (B) and HU-induced HbF levels.
Abbreviations: HU, hydroxyurea; HbF, fetal hemoglobin; SCD, sickle cell disease.
Results
Response of HbF to HU is associated with hematologic improvements in SCD patients
Our previous study showed that reduced leukocyte counts in response to HU therapy is critical for efficient HU-mediated HbF induction.Citation17 To further investigate the mechanisms by which HbF response is regulated in SCD, we first examined whether HbF response was associated with hematologic improvements in a retrospective study of a cohort of SCD patients described previously.Citation17 Of these, 82 HU responders were selected. There were substantial correlations between the levels of HbF induction by HU and increases in the total hemoglobin levels, MCV, and MCHC (). This suggests that HU therapy significantly improves RBC parameters, suggesting its clinical effectiveness for SCD. Our cohort of SCD patients may be pathophysiologically comparable to those reviewed recently.Citation24
HU-induced HbF levels are not correlated with reticulocyte or platelet counts
Next, we investigated whether HU-mediated HbF levels correlated with reticulocyte and platelet counts, both of which are assumed to play a role in modulating the pathophysiology of SCD.Citation25,Citation26 However, we saw no significant correlations between HU-induced HbF levels and reticulocyte or platelet counts (), suggesting that the mechanisms controlling the numbers of these lineage cells in peripheral blood may not be relevant to the mechanisms by which HbF response is regulated by HU.
Associations between HU-induced HbF levels and leukocyte counts
We had previously found that HU-induced HbF correlates with reduced leukocyte counts,Citation17 suggesting that the mechanisms by which HU regulates HbF induction in SCD may be relevant to those controlling leukocyte numbers in peripheral blood. Charache et al demonstrated that predictors of HbF response include pre-therapy leukocyte counts and HbF levels.Citation14 However, we found that HU-mediated HbF induction levels did not correlate with pre-therapy leukocyte counts (=0.056) but did correlate with post-therapy leukocyte counts (<0.004). The HbF increase was not influenced by pre-therapy HbF levels (, NS), a result that was inconsistent with Charache et al’s study.Citation14 This might reflect genetic and cellular differences in the cohorts studied.
To investigate the cell lineages of leukocytes involved in determining HbF response to HU therapy, we compared HU-mediated HbF increases and reductions of neutrophil or lymphocyte/monocyte counts (). The correlation value between the HbF increases and the reduction levels of neutrophil counts (P<0.00268) was the almost the same as that between the HbF increases and lymphocyte/monocyte reductions (P<0.00267; ). This suggests that both cell lineages are involved in determining HbF levels induced by HU.
HbF silencing factors that are present in blood serum of SCD patients
Importantly, the results shown in , together with our previous finding that leukocyte counts of steady-state SCD patients with high HbF levels demonstrated a strong inversely correlation with HbF levels (N = 47, RCitation2 = 0.229, P<0,0006),Citation17 has led us to hypothesize that leukocytes may have a role in downregulating HbF expression in erythroid-lineage cells by secreting HbF silencing factors.
Figure 5 Presence of HbF silencing factors in serum of SCD patients.
Abbreviations: HbF, fetal hemoglobin; SCD, sickle cell disease; FBS, fetal bovine serum; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HU, hydroxyurea; GM-CSF, granulocyte-macrophage colony-stimulating factor; RT-PCR, real-time PCR.
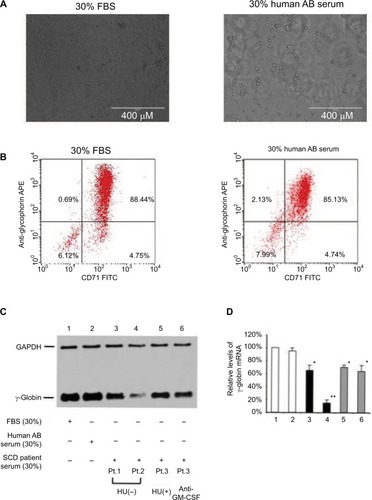
To detect the HbF silencing factors present in serum of SCD patients, we first determined whether CD34+ cells cultured with 30% human AB serum can be differentiated to erythroblasts as our previous studies had employed both FBS and human AB serum.Citation19,Citation20,Citation27 As shown in , 30% human AB serum permitted CD34+ cells to differentiate to erythroid-lineage cells to a degree similar to that by 30% FBS, as analyzed by flow cytometry on the basis of expression of glycophorin A and CD71. Next, we investigated the expression of both γ-globin and γ-globin mRNA in CD34+-derived erythroblasts incubated with FBS, normal human AB serum, or SCD patient serum (). The leukocyte counts of SCD patients studied here were as follows: Pt.1, 7,800/µL; Pt. 2, 15,300/µL; Pt. 3, 11,800/µL; the counts represent average numbers for 3–6 months. Although CD34+-derived erythroblasts (lane 2) cultured with 30% human AB serum expressed γ-globin at a level similar to that of cells cultured with 30% FBS (lane 1), the cells that were cultured with the serum of SCD patients demonstrated significantly lower levels of γ-globin expression (lanes 3–6). It is of note that the levels of γ-globin expression was substantially varied among steady-state SCD patients without HU (lanes 3 and 4), and serum from a SCD patient who was receiving HU revealed a weaker HbF silencing effect (lane 5). Because we previously showed that the proinflammatory cytokine GM-CSF has an inhibitory effect on HbF expression in erythroid-lineage cells,Citation17 it is confirmed that such HbF silencing is not blocked by the addition of anti-GM-CSF antibody (lane 6), indicating that HbF silencing factors are distinct from GM-CSF. Next, we examined γ-globin mRNA expression levels in these erythroblast preparations by RT-PCR (). The results of γ-globin mRNA expression were in accord with those of immunoblotting shown in . Thus, it is likely that in SCD, leukocytes may secrete HbF silencing factors that negatively affect HbF expression in erythroid cells, and that HU may reduce the serum levels of such factors, which may be relevant to HU-mediated HbF induction in SCD patients.
Discussion
It is of paramount importance to elucidate the mechanisms by which HU regulates HbF expression in the context of SCD because the magnitude of HbF response to HU in SCD is remarkably heterogeneous and a large subset of patients are non-responders.Citation28 Furthermore, HU-induced HbF increases are eventually lost even in SCD patients who initially responded to HU and these underlying mechanisms also remain unknown.Citation15,Citation18 A number of SNPs associated with HbF responders in SCD patients have been identified;Citation29 however, the significance and the implications of such SNPs are yet to be established.
We recently reported that HU-induced HbF levels correlate with reduced leukocyte counts.Citation17 This suggests that the mechanisms mediating HU-induced HbF expression may be relevant to those that control leukocyte counts. In this study, we performed a retrospective analysis of SCD patients for whom clinical data of pre-HU and post-HU therapy were available. We found that the levels of HbF induction following HU therapy are associated with the improvements of hematological parameters such as total hemoglobin, MCV, and MCHC (), suggesting that HbF response to HU is a legitimate marker for confirming the clinical effectiveness of HU therapy. By contrast, there were no significant correlations between HU-induced HbF levels and reticulocyte or platelet counts (), suggesting that these lineage cells are unlikely to play a role in determining HbF response to HU; rather, their changes reflect secondary effects of HU therapy.
Previous studies have shown correlations between HU-induced HbF expression and leukocyte counts.Citation15,Citation17,Citation30 We also found that HbF expression levels of steady-state SCD patients with high HbF levels due to a mutation in the Gγ-globin gene promoter inversely correlate with leukocyte counts.Citation17 Also, a notion that HU-induced HbF expression may be relevant to the mechanisms controlling leukocyte counts is supported by an earlier report by Steinberg that SCD patients with high baseline leukocyte counts who exhibit a great reduction in leukocytes with HU therapy have more robust increases in HbF.Citation31 Thus, multiple lines of clinical evidence strongly suggest that HbF expression is significantly influenced by the mechanisms controlling leukocyte count in the context of SCD.
The molecular and cellular mechanisms by which HU-induced HbF levels are associated with reduced leukocyte counts are not yet clear. Based on our study results, we hypothesize that leukocytes may secrete protein factors that bind to erythroid cells and inhibit HbF expression. Our in vitro studies () have demonstrated the presence of HbF silencing factors in the serum of SCD patients, whether or not they are receiving HU therapy. This is the first demonstration of HbF silencing factors in the serum of SCD patients. Interestingly, the HbF silencing activity in the serum varied significantly among SCD patients (, lanes 3 and 4). However, it can be argued that such factors may not in fact be secreted by leukocytes. Leukocyte numbers usually decrease in response to HU therapy in SCD because HU is a ribonucleotide reductase inhibitor.Citation32 Consistent with this clinical evidence, HbF silencing activities are also concomitantly reduced in someone with SCD who is receiving HU ( lane 5). Further rigorous scrutiny is required to verify that HbF silencing factors derive from leukocytes. Importantly, such HbF silencing factors were not absorbed by anti-GM-CSF antibody (), suggesting that HbF silencing factors are distinct from GM-CSF, which we showed has a negative consequence on HbF expression.Citation17
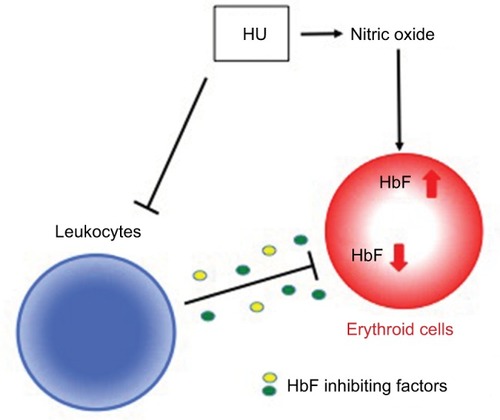
Our current model of the role of leukocytes in HU-mediated HbF expression in erythroid cells is summarized in . We and others have shown that the soluble guanylate cyclase (sGC)-cGMP pathway plays a role in HU-induced HbF expression.Citation33–Citation35 HU is assumed to augment HbF levels at least in part by generating nitric oxide and through the sGC-cGMP pathway. Although the sGC-cGMP pathway is also implicated in erythroid cells as a signaling mechanism for chemically induced HbF expression, it is still unknown whether extracellular signals from other lineage cells are transduced to erythroid cells and whether such signals are capable of modulating HbF expression by intracellular sig-naling pathways. In this study, we have clearly shown that HbF levels in SCD patients, whether they are receiving HU treatment, or not, are closely affected by leukocyte count in peripheral blood, presumably because of HbF silencing factors. As leukocyte count generally decreases in response to HU administration in SCD patients, it is reasonable to speculate that in addition to the nitric oxide/sGC-cGMP axis, HU might also increase HbF expression in SCD patients by inhibiting the secretion of HbF silencing factors from leukocytes. Collectively, there may be at least three groups of HbF regulatory proteins or pathways involved in HbF expression are possibly involved in inducing vaso-occlusive crisis therapy. One is GM-CSF, which is supposed to cause leukocytosis in SCDCitation16 and has a negative consequence on HbF expression.Citation17 Myeloid cytokines such as G-CSF or GM-CSF are possibly involved to induce vaso-occlusive crisis.Citation36,Citation37 Second, HU is shown to stimulate HbF expression at least in part through the nitric oxide/sGC-cGMP axis.Citation33–Citation35 This is consistent with prior vitro and in vivo findings showing that nitric oxide is generated from HU.Citation38,Citation39 A third mechanism includes HbF silencing factors that are presumably released mainly from leukocytes, as reported in this study (). HbF silencing factors may be cytokines or chemokines; further studies are necessary to precisely characterize HbF silencing factors.
Figure 6 Possible model for the mechanisms that regulate HU-induced HbF expression.
Abbreviations: HU, hydroxyurea; HbF, fetal hemoglobin; sGC, soluble guanylate cyclase.
Conclusion
This study has shown that HU-induced HbF expression is regulated at least in part by the mechanisms controlling leukocyte counts, and that HbF silencing factors that are secreted, possibly by leukocytes, may be involved in HU-regulated HbF expression in SCD. Thus, this study provides an important clue to the mechanisms by which HbF expression is regulated in the context of SCD patients receiving HU. It would be interesting to compare serum levels of cytokines or chemokines in SCD patients, both with and without HU treatment. Further insight into HbF silencing factors might help us clarify the mechanisms underlying HU resistance as seen in a subset of SCD patients.Citation15,Citation18
Acknowledgments
This study was supported in part by National Institutes of Health grants (DK61806, HL73452, and P20 MD003383 to TI) and the American Heart Association USA (15GRNT25710387 to TI). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
- PaulingLItanoHASingerSJWellsICSickle cell anemia a molecular diseaseScience1949110286554354815395398
- CharacheSBartonFBMooreRDHydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell AnemiaMedicine (Baltimore)19967563003268982148
- KaulDKHebbelRPHypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal miceJ Clin Invest2000106341142010930444
- ReiterCDWangXTanus-SantosJECell-free hemoglobin limits nitric oxide bioavailability in sickle-cell diseaseNat Med200181213831389
- MillerBASalamehMAhmedMHigh fetal hemoglobin production in sickle cell anemia in the eastern province of Saudi Arabia is genetically determinedBlood1986675140414102421808
- CraigJERochetteJFisherCADissecting the loci controlling fetal haemoglobin production on chromosomes 11p and 6q by the regressive approachNat Genet199612158648528252
- CloseJGameLClarkBBergouniouxJGerovassiliATheinSLGenome annotation of a 1.5 Mb region of human chromosome 6q23 encompassing a quantitative trait locus for fetal hemoglobin expression in adultsBMC Genomics200451334615169551
- YavarianMKarimiMBakkerEHarteveldCLGiordanoPCResponse to hydroxyurea treatment in Iranian transfusion-dependent beta-thalassemia patientsHaematologica200489101172117815477200
- LettreGSankaranVGBezerraMADNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell diseaseProc Natl Acad Sci U S A200810533118691187418667698
- BoggsDRHydeFSrodesCAn unusual pattern of neutrophil kinetics in sickle cell anemiaBlood197341159654682080
- PlattOSBrambillaDJRosseWFMortality in sickle cell disease. Life expectancy and risk factors for early deathN Engl J Med199433023163916447993409
- MillerSTSleeperLAPegelowCHPrediction of adverse outcomes in children with sickle cell diseaseN Engl J Med20003422838910631276
- BrawleyOWCorneliusLJEdwardsLRNational Institutes of HealthConsensus Development Conference Statement: hydroxyurea treatment for sickle cell diseaseAnn Intern Med20081481293293818458271
- CharacheSDoverGJMooreRDHydroxyurea: effects on hemoglobin F production in patients with sickle cell anemiaBlood19927910255525651375104
- WareREEgglestonBRedding-LallingerRPredictors of fetal hemoglobin response in children with sickle cell anemia receiving hydroxyurea therapyBlood2002991101411756146
- ConranNSaadSTCostaFFIkutaTLeukocyte numbers correlate with plasma levels of granulocyte-macrophage colony-stimulating factor in sickle cell diseaseAnn Hematol200786425526117205286
- IkutaTAdekileADGutsaevaDRThe proinflammatory cytokine GM-CSF downregulates fetal hemoglobin expression by attenuating the cAMP-dependent pathway in sickle cell diseaseBlood Cells Mol Dis201147423524221945571
- SteinbergMHLuZHBartonFBTerrinMLCharacheSDoverGJFetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter Study of HydroxyureaBlood1997893107810889028341
- BaileyLKuroyanagiYFranco-PenteadoCFExpression of the gamma-globin gene is sustained by the cAMP-dependent pathway in beta-thalassaemiaBr J Haematol2007138338239517614826
- KuroyanagiYKanekoYMutaKcAMP differentially regulates gamma-globin gene expression in erythroleukemic cells and primary erythroblasts through c-Myb expressionBiochem Biophys Res Commun200634431038104716631597
- IkutaTKanYWSwerdlowPSFallerDVPerrineSPAlterations in protein-DNA interactions in the gamma-globin gene promoter in response to butyrate therapyBlood1998928292429339763579
- SuzukiYTakedaYIkutaTImmunoblotting conditions for human hemoglobin chainsAnal Biochem2008378221822018445469
- GhoshalPRajendranMOdoNIkutaTGlycosylation inhibitors efficiently inhibit P-selectin-mediated cell adhesion to endothelial cellsPLoS One201496e9936324945938
- LanzkronSStrouseJJWilsonRSystematic review: hydroxyurea for the treatment of adults with sickle cell diseaseAnn Intern Med20081481293995518458272
- BorbaRLimaCSGrottoHZReticulocyte parameters and hemoglobin F production in sickle cell disease patients undergoing hydroxyurea therapyJ Clin Lab Anal2003172667212640630
- LeeSPAtagaKIOrringerEPPhillipsDRPariseLVBiologically active CD40 ligand is elevated in sickle cell anemia: potential role for platelet-mediated inflammationArterioscler Thromb Vasc Biol20062671626163116601237
- UddinSAh-KangJUlaszekJMahmudDWickremaADifferentiation stage-specific activation of p38 mitogen-activated protein kinase isoforms in primary human erythroid cellsProc Natl Acad Sci U S A2004101114715214694199
- RodgersGPDoverGJNoguchiCTSchechterANNienhuisAWHematologic responses of patients with sickle cell disease to treatment with hydroxyureaN Engl J Med199032215103710451690857
- MaQWyszynskiDFFarrellJJFetal hemoglobin in sickle cell anemia: genetic determinants of response to hydroxyureaPharmacogenomics J20077638639417299377
- CharacheSDoverGJMoyerMAMooreJWHydroxyurea-induced augmentation of fetal hemoglobin production in patients with sickle cell anemiaBlood19876911091162431728
- SteinbergMHManagement of sickle cell diseaseN Engl J Med1999340131021103010099145
- YarbroJWMechanism of action of hydroxyureaSemin Oncol1992193 Suppl 9110
- IkutaTAusendaSCappelliniMDMechanism for fetal globin gene expression: role of the soluble guanylate cyclase-cGMP-dependent protein kinase pathwayProc Natl Acad Sci U S A20019841847185211172039
- CokicVPSmithRDBeleslin-CokicBBHydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclaseJ Clin Invest2003111223123912531879
- IkutaTCappelliniMDA novel mechanism for fetal globin gene expression: role of the soluble guanylate cyclase-cyclic GMP pathway [abstract]Blood199994615a
- PietersRCRojerRASalehAWSalehAEDuitsAJMolgramostim to treat SS-sickle cell leg ulcersLancet19953458948528
- AbboudMLaverJBlauCAGranulocytosis causing sickle-cell crisisLancet19983519107959
- JiangJJordanSJBarrDPGuntherMRMaedaHMasonRPIn vivo production of nitric oxide in rats after administration of hydroxyureaMol Pharmacol1997526108110869415718
- GloverREIvyEDOrringerEPMaedaHMasonRPDetection of nitrosyl hemoglobin in venous blood in the treatment of sickle cell anemia with hydroxyureaMol Pharmacol19995561006101010347241