
Abstract
From clinical and laboratory studies of specific coagulation defects induced by injury, damage control resuscitation (DCR) emerged as the most effective management strategy for hemorrhagic shock. DCR of the trauma patient who has sustained massive blood loss consists of 1) hemorrhage control; 2) permissive hypotension; and 3) the prevention and correction of trauma-induced coagulopathies, referred to collectively here as acute coagulopathy of trauma (ACOT). Trauma patients with ACOT have higher transfusion requirements, may eventually require massive transfusion, and are at higher risk of exsanguinating. Distinct impairments in the hemostatic system associated with trauma include acquired quantitative and qualitative platelet defects, hypocoagulable and hypercoagulable states, and dysregulation of the fibrinolytic system giving rise to hyperfibrinolysis or a phenomenon referred to as fibrinolytic shutdown. Furthermore, ACOT is a component of a systemic host defense dysregulation syndrome that bears several phenotypic features comparable with other acute systemic physiological insults such as sepsis, myocardial infarction, and postcardiac arrest syndrome. Progress in the science of resuscitation has been continuing at an accelerated rate, and clinicians who manage catastrophic blood loss may be incompletely informed of important advances that pertain to DCR. Therefore, we review recent findings that further characterize the pathophysiology of ACOT and describe the application of this new information to optimization of resuscitation strategies for the patient in hemorrhagic shock.
Introduction
Massive hemorrhage from extensive trauma may rapidly overwhelm hemostatic systems that, arguably, evolved in response to lesser amounts of bleeding from more confined areas of injury.Citation1 Many patients suffering a catastrophic loss of blood will develop a complex multisystem dysfunction syndrome, which includes specific defects in the coagulation system referred to collectively as acute coagulopathy of trauma (ACOT).Citation2
ACOT is defined by clinical and laboratory findings suggesting acquired coagulopathy in patients with severe trauma. This pathophysiologic entity appears early after injury typically before any intervention has been instituted. This characteristic separates ACOT from coagulopathies secondary to dilution or consumption known to develop in trauma patients, particularly during resuscitation (referred to as resuscitation-induced coagulopathies.) ACOT is estimated to occur in ~25% of severely injured patients (Injury Severity Score [ISS] > 25). In one prospective, observational study ACOT was found in 16.3% of all admitted trauma patients, including 11% of mildly injured patients (patients without physiological derangement or blood product administration).Citation3
Coagulopathy in trauma patients is suggested by nonsurgical bleeding that appears, for example, from superficial abrasions, lacerations, and sites of vascular puncture, or by an increase in far more problematic surgical bleeding from sites of hemorrhage within major injuries. ACOT may manifest very soon after injury and is often present in the trauma patient prior to arriving at the hospital. Predictably, hemostatic failure in these particular circumstances substantially complicates management and significantly increases rates of adverse outcomes, including a fourfold increase in mortality.Citation4
Hemostatic failure in the trauma patient must be anticipated and promptly diagnosed. Furthermore, the resuscitation of a patient in hemorrhagic shock incorporates hemostatic resuscitation strategies designed to rapidly correct coagulation system abnormalities and prevent further deterioration in the patient’s capacity to form clot. Hemostatic resuscitation exists within the broader context of damage control resuscitation (DCR), which is characterized by certain additional clinical aspects.Citation5 First, DCR involves rapid and often massive infusion of packed red blood cells (pRBCs) to expeditiously restore global oxygen delivery. Transfusion of this magnitude (referred to as “massive transfusion” or MT) has been arbitrarily characterized as the transfusion of ≥10 units of pRBCs within a 24-h period.Citation6 However, this definition is not considered a valid surrogate for severe hemorrhageCitation7 since it does not accurately reflect the intensity of blood loss and fails to capture exsanguinating patients who die before receiving 10 units (survival bias).Citation8 More recently, MT has been characterized as transfusion of ≥5 units of RBCs over 4 hCitation9 or ≥6 units of RBC in 6 h. However, an international forum on the treatment of trauma coagulopathy identified no fewer than 12 different definitions for MT.Citation10 In general, newer definitions of MT delineate use of blood products within a more narrow time frame.Citation6 For example, the critical administration threshold is defined as the transfusion of at least 3 units of pRBCs in any 1-h period of time, within the first 24 h.Citation11 Second, DCR is carried out accepting a lower perfusion pressure as a resuscitation end point (referred to as permissive hypotension). Third, DCR protocols specify prompt recognition and management of profound homeostatic imbalances (eg, hypocalcemia and hypothermia) that arise in the course of resuscitation or in the aftermath of this intervention. And last, DCR is initiated while anticipating immediate hemorrhage control, which may be achieved by emergent surgical (including Resuscitative Endovascular Balloon Occlusion of the Aorta placement), angiographic, or endoscopic approaches.
A coordinated effort of multiple disciplines has resulted in the development of DCR protocols that are associated with an increase in survival of the severely injured in concert with a reduction in costly waste of blood bank products.Citation12–Citation17 Trauma center verification (in particular, of Level 1 and Level 2 centers) by the American College of Surgeons Committee on Trauma is predicated on demonstration of both an organized capability to carry out DCR and a commitment to the process.
Although trauma patients account for 90% of those receiving this complex, resource-intensive, and potentially dangerous intervention, a specific discussion of DCR for trauma may be extended to treatment of significant blood loss for other reasons, including massive gastrointestinal bleeding, gynecological hemorrhagic crises, and intraoperative and postoperative hemorrhage complicating vascular, cardiovascular, and other complex surgical procedures.Citation18,Citation19 For example, treatment of severe postpartum hemorrhage (PPH) complicating high-risk obstetrical cases may be optimized when carried out by a multidisciplinary, rapid response team well-versed in the pathophysiology of hemorrhagic shock and the principles of DCR.Citation20,Citation21
Here, we describe processes that destabilize the balance of interdependent effectors and modulators of coagulation and fibrinolysis associated with the acquisition of ACOT. Furthermore, we review therapeutic principles upon which correction of these pathological processes is achieved. However, the pathophysiology of hemorrhagic shock is not yet completely understood, and it is expected that the management of massive blood loss will continue to be significantly modified as further information derived from original investigation emerges.
Cell-based model of clot formation
Hemostasis is an exact balance between opposing pathways of coagulation and fibrinolysis.Citation22 In response to injury, formation of thrombin occurs in four corresponding phases:Citation23,Citation24 1) “initiation”, during which small amounts of coagulation factor (F) IX and FX are activated by exposed subendothelial tissue factor (TF) in the presence of constitutively activated FVII (FVIIa); 2) “amplification”, in which FXa produces a small amount of thrombin that in turn activates FV, FVIII, FIX, and resting platelets; 3) “propagation”, by which activated coagulation factors assemble on aggregating platelets to mediate formation of thrombin in amounts sufficient to convert fibrinogen into long noncovalently linked fibers (fibrin); and 4) stabilization through which fibrin is converted to a covalently linked polymerCitation23 (). Covalent cross-linking of fibrin fibers by thrombin-activated FXIII increases the elasticity of individual fibers, establishing the overall viscoelastic properties of a fibrin clot.Citation25 A blood clot is a viscoelastic polymer that exhibits both the elastic properties of a spring and the viscous properties of a fluid.Citation26 Furthermore, thrombin promotes resistance of formed clot to fibrinolytic degradation through activation of a plasminogen-bound carboxypeptidase, thrombin-activatable fibrinolysis inhibitor (TAFI).Citation27
Figure 1 Normal hemostasis involves both coagulation and fibrinolysis regulated by several physiologic inhibitors. (A) Phases of a cell-based model of coagulation include “initiation” that occurs after exposure of TF in the presence of VIIa, a small amount of which is normally present in the circulation, followed by “priming” in which FXa produces a small amount of thrombin to activate FVIII, FV; and platelets, resulting in “propagation”, involving assembly of activated coagulation factors on aggregating platelets to produce a thrombin burst on the surface of platelets that mediate conversion of fibrinogen to fibrin. Three endogenous anticoagulation systems modulate coagulation to prevent clot formation in excess (thrombosis) beyond the site of injury. These are predominately localized to expression on vascular endothelial surfaces. Trauma-associated coagulopathy is related to excess thrombomodulin (TM) expression resulting in high concentrations of activated protein C (aPC) and possibly to disruption of the endothelial glycocalyx with release of heparin-like proteoglycans that interact with antithrombin in a process referred to as auto-heparinization. A role for TFPI in trauma-associated coagulopathy has not been identified; however, experimentally, depletion of TFPI predisposes to disseminated intravascular coagulation, which has been suggested to be a mechanism of coagulopathy following trauma. (B) Fibrinolytic pathway and endogenous mediators that regulate fibrinolysis. Plasmin is generated from plasminogen by tPA and is controlled by several inhibitors, principally PAI-1. Upregulation of TM and thrombin–TM interactions increases thrombin-mediated activation of TAFI 1,000-fold; elevated levels of aPC may promote fibrinolysis by limiting thrombin production and consequently TAFI activation. Hyperfibrinolysis associated with trauma is thought to occur after endothelial cell perturbation following injury with trauma-induced release of large amounts of tPA in association with downregulation of PAI-1. Fibrinolysis shutdown is thought to occur through trauma-mediated dysregulation of PA-1 expression.
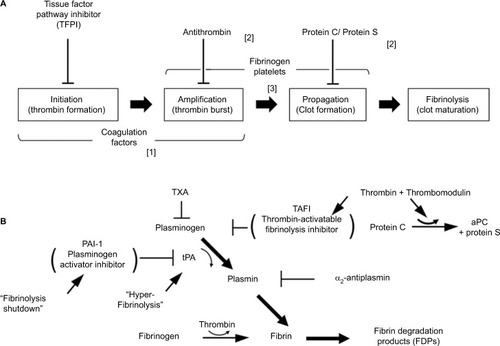
Plasmin is the critical mediator of fibrinolysis. The accumulation of fibrin promotes activation of the fibrinolytic system by enhancing tissue plasminogen activator (tPA)-mediated conversion of plasminogen to plasmin. Fibrinogen also promotes plasminogen activation but at a rate that is ~100-fold less than fibrin, which tends to concentrate fibrinolytic activity to fibrin clot. In addition to limiting extension of clots beyond sites of injury, plasmin mediates subsequent dissolution of clots once repair of injured tissue is established ().
Several endogenous systems regulate coagulation and fibrinolysis. Tissue factor pathway inhibitor (TFPI) modulates three cofactor complexes, TF–FVIIa, FXa–FVa, and FIXa–FVIIIa, depending on TFPI isoform.Citation28,Citation29 Inhibition of prothrombinase by TFPIα may have direct clinical relevance,Citation30 although TFPI modulation of TF–VIIa complexes may be insufficient to control coagulation in extreme conditions.Citation31 Activated protein C (aPC)Citation32 in the presence of protein S, sphingolipids, and/or high-density lipoproteins inactivates FVa and FVIIIa. Consequently, tenase complex and prothrombinase complex formation diminish, and the conversion of prothrombin to thrombin is attenuated. aPC also promotes clot breakdown by blocking plasminogen activation inhibitor-1 (PAI-1)-mediated inhibition of fibrinolysis.Citation33 Antithrombin (AT) inactivates thrombin, and to a lesser extent inhibits the activities of FXa, FIXa, FXIa, and FXIIa. The inactivation of thrombin by AT is accelerated several fold by binding of glycosaminoglycans, including heparin and heparin sulfate to AT, which induces a specific allosteric change in AT.Citation34 AT, TFPI, and aPC suppress proinflammatory mediators as well.Citation35
Thrombomodulin is an endothelial cell surface protein that couples the downregulation of coagulation and fibrinolysis by functioning as an essential cofactor in the activation of PC and TAFI. TAFI blocks fibrinolysis by cleaving plasmin-exposed lysine residues on fibrin clot, which limits tPA binding to fibrin, thereby reducing any further conversion of plasminogen to plasmin. Furthermore, TAFI inhibits complement anaphylatoxins C3a and C5a indicating a potentially important anti-inflammatory role for this carboxypeptidase in addition to a procoagulant function.Citation36–Citation38 Thrombin binding to thrombomodulin (TM) converts the substrate specificity of thrombin from fibrinogen to PC and TAFI. Thus, during downregulation of hemostasis under nonpathological conditions, the balance between coagulation and fibrinolysis is maintained. However, in trauma patients a systemic injury to the vascular endothelium results in the release of angiopoetin-2 (Ang-2) from endothelial cell Weibel-Palade bodies.Citation39 Ang-2 has recently been demonstrated to bind to endothelial cell TM and thereby block thrombin–TM interactions.Citation40 Whether Ang-2 binds soluble TM under these conditions has not yet been established. Loss of thrombin-mediated activation of PC and TAFI, in effect, decouples inhibition of coagulation and fibrinolysis. The persisting activity of these two pathways would then be determined by the relative effectiveness of other specific inhibitors.
In an unperturbed state, endothelial Ang-1 and Ang-2 maintain vascular homeostasis through tightly regulated interaction with the kinase receptor, Tie2 expressed on quiescent endothelial cells. In contrast, Ang-2, released from activated cells ECs, destabilizes vascular function.Citation41 Ang-2–Tie2 interactions activate the conical NF-κB signaling pathway leading to expression of proteins that mediate leukocyte firm adhesion and transmigration across the endothelial barrier. NF-κB nuclear translocation under these conditions also promotes expression of IL-6 and TNF-α in both plasma and lung.Citation42,Citation43 These systemic auto-inflammatory processes may be associated mechanistically with the development of trauma-induced acute respiratory distress syndrome (ARDS).Citation44 Damage to the alveolar epithelium is considered the principle mechanism causing increased pulmonary permeability, permitting accumulation of edema fluid containing high concentrations of macromolecules in the alveoli.Citation45 The breakdown of the alveolar–epithelial barrier is a consequence of multiple factors that include dysregulated inflammation, intense leukocyte infiltration, activation of procoagulant processes, cell death, and mechanical stretch.Citation46
Rapid assessment of hemostasis
Conventional coagulation tests, such as prothrombin time, activated partial thromboplastin time, international normalized ratio, fibrinogen concentration, platelet count, and assays to detect fibrinolysis are performed on cell-free plasma samples at 37°C. Typically, assays require up to 60 min to complete even though clinical conditions may be changing rapidly, and conventional tests of coagulation have never been validated in bleeding trauma patients.Citation47
The plasma half-life of thrombin is very short, which precludes measurement of plasma concentrations; therefore, other parameters have been used as evidence of thrombin generation in the systemic circulation, such as thrombin-AT complex levels and soluble fibrin levels. Moreover, conventional evaluations of coagulation does not generally include an assessment of platelet function.Citation48,Citation49 In contrast, rheology-based coagulation tests quantify the viscoelasticity of clots in real time and may provide more clinically relevant data within 5–10 min. In addition, these assays are readily repeated, and by superimposing tracings upon one another, we are able to detect important trends in the results of multiple decisions made during the course of a resuscitation that otherwise would not be apparent.
Viscoelastic hemostatic assays essentially integrate assessments of the kinetics of clot formation, the physical properties of the forming clot, which includes the clot elastic modulus (CEM), and the extent of fibrinolytic activity. CEM is a critical parameter defining clot flexibility to shear force.Citation50 Since whole blood is used in the assay, clots are a more realistic composition of platelets, fibrin, and erythrocytes compared with clots formed in conventional test done on plasma only.
A standard assay measures five parameters, which correlate with various phases of coagulationCitation51–Citation53 and certain coagulopathies (). Viscoelastic hemostatic assays do not include a platelet count, and an evaluation of platelet function requires additional assay modules. Two systems, thromboelastography (TEG®; Haemonetics Corp, Niles, IL, USA) and rotational thromboelastometry (ROTEM®; Tem International GmbH, Munich, Germany), are used in clinical laboratory or point-of-care (POC) coagulation testing. TEG and ROTEM both examine properties of hemostasis under low shear stress conditions. However, neither TEG nor ROTEM assesses the contribution of the endothelium to coagulation.
Figure 2 Thromboelastography (TEG®). (A) Schematic presentation of different viscoelastic tracings reflecting states of the coagulation system compared with normal. (B) Basic viscoelastic tracing with measured parameters and limits of normal for thromboelastography, correlated with different elements of the coagulation system (R = reaction time, K = clot formation time, angle, MA = maximum amplitude, Ly30 = percent clot lysis 30 m after MA). Viscoelastic k-time and angle correlate to some degree with fibrinogen concentration. However, the agreement between these parameters and fibrinogen levels determined by standard von Clauss assay is not sufficiently strong to be useful clinically.Citation174–Citation176 To overcome this limitation with TEG, the specific contributions of fibrinogen and platelets to clot strength can be determined with additional reagents (TEG; Functional Fibrinogen [Haemonetics Corp, Niles, IL, USA]).Citation177 Using TEG, additional measures of clot strength can be computed. Coagulation index (CI; black arrow) is derived from R, k-time, angle, and MA, with a CI > +3.0 suggesting a hypercoagulable state and CI <–3.0 suggesting coagulopathy. The shear elastic module strength, designated G, is a computer-generated quantity that reflects an integrated measure of clot strength. Conceptually, G is considered the most informative parameter of clot strength because it reflects the contributions of the enzymatic and platelet components of hemostasis.Citation178,Citation179 (C) Thrombus generation velocity curve is a mathematical first derivative of the TEG tracing, which provides additional information with respect to both thrombus formation and lysis.Citation179 For example, velocity curve measures of fibrinolysis are stronger predictors of early transfusion of blood components, bleeding, and mortality after trauma compared with conventional rTEG values. In addition, the maximal rate of lysis is more rapidly available after arrival, which may facilitate earlier diagnosis and treatment of clinically significant hyperfibrinolysis.Citation180
![Figure 2 Thromboelastography (TEG®). (A) Schematic presentation of different viscoelastic tracings reflecting states of the coagulation system compared with normal. (B) Basic viscoelastic tracing with measured parameters and limits of normal for thromboelastography, correlated with different elements of the coagulation system (R = reaction time, K = clot formation time, angle, MA = maximum amplitude, Ly30 = percent clot lysis 30 m after MA). Viscoelastic k-time and angle correlate to some degree with fibrinogen concentration. However, the agreement between these parameters and fibrinogen levels determined by standard von Clauss assay is not sufficiently strong to be useful clinically.Citation174–Citation176 To overcome this limitation with TEG, the specific contributions of fibrinogen and platelets to clot strength can be determined with additional reagents (TEG; Functional Fibrinogen [Haemonetics Corp, Niles, IL, USA]).Citation177 Using TEG, additional measures of clot strength can be computed. Coagulation index (CI; black arrow) is derived from R, k-time, angle, and MA, with a CI > +3.0 suggesting a hypercoagulable state and CI <–3.0 suggesting coagulopathy. The shear elastic module strength, designated G, is a computer-generated quantity that reflects an integrated measure of clot strength. Conceptually, G is considered the most informative parameter of clot strength because it reflects the contributions of the enzymatic and platelet components of hemostasis.Citation178,Citation179 (C) Thrombus generation velocity curve is a mathematical first derivative of the TEG tracing, which provides additional information with respect to both thrombus formation and lysis.Citation179 For example, velocity curve measures of fibrinolysis are stronger predictors of early transfusion of blood components, bleeding, and mortality after trauma compared with conventional rTEG values. In addition, the maximal rate of lysis is more rapidly available after arrival, which may facilitate earlier diagnosis and treatment of clinically significant hyperfibrinolysis.Citation180](/cms/asset/2249eafb-d80b-4c52-8fd5-5b1a7f95b8e1/djbm_a_165394_f0002_b.jpg)
Rapid TEG® (r-TEG) can be completed within 15 min compared with an average 30–45 min processing time for a standard TEG assay. In an r-TEG assay, samples are activated with TF in addition to kaolin which yields an activated clotting time, or TEG-ACT, in place of the reaction time (R-time). TEG-ACT may predict abnormalities that will appear later in the tracing. For example, ACT >140 s predicts a flattened angle and diminished MA, indicating almost immediately a requirement for cryoprecipitate and platelet transfusions that would not have been known.Citation54 Data from a recent pragmatic, randomized clinical trial indicate now that utilization of a TEG-guided massive transfusion protocol (referred to as a “goal-directed” protocol) to resuscitate severely injured patients improves survival compared with massive transfusion guided by conventional coagulation assays and utilizes less plasma and platelet transfusions during the early phase of resuscitation.Citation55 In addition, the fibrinogen assay component of TEG may detect fibrinolysis much earlier permitting more timely and appropriate utilization of antifibrinolytic therapy.Citation56
Acute coagulopathy of trauma
Distinct impairments in the coagulation system characterize ACOT. These include acquired quantitative and qualitative platelet defects, hypocoagulable and hypercoagulable states, and dysregulation of the fibrinolytic system giving rise to HF or a phenomenon referred to as fibrinolytic shutdown.Citation57 Of interest, ACOT is a component of a systemic host defense dysregulation syndrome that bears several phenotypic features comparable with those of other acute systemic physiological insults, including sepsis, myocardial infarction, and postcardiac arrest syndrome.Citation58
Defects in platelet function may be caused by injury,Citation59 particularly traumatic brain injury (TBI),Citation60–Citation63 secondary to downregulation of platelet P2Y12 and arachidonic acid receptors.Citation60,Citation64 The P2Y12 receptor binds the platelet agonist ADP (which is blocked by a frequently prescribed anticoagulant, clopidogrel [Plavix™; Bristol-Myers Squibb/Sanofi Pharmaceuticals Partnership Bridgewater, NJ, USA]). P2Y12 inhibition correlates with the severity of TBI as well as TBI-related mortality. The mechanism responsible for TBI-induced P2Y12 downregulation is not clearly defined, and this type of platelet dysfunction is often overlooked in the TBI patient who is bleeding, particularly when the platelet count is normal.
Two separate (yet conceivably overlapping) pathophysiological processes are proposed as the principal cause of coagulopathy in patients who are severely injured (ISS ≥25). Disseminated intravascular coagulation (DIC) with a fibrinolytic phenotype is suggested by thrombocytopenia, increased fibrinogen/fibrin degradation product (FDP) levels, increased measured D-dimer, higher FDP:D-dimer ratios,Citation5 increased prothrombin times, and diminished levels of endogenous inhibitors of intravascular coagulation and fibrinolysis (eg, AT and α2-antiplasmin, respectively).Citation5,Citation65 DIC-mediated coaguloapthy in the trauma patient requires diffuse intra-vascular generation of procoagulants induced by reperfusion injury in the setting of hemorrhagic shock.Citation31,Citation65 These procoagulants include microparticles (MPs) expressing TF, which are derived from platelets,Citation66,Citation67 erythrocytes,Citation68 possibly brain tissue,Citation69,Citation70 free circulating mitochondrial DNA,Citation71 histone-complexed DNA fragments,Citation72 and HMGB1.Citation73,Citation74 Intravascular procoagulant activity is also augmented by decreased AT levels and diminished aPC pathway activity.Citation75
In response to intravascular activation of coagulation, HF (in this setting referred to as secondary HF) with collateral fibrinogenolysis may occur in trauma patients as suggested by the elevated levels of plasmin–α2-antiplasmin complexes, D-dimer and FDPs.Citation76–Citation79 This is not a specific pathological process, but rather a compensatory process to counter intravascular clot formation. However, primary HF (and hyperfibrinogenolysis) due to a massive shock-induced release of tPA from vascular endothelium, which rapidly overwhelms PAI-1, may occur in this setting.Citation80 Within minutes of arrival, an unsurvivable subtype of primary HF can be recognized on r-TEG tracing as a characteristic diamond-shaped pattern.Citation81 Intravascular coagulation in concert with either primary or secondary HF consumes endogenous inhibitors leading to further coagulofibrinolytic dysregulation, a defining characteristic of DIC. DIC results in consumption of platelets and coagulation factors,Citation82 notably fibrinogen, which manifests clinically in the trauma patient as coagulopathy.
An alternative conjecture, shock-induced endotheliopathy (SHINE)Citation83 posits diffuse injury to the endothelium and the endothelial glycocalyx due to whole-body ischemia–reperfusion (I/R) injury and massive sympatho-adrenergic discharge (the endothelium weighs ~1 kg and covers a surface area of the vascular tree of ~5,000 m2, which is equivalent to the surface area of an American football field). A systemic hypoxia may also contribute to endothelial injury causing destabilization of homeostasis and barrier disruption by promoting release of angiopoietins, TF, PAI-1, soluble TM, vWF, and tPA.Citation84 In severely injured patients, high levels of a marker of significant glycocalyx disruption, syndecan-1, can be detected in ~5% of patients with ACOT.Citation85 After adjusting for ISS, syndecan-1 independently predicts increased mortality.Citation86 Damage to the luminal glycocalyx presumes release of constituents of this layer, such as proteoglycans, which in effect heparinize the bleeding trauma patient. Systemic endothelial cell activation is associated with disruption of TFPI, and aPC-mediated regulation of coagulation in concert with endothelial release of tPA and urokinase-type plasminogen activator (uPA). Evidence also exists for downregulation of other endogenous anticoagulant and anti-inflammatory pathways in this syndrome.Citation5,Citation87–Citation89
In trauma, fibrinolytic system dysfunction develops as either HF or fibrinolysis shutdown (also referred to as postinjury fibrinolysis resistance). Neither HF nor fibrinolysis shutdown necessarily correlates with ISS or the magnitude of shock, although the development of either one of these two hemostatic abnormalities is associated with increasing mortality. Unregulated release of tPA from activated endothelial cells is suggested as the cause of HF,Citation81,Citation90–Citation92 whereas massive release of PAI-1 from activated plateletsCitation93 may be the pathological basis of fibrinolysis shutdown. Also, impaired tPA generation due to SHINE is suggested as a contributing mechanism to fibrinolytic shutdown. Fibrinolytic shutdown is the most common form of fibrinolytic dysfunction occurring in ~65% of trauma patients, whereas systemic HF develops in a much smaller percentage (18%–20%).Citation93 However, HF has been reported to develop in up to 53% of a subgroup of severely injured patients who progress to massive transfusion.Citation78,Citation91,Citation94,Citation95 These observations argue against the early empiric use (within 3 h of injury) of tranexamic acid (TXA) in most acutely injured patients with the exception of patients undergoing massive transfusion. Empiric administration of TXA 3 h after injury is also proscribed by CRASH-2,Citation96 although TXA is potentially permitted after 3 h from time of injury if viscoelastic assay demonstrates ongoing HF.Citation97
Furthermore, a number of studies have been completed, or are ongoing that examine the use of TXA in other settings of massive hemorrhage. For example, antifibrinolytic drugs have been shown to reduce bleeding and the requirement for blood transfusions in patients who develop PPH, prompting the World Health Organization to recommend TXA for patients with established PPH refractory to conventional therapy. However, little is known about the prophylactic use of TXA to prevent PPH. A pragmatic, single-centered, double-blinded, randomized controlled pilot trial will assess the feasibility of administering a prophylactic dose of TXA to prevent the onset of PPH and is expected to be completed by December 2018.Citation98
HALT-IT is a much larger pragmatic, randomized, double-blind, placebo controlled trial that will determine the effect of TXA on mortality, morbidity (rebleeding, nonfatal vascular events), blood transfusion, surgical intervention, and health status in patients with acute gastrointestinal bleeding. Replicating the power of the CRASH-2 study, HALT-IT involves ~200 centers in 40 countries, which will enroll 12,000 subjects. HALT-IT is expected to finish in 2020.Citation99
TXA also inhibits platelet activation induced by plasmin and plasmin-mediated proinflammatory processes that may lead to multiple organ failure.Citation100 Whether these associated effects of TXA impact survival are not known.
Hypofibrinogenemia is common in trauma but associations of trauma-induced hypofibrinogenemia with specific outcomes remain incompletely defined.Citation101 Maintaining a fibrinogen level >150–200 mg/dL is recommended for bleeding patients.Citation102 Hypofibrinogenemia is observed more often in patients with severe extremity or pelvic fractures. Also, patients who are deeply acidotic or experience long delays before arriving at definitive care are more likely to have a low fibrinogen concentration.Citation103 Low fibrinogen levels may be present prior to injury because of liver disease or develop secondary to severe liver injury or hypoxic liver injury (shock liver).Citation104 Furthermore, acidosis and hypothermia may exacerbate injury-induced consumption of fibrinogen.Citation105,Citation106 FDPs interfere with fibrin covalent polymerization and clot stability and block ADP-induced platelet aggregation.Citation107,Citation108
Acquired FXIII deficiency secondary to hemorrhage or dilution by massive transfusion has been reported,Citation109 and we consider this coagulation factor (which requires activation by thrombin and can be found concentrated in cryoprecipitate) to be crucial for correction of ACOT.Citation1,Citation110
Coagulation and inflammation
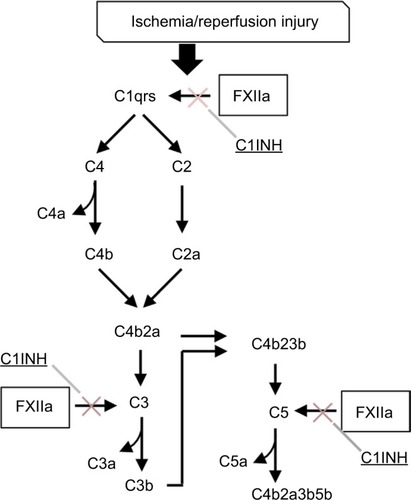
Although implementation of DCR principles has decreased overall mortality from exsanguination, ARDS and multiple organ dysfunction syndrome, two delayed complications of hemorrhagic shock, may be increasing as an unintended consequence. Hemorrhage followed by rapid reperfusion produce hepatic, renal, cardiovascular, and pulmonary dysfunction accompanied by acidosis and lactate accumulation.Citation111–Citation114 Upon restoration of blood flow, several plasma protein cascades are activated, including in addition to coagulation and fibrinolysis, complement and the kinin–kallikrein system, which mediate reperfusion-triggered inflammatory processes. For example, coagulation FXIIa integrates the contact activation plasma defense system with complement activation and the generation of proinflammatory anaphylatoxins, C3a, C4a, and C5a. This interaction is regulated by C1 inhibitor (). Conceivably, individuals who express lower levels of this inhibitor, yet still within a normal range of phenotypic expression, would be more susceptible to I/R injury. In addition, the formation of neutrophil extracellular traps (NETs) has been suggested as an important mechanism in the development of a systemic auto-inflammatory reaction.Citation115
Figure 3 Complement–contact activation system interactions. Coagulation factor XII-induced increases in vascular permeability and proinflammatory activity are mediated by complement activation and are regulated in part by C1 inhibitor (C1INH).Citation181 Attenuation of inflammation by C1INH has been described in several animal models of ischemia-reperfusion injury.Citation182
HMGB1, the prototype damage-associated molecular pattern (DAMP) molecule, is a ubiquitous DNA-binding protein found in abundant amounts in human cells. HMGB1 is released from damaged tissue and circulates beyond the site of injury, engaging specific cell receptors, RAGE (receptor for advanced glycation end products), TLR-4 (toll-like receptor-4) and TLR-2 expressed on endothelial cells, and circulating mononuclear phagocytes.Citation116,Citation117 HMGB1 serum levels increase in hemorrhagic shock and trauma,Citation118,Citation119 suggesting plausibly that HMGB1 mediates in part pathophysiologic consequences of rapid reperfusion of ischemic tissue. Plasma levels of HMGB1 are increased within 30 min after severe trauma in humans and correlate with the severity of injury, extent of tissue hypoperfusion, early posttraumatic coagulofibrinolytic abnormalities, and auto-inflammatory responses.Citation73,Citation120,Citation121 Nonsurviving trauma patients have significantly higher plasma levels of HMGB1 than survivors, and higher levels of HMGB1 are detectable in survivors who later develop organ injury such as acute lung injury and acute renal failure after DCR. Of note, HMGB1 demonstrates several interactions with the hemostatic system that may be the basis of the posttraumatic modulation of coagulation by hypoperfusion, which is characterized by TM upregulation, PC activation, inhibition of thrombin generation, and aPC-mediated consumption of PAI-1.Citation77,Citation78 Thus, HMGB1 has been considered as both a clinical markerCitation121 and a mediator Citation121–Citation123 of trauma-associated SIRS and organ failure.
RBCs stored for long periods release free hemoglobin-containing DAMP structures, which may contribute in unexpected ways to the adverse effects of massive transfusion.Citation124
Recent observations have also shown that trauma leads to significant mitochondrial injury by a PI3-K-dependent pathway. This type of subcellular injury causes the release of mtDNA fragmentsCitation125 into the local and systemic circulation, and that circulating levels of mtDNA may represent a highly sensitive (although not specific) indicator of the degree of systemic cellular injury following trauma associated with hemorrhagic shock.Citation126 Furthermore, mtDNAs have also been shown to have significant proinflammatory properties as DAMPs leading to postresuscitation systemic auto-inflammatory disorders, which may be attenuated by PI3K pathway inhibition.Citation127
Damage control resuscitation
In the hemorrhaging trauma patient, the decision to initiate DCR is predicated on apparent or anticipated life-threatening blood loss. Specifically, measured blood loss of a magnitude requiring management with this intervention is often associated with hypotension (SBP <90 mmHg) and tachycardia (numerically a heart rate > SBP). (Shock Index [SI] is defined as heart rate/SBP. For example, a trauma patient with a blood pressure of 94/60 mmHg and heart rate of 110 beats/min exhibits a SI=110/94=1.2. Hemorrhagic shock is arbitrarily defined as SI≥0.9.) This clinical state represents decompensated hemorrhagic shock, a relatively late finding in a progression to exsanguination. A clinical state more responsive to resuscitation exists when hemorrhage can be recognized before the volume of blood lost becomes life threatening.Citation128–Citation130 However, a diagnosis of compensated hemorrhagic shock in a patient may be challenging.
In 2013, a survey of American College of Surgeons Trauma Quality Improvement Program participants (which included 82 Level 1 and 42 Level 2 trauma centers) indicated that virtually all the sites surveyed relied on trauma surgeon discretion to activate a DCR protocol. The presence of hypotension and whether the immediate administration of uncrossed blood was required are somewhat more objective triggers for activation.Citation131 Several scoring systems or algorithms have been developed to guide clinicians responsible for initiating DCR.Citation6,Citation132 Noninvasive measurement of muscle oxygenation based on optical spectroscopy (in contrast to near-infrared spectroscopy that determines transcutaneous oxygen saturation, an optical spectroscopy measurement does not require assumptions about the ratio of arterial to venous blood) may provide the most objective measure of shock and the best predictor of DCR.Citation133–Citation136
Incentives for refinement of policy in trauma centers regarding DCR can be demonstrated. As an example, an observational study comparing two time periods before (2005–2007) and after (2012–2014) the implementation of changes in trauma management protocols which included: use of goal-directed coagulation management, permissive hypotension, restrictive fluid resuscitation, and administration of TXA. This study showed reduction in incidence of massive transfusion and a reduction in the transfusion of RBCs and fresh frozen plasma (FFP) after implementation.
In addition to massive transfusion of pRBCs, DCR includes the balanced transfusion of plasma and platelets (plasma:platelets:RBC = 1:1:1). For example in the Pragmatic, Randomized Optimal Platelet and Plasma Ratios trial, mortality from exsanguination decreased by ~35% in patients transfused with a 1:1:1 ratio compared with a lower 1:1:2 ratio.Citation137 DCR also can be “goal-directed” by applying POC viscoelastic coagulation testing.Citation138 In practical terms, resuscitation begins with empiric transfusion of components in a fixed 1:1:1 ratio and is followed by transfusions of specific blood components, in certain amounts, at distinct time intervals, all determined by the patient’s coagulation profile. The supposition that goal-directed DCR is superior to empiric fixed component ratio-directed DCR with respect to both patient outcome and resource utilization was recently examined in a randomized controlled trial. Compared with DCR of patients characterized by conventional tests of coagulation, TEG-guided goal-directed DCR improves survival and utilizes less plasma and platelet transfusions during the early phase of resuscitation.Citation55
Platelet transfusions are an essential element of DCR, although under these conditions, the efficacy of empiric platelet transfusions have not yet been proven.Citation139 Platelet transfusions pose distinct challenges including variable availability, contamination risks, eventual immunological sensitization, and others.Citation140 Platelet storage at room temperature (24°C) increases the chance of bacterial contamination (estimated to occur in one of every 3,000 platelet units) and increases the risk of transfusion-associated sepsis in recipients.Citation141 Even though techniques to reduce pathogens contaminating platelet transfusion packs have proven to be quite effective, the success of this advance was lessened after extensive damage done to platelet function under these conditions became apparent.Citation142
One unit of platelets obtained by apheresis contains ~6 × 1011 platelets and will increase a platelet count by ~30,000–60,000/µL in a 70 kg nonhemorrhaging patient.Citation110 Platelets are suspended in plasma. Thus, one unit provides 500 mg of fibrinogen as well as other coagulation factors but also adds a risk of plasma-mediated febrile nonhemolytic transfusion and allergic reactions.Citation143
Plasma as a blood bank component may be supplied as FFP; FP-24; so-called liquid, never frozen plasma; pooled detergent-treated plasma; or lyophilized plasma. The difference between FFP and FP-24, the two most common forms of plasma used, is the length of time between donor phlebotomy and when FFP and FP-24 are placed into –18°C storage. In the USA and Canada, the designations FFP and FP-24 mean plasma is frozen within 8 h or within 24 h, respectively. In Australia, plasma obtained by apheresis can be considered FFP only if frozen within 6 h, whereas plasma separated from whole blood can be considered FFP if frozen within 18 h.Citation144
FVIII is reduced in FP-24 compared with FFP but remains above 30%, which is sufficient to support coagulation. FP-24 is associated with a lower incidence of transfusion-related acute lung injury (TRALI) compared with FFP, and FP-24 is favored due to often complex logistics of transporting blood donations to blood centers from remote donation sites. FFP and FP-24 may be prepared by separation from a single unit donation of whole blood, yielding 200–250 mL, or from apheresis of a single donor (400–600 mL). FFP or FP-24 is prepared for transfusion by thawing over several minutes at 34°C–37°C. If these components are not transfused, they are not refrozen but are stored thawed 4°C for up to 4 days more. FVIII decays to ~40% of normal; the decline in other coagulation factors by 5 days is comparable between thawed FFP and FP-24, although there is a more significant fall in FVII in thawed FP-24.Citation145 Most trauma centers use thawed plasma for hemostatic resuscitation to be able to begin plasma transfusion immediately. Citation146
Solvent-/detergent-treated pooled human plasma (octaplasLG®; Octapharma USA Inc, Hoboken, NJ, USA) is manufactured by pooling plasma of the same blood type from ~650–1,500 individuals at a time. Plasma is treated with detergent to inactivate enveloped viruses, and prions are removed by affinity chromatography. Pooled plasma treated in this way has a lower rate of pathogen transmission, and due to dilution of any prevailing anti-HLA and anti-HNA antibodies, these plasma preparations when compared with FFP or FP-24 are associated with smaller risks of TRALI and posttransfusion thrombocytopenia.Citation147 Both Protein S and α2-antiplasmin are susceptible to detergent degradation, which potentially increases, respectively, the thrombotic and fibrinolytic activity of plasma prepared in this manner.Citation148 Coagulation factor levels after 5 days of storage of thawed solvent-/detergent-treated pooled plasma are essentially the same as found in thawed single-donor plasma.Citation149 In the US, the FDA currently approves use of octaplast for acquired factor-deficiency coagulopathies due to liver disease or that develop during the course of cardiovascular surgical procedures and liver transplantation.Citation150
Until a recipient’s blood type is determined, group AB plasma is considered the “universal donor” plasma type to use empirically. Blood bank inventories of AB plasma, however, are routinely sparse. Therefore, plasma of this blood group type is never maintained thawed and consequently never immediately available at the beginning of resuscitation.Citation146 Thus, in place of AB plasma, thawed group A plasma is transfused empirically when resuscitation is initiated.Citation151–Citation153 The risk of ABO blood group mismatch transfusion reaction is decreased by the fact that the most common encountered blood group in a recipient will be group A, and anti-B antibodies in group A donated plasma, which can be screened for (but typically are not), generally are low, particularly in plasma of North American males >50 years of age.
Reconstituted lyophilized plasma is another source of this blood component that may be used immediately for a recipient of any blood type because of dilution and neutralization of anti-A and anti-B hemagglutinins. This type of plasma preparation is useful logistically to medical support systems of armed services.
Because fibrinogen plays a critical role in traumatic hemorrhage, early repletion of fibrinogen is advocated.Citation154,Citation155 Several international guidelines for DCR specify fibrinogen infusion for plasma fibrinogen levels <150–200 mg/dL or for TEG or ROTEM signs of hypofibrinogenemia (or possibly acquired dysfibrinogenemia). In the USA, cryoprecipitate (cryo) is the principal source of fibrinogen.Citation156 Other coagulation factors enriched in cryo include von Willebrand factor, FVIII, and FXIII, and these plausibly contribute to hemostatic resuscitation.
Cryo is prepared by centrifugation of a slowly thawed unit of FFP and resuspension of the precipitate in a smaller volume of plasma, which is then frozen again at –18°C. This represents ~30% of the fibrinogen (200–300 mg) from the original unit of FFP, but in much higher concentration because of a much smaller volume of plasma (10–20 mL). For transfusion, this preparation is rethawed and pooled with multiple donors. Typically, five single units are pooled into one before issue. An adult dose comprises two pools (10 single units total), which should increase plasma fibrinogen levels by ~100 mg/dL. Platelet membrane MPs, generated by the plasma freeze-thaw cycling, are concentrated in the precipitate,Citation157 although it is not known if MPs formed under these conditions retain thrombotic or inflammatory potential.
In Europe, saline-reconstituted purified fibrinogen concentrate has largely replaced cryo in hemostatic resuscitation protocols for hemorrhagic shock. Although several advantages to fibrinogen concentrate over cryo in this setting appear evident,Citation156 to date a significant clinical benefit or measurable cost-effectiveness of fibrinogen concentrate compared with cryo remain unproven. The Fibrinogen Early In Severe Trauma studY (FEISTY),Citation155 a multicenter, randomized controlled trial comparing fibrinogen concentrate to cryo for fibrinogen supplementation in traumatic hemorrhage, is expected to finish enrollment by June 2018. Whether the coadministration of FXIII concentrateCitation109,Citation158–Citation161 potentiates fibrinogen concentrate also is unknown.
Prothrombin complex concentrates (PCCs) include vitamin K-dependent factors, FII, FVII, FIX, and FX. PCC products are considered either three-factor PCCs (containing a negligible concentration of FVII) or four-factor PCCs (with a higher concentration of FVII). Most newer PCC products should be considered six-factor PCCs because these contain two additional vitamin K covalently modified proteins, protein C and protein S. Thrombotic complications associated with PCCs include venous thromboembolism, DIC, microvascular thrombosis, and myocardial infarction.Citation162 Although these agents have specific indications for reversal of pharmacological anticoagulation in trauma patients, the use and timing of PCCs for management of ACOT remains to be defined.
Empiric treatment of patients with blunt and penetrating trauma with high doses of human recombinant FVIIa (rFVIIa) does not decrease mortality significantly but increases the risk of arterial thromboembolic events (eg, coronary artery thrombosis), especially among the elderly.Citation163
Acidosis and hypothermia
Hypoperfusion of tissue and organs due to hemorrhagic shock produces lactic acidosis. Acidemia diminishes platelet activation, decreases coagulation factor enzymatic activity, and depletes substrate through enhanced fibrinogenolysis.Citation106 In addition, the presence of excess hydrogen ion destabilizes crucial interactions between phospholipid platforms in platelets that are activated and coagulation factor complexes that have formed, further diminishing coagulation potential in the patient suffering from hemorrhagic shock.Citation164
Reversal of lactic acidosis is frequently used as an end point indicating when resuscitation should be terminated. During DCR of a patient in whom there exists an expected elevated anion gap (AG) metabolic acidosis, the relationship between change (∆) in AG and serum bicarbonate levels (∆AG:∆HCO3) may reveal unsuspected mixed metabolic acid–base disorders,Citation165 which distort lactic acidosis as an end point. ∆AG:∆HCO3– will fall between 0.8 and 1.2 if no other complicating metabolic process is occurring. A ∆AG:∆HCO3– <0.7 suggests an additional hyperchloremic, nongap metabolic acidosis that often develops, for example, in patients treated inappropriately with large volumes of normal saline. Conversely, a ∆AG:∆HCO3– >1.2 may indicate the presence of metabolic alkalosis (eg, lactic acidosis in a patient with preexisting COPD). A very high ∆AG:∆HCO3– ratio should suggest rhabdomyolysis complicating hemorrhagic shock-induced lactic acidosisCitation166 or severe hypophosphatemia.Citation167 We cautiously treat acidosis (pH<7.2) with NaHCO3 to reverse impairment in myocardial contractility. This improves global oxygen delivery and accelerates correction of a base deficit.
Hypothermia frequently complicates coagulopathy in trauma patients. A decrease in core body temperature suppresses the enzymatic specificity constants of coagulation factors in the hemostatic pathway. Thus, each 1°C decrease in core temperature reduces coagulation factor activity by ~10%–15%, which is exacerbated by factor depletion secondary to dilution or consumption.Citation168 However, hypothermia also inhibits endogenous anticoagulant activity (eg, thrombin-TM-protein C pathway) as well as coagulation factor activity. A more important explanation for hypothermia-related coagulopathy is inhibition of von Willebrand factor–platelet GPIb-IX-V interactions, which mediate platelet adherence to damaged subendothelium.Citation169
Hypothermia can be prevented by infusion of warm fluids both during the prehospital phase and resuscitation phase of treatment of a patient in hemorrhagic shock. Restoring perfusion and oxygen delivery will also reestablish endogenous heat generation. Resuscitation and damage control surgery should be performed in shock rooms and operating rooms, respectively, that are maintained at very warm temperatures, notwithstanding discomfort for providers.
Resuscitation-induced coagulopathy
During DCR, reduced circulating ionized calcium and magnesium develop from citrate toxicity that occurs as a result of transfusion of citrate-containing blood components. A healthy adult liver metabolizes up to 3 g of citrate every 5 min (approximately the amount in two units of pRBCs). However, exceptionally high transfusion rates, often reached during DCR, exceed this unimpaired capacity to clear citrate, while preexisting liver disease, impaired hepatic function from hypothermia, or severe traumatic liver injury may reduce citrate metabolism.Citation170 Citrate toxicity-mediated divalent cation deficiencies are exacerbated by dilution of the patients circulating ionized calcium.Citation171 Furthermore, preexisting liver disease or hypothermia-induced impairment of hepatic function may reduce citrate metabolism and exacerbate hypocalcemia and hypomagnesemia. Citrate accumulation during resuscitation and citrate-mediated hypocalcemia are made worse by dilution of calcium by resuscitation fluids that do not contain this divalent cation. Hypocalcemia reduces myocardial contractility and impairs maintenance of vasomotor tone. Hypomagnesemia also develops due to citrate accumulation and dilution. Hypomagnesemia and hypocalcemia are associated with disruption of myocardial repolarization characterized by a prolonged QT interval and a risk for development of “torsades de pointes”.Citation1 We administer one ampule of intravenous calcium chloride empirically when we activate our DCR protocol. We administer additional calcium chloride throughout resuscitation as massive transfusion of citrate-containing components continues.
Conclusion
Hemostasis and inflammation are critical elements of host defense. The activation, regulation, and coordinated interaction of these systems are controlled predominately by mediators expressed by vascular endothelium. An extensive number of careful laboratory and clinical investigations identify endothelial cell dysfunction as the principal cause of a systemic physiologic dysfunction syndrome, which contributes, in part, to ACOT. ACOT substantially increases rates of trauma mortality and is an independent predictor of other adverse outcomes, including higher amounts of blood component transfusion. ACOT cannot be considered a single entity, however, and hemostatic failure associated with serious injury and hemorrhagic shock is arguably the sum of potentially several disturbed host mechanisms. Research agendas that focus on these undeniably complex hematologic and auto-inflammatory pathologies in the trauma patient () have been assigned the highest priority by experts in trauma critical care.Citation172 For example, we believe the adoption by the FDA of acquired hypofibrinogenemia or dysfibrinogenemia as an indication for fibrinogen concentrate will have a substantial positive effect on DCR in the future. Also, we predict further examination of the different changes in the fibrinolytic system in trauma patients, and how these changes modify the activity of other plasma host defense systems (complement, contact activation, and NET formation) may suggest additional targets for intervention during DCR. And, we anticipate that advances in the science of transfusion medicine will have a substantial impact on future DCR protocols. For example, a remarkable synthetic platelet surrogate nearly at hand, SynthoPlate™, which is based on expression of clinically relevant biocompatible liposomal platforms, promises to relieve many of the general problems that plague platelet transfusions presently.Citation173 Development of novel therapies based on results from these investigations and many others will continue to be the basis for further optimization of DCR for catastrophic blood loss.
Table 1 Representative examples of recently completed or ongoing clinical trials in trauma and critical care
Acknowledgments
The authors thank Dr Pradeesh George for helpful suggestions and insightful discussions during drafting of the manuscript. His extensive knowledge of the subject substantially focused this review.
Disclosure
The authors report no conflicts of interest in this work.
References
- PohlmanTHWalshMAversaJHutchisonEMOlsenKPLawrence ReedRDamage control resuscitationBlood Rev201529425126225631636
- ChangRCardenasJCWadeCEHolcombJBAdvances in the understanding of trauma-induced coagulopathyBlood201612881043104927381903
- MacLeodJBWinklerAMMcCoyCCHillyerCDShazBHEarly trauma induced coagulopathy (ETIC): prevalence across the injury spectrumInjury201445591091524438827
- SchochlHSchlimpCJVoelckelWPotential value of pharmacological protocols in traumaCurr Opin Anaesthesiol201326222122923321555
- OshiroAYanagidaYGandoSHenzanNTakahashiIMakiseHHemostasis during the early stages of trauma: comparison with disseminated intravascular coagulationCrit Care2014182R6124708802
- ChangRHolcombJBOptimal fluid therapy for traumatic hemorrhagic shockCrit Care Clin2017331153627894494
- RahbarEFoxEEdel JuncoDJEarly resuscitation intensity as a surrogate for bleeding severity and early mortality in the PROMMTT studyJ Trauma Acute Care Surg2013751 Suppl 1S162323778506
- RahbarMHdel JuncoDJHuangHA latent class model for defining severe hemorrhage: experience from the PROMMTT studyJ Trauma Acute Care Surg2013751 Suppl 1S828823778516
- OldroydJCVenardosKMAokiNJImproving outcomes for hospital patients with critical bleeding requiring massive transfusion: the Australian and New Zealand Massive Transfusion Registry study methodologyBMC Res Notes20169145727716381
- LeviMFriesDGombotzHPrevention and treatment of coagulopathy in patients receiving massive transfusionsVox Sang2011101215417421749403
- SavageSAZarzaurBLCroceMAFabianTCRedefining massive transfusion when every second countsJ Trauma Acute Care Surg201374239640023354230
- BrohiKCohenMJGanterMTAcute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysisJ Trauma20086451211121718469643
- BrohiKSinghJHeronMCoatsTAcute traumatic coagulopathyJ Trauma20035461127113012813333
- HessJRDuttonRBHolcombJBScaleaTMGiving plasma at a 1:1 ratio with red cells in resuscitation: who might benefit?Transfusion20084881763176518482190
- HolcombJBJenkinsDRheePDamage control resuscitation: directly addressing the early coagulopathy of traumaJ Trauma200762230731017297317
- McLaughlinDFNilesSESalinasJA predictive model for massive transfusion in combat casualty patientsJ Trauma2008642 SupplS57S6318376173
- NilesSEMcLaughlinDFPerkinsJGIncreased mortality associated with the early coagulopathy of trauma in combat casualtiesJ Trauma20086461459146318545109
- MesarTLarentzakisADzikWChangYVelmahosGYehDDAssociation between ratio of fresh frozen plasma to red blood cells during massive transfusion and survival among patients without traumatic injuryJAMA Surg2017152657458028273299
- FarooqNGaliatsatosPAulakhJKHigginsCMartinezAMassive transfusion practice in non-trauma related hemorrhagic shockJ Crit Care201843656928846895
- American College of Obstetricians and Gynecologists Committee on Patient Safety and Quality ImprovementCommittee Opinion No. 590: preparing for clinical emergencies in obstetrics and gynecologyObstet Gynecol2014123372272524553170
- WeisbrodABSheppardFRChernofskyMREmergent management of postpartum hemorrhage for the general and acute care surgeonWorld J Emerg Surg200944319939251
- MatsumotoTNogamiKShimaMSimultaneous measurement of thrombin and plasmin generation to assess the interplay between coagulation and fibrinolysisThromb Haemost2013110476176824072166
- HoffmanMMonroeDM3rdA cell-based model of hemostasisThromb Haemost200185695896511434702
- MonroeDMHoffmanMRobertsHRPlatelets and thrombin generationArterioscler Thromb Vasc Biol20022291381138912231555
- WolbergASThrombin generation and fibrin clot structureBlood Rev200721313114217208341
- HuangCCShihCCLiuTYLeePYAssessing the viscoelastic properties of thrombus using a solid-sphere-based instantaneous force approachUltrasound Med Biol201137101722173321821355
- Van de WouwerMCollenDConwayEMThrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammationArterioscler Thromb Vasc Biol20042481374138315178554
- WoodJPElleryPEMaroneySAMastAEBiology of tissue factor pathway inhibitorBlood2014123192934294324620349
- MastAETissue factor pathway inhibitor: multiple anticoagulant activities for a single proteinArterioscler Thromb Vasc Biol201636191426603155
- MaroneySAMastAENew insights into the biology of tissue factor pathway inhibitorJ Thromb Haemost201513Suppl 1S200S20726149025
- GandoSOtomoYLocal hemostasis, immunothrombosis, and systemic disseminated intravascular coagulation in trauma and traumatic shockCrit Care2015197225886801
- MartinFAMurphyRPCumminsPMThrombomodulin and the vascular endothelium: insights into functional, regulatory, and therapeutic aspectsAm J Physiol Heart Circ Physiol201330412H1585H159723604713
- ChristiaansSCWagenerBMEsmonCTPittetJFProtein C and acute inflammation: a clinical and biological perspectiveAm J Physiol Lung Cell Mol Physiol20133057L455L46623911436
- Schedin-WeissSRichardBOlsonSTKinetic evidence that allosteric activation of antithrombin by heparin is mediated by two sequential conformational changesArch Biochem Biophys2010504216917620816747
- LevyJHSniecinskiRMWelsbyIJLeviMAntithrombin: anti-inflammatory properties and clinical applicationsThromb Haemost2016115471272826676884
- CampbellWDLazouraEOkadaNOkadaHInactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase NMicrobiol Immunol200246213113411939578
- MylesTNishimuraTYunTHThrombin activatable fibrinolysis inhibitor, a potential regulator of vascular inflammationJ Biol Chem200327851510595106714525995
- NishimuraTMylesTPiliponskyAMKaoPNBerryGJLeungLLThrombin-activatable procarboxypeptidase B regulates activated complement C5a in vivoBlood200710951992199717105819
- WengHBLiSEarly changes of plasma angiopoietin-2 in patients with multiple traumaWorld J Emerg Med20112428729025215025
- DalyCQianXCastanaroCAngiopoietins bind thrombomodulin and inhibit its function as a thrombin cofactorSci Rep20188150529323190
- Lomas-NeiraJVenetFChungCSThakkarRHeffernanDAyalaANeutrophil-endothelial interactions mediate angiopoietin-2-associated pulmonary endothelial cell dysfunction in indirect acute lung injury in miceAm J Respir Cell Mol Biol201450119320023980650
- HughesDPMarronMBBrindleNPThe antiinflammatory endothelial tyrosine kinase Tie2 interacts with a novel nuclear factor-kappaB inhibitor ABIN-2Circ Res200392663063612609966
- TadrosAHughesDPDunmoreBJBrindleNPABIN-2 protects endothelial cells from death and has a role in the antiapoptotic effect of angiopoietin-1Blood2003102134407440912933576
- TerpstraMLAmanJvan Nieuw AmerongenGPGroeneveldABPlasma biomarkers for acute respiratory distress syndrome: a systematic review and meta-analysis*Crit Care Med201442369170024158164
- ZinterMSSpicerAOrwollBOPlasma angiopoietin-2 outperforms other markers of endothelial injury in prognosticating pediatric ARDS mortalityAm J Physiol Lung Cell Mol Physiol20163103L224L23126660787
- HerreroRSanchezGLorenteJANew insights into the mechanisms of pulmonary edema in acute lung injuryAnn Transl Med2018623229430449
- HajjarianZTripathiMMNadkarniSKOptical Thromboelastography to evaluate whole blood coagulationJ Biophotonics20158537238124700701
- JohanssonPICoagulation monitoring of the bleeding traumatized patientCurr Opin Anaesthesiol201225223524122193152
- WeederPDPorteRJLismanTHemostasis in liver disease: implications of new concepts for perioperative managementTransfus Med Rev201428310711324721432
- WeiselJWBiophysics. Enigmas of blood clot elasticityScience2008320587545645718436761
- JohanssonPIStissingTBochsenLOstrowskiSRThrombelastography and tromboelastometry in assessing coagulopathy in traumaScand J Trauma Resusc Emerg Med2009174519775458
- JohanssonPISvendsenMSSaladoJBochsenLKristensenATInvestigation of the thrombin-generating capacity, evaluated by thrombogram, and clot formation evaluated by thrombelastography of platelets stored in the blood bank for up to 7 daysVox Sang200894211311818067490
- KawasakiJKatoriNKodakaMMiyaoHTanakaKAElectron microscopic evaluations of clot morphology during thrombelastographyAnesth Analg20049951440144415502045
- GonzalezEMooreEEMooreHBManagement of trauma-induced coagulopathy with thrombelastographyCrit Care Clin201733111913427894492
- GonzalezEMooreEEMooreHBGoal-directed hemostatic resuscitation of trauma-induced coagulopathy: a pragmatic randomized clinical trial comparing a viscoelastic assay to conventional coagulation assaysAnn Surg201626361051105926720428
- HarrJNMooreEEChinTLViscoelastic hemostatic fibrinogen assays detect fibrinolysis earlyEur J Trauma Emerg Surg2015411495626038165
- MooreEEMooreHBGonzalezEPostinjury fibrinolysis shutdown: rationale for selective tranexamic acidJ Trauma Acute Care Surg2015786 Suppl 1S65S6926002266
- WadaTCoagulofibrinolytic changes in patients with post-cardiac arrest syndromeFront Med (Lausanne)2017415629034235
- RamseyMTFabianTCShahanCPA prospective study of platelet function in trauma patientsJ Trauma Acute Care Surg201680572673226895088
- DavisPKMusunuruHWalshMPlatelet dysfunction is an early marker for traumatic brain injury-induced coagulopathyNeurocrit Care201318220120822847397
- WindelovNASorensenAMPernerAPlatelet aggregation following trauma: a prospective studyBlood Coagul Fibrinolysis2014251677323945060
- WohlauerMVMooreEEDrozNMHemodilution is not critical in the pathogenesis of the acute coagulopathy of traumaJ Surg Res20121731263021696767
- WohlauerMVMooreEEThomasSEarly platelet dysfunction: an unrecognized role in the acute coagulopathy of traumaJ Am Coll Surg2012214573974622520693
- CastellinoFJChapmanMPDonahueDLTraumatic brain injury causes platelet adenosine diphosphate and arachidonic acid receptor inhibition independent of hemorrhagic shock in humans and ratsJ Trauma Acute Care Surg20147651169117624747445
- GandoSSawamuraAHayakawaMTrauma, shock, and disseminated intravascular coagulation: lessons from the classical literatureAnn Surg20112541101921368657
- ParkMSOwenBABallingerBAQuantification of hypercoagulable state after blunt trauma: microparticle and thrombin generation are increased relative to injury severity, while standard markers are notSurgery2012151683183622316436
- ParkMSXueASpearsGMThrombin generation and procoagulant microparticle profiles after acute trauma: a prospective cohort studyJ Trauma Acute Care Surg201579572673126496097
- MatijevicNWangYWWadeCECellular microparticle and thrombogram phenotypes in the Prospective Observational Multicenter Major Trauma Transfusion (PROMMTT) study: correlation with coagulopathyThromb Res2014134365265825086657
- TianYSalsberyBWangMBrain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injuryBlood2015125132151215925628471
- NekludovMMobarrezFGrythDBellanderBMWallenHFormation of microparticles in the injured brain of patients with severe isolated traumatic brain injuryJ Neurotrauma201431231927193324956150
- ZhangQRaoofMChenYCirculating mitochondrial DAMPs cause inflammatory responses to injuryNature2010464728510410720203610
- JohanssonPIWindelovNARasmussenLSSorensenAMOstrowskiSRBlood levels of histone-complexed DNA fragments are associated with coagulopathy, inflammation and endothelial damage early after traumaJ Emerg Trauma Shock20136317117523960372
- CohenMJBrohiKCalfeeCSEarly release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusionCrit Care2009136R17419887013
- GiannoudisPVMallinaRHarwoodPPerrySSanteEDPapeHCPattern of release and relationship between HMGB-1 and IL-6 following blunt traumaInjury201041121323132720887988
- TreutigerCJMullinsGEJohanssonASHigh mobility group 1 B-box mediates activation of human endotheliumJ Intern Med2003254437538512974876
- LevratAGrosARugeriLEvaluation of rotation thrombelastography for the diagnosis of hyperfibrinolysis in trauma patientsBr J Anaesth2008100679279718440953
- SchochlHFrietschTPavelkaMJamborCHyperfibrinolysis after major trauma: differential diagnosis of lysis patterns and prognostic value of thrombelastometryJ Trauma200967112513119590321
- SchochlHVoelckelWMaegeleMSolomonCTrauma-associated hyperfibrinolysisHamostaseologie2012321222722009115
- LongstaffCKolevKBasic mechanisms and regulation of fibrinolysisJ Thromb Haemost201513Suppl 1S9810526149056
- CottonBAHarvinJAKostousouvVHyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administrationJ Trauma Acute Care Surg201273236537022846941
- ChapmanMPMooreEEMooreHBThe “Black Diamond”: Rapid thrombelastography identifies lethal hyperfibrinolysisJ Trauma Acute Care Surg201579692592926488324
- AllardSGreenLHuntBJHow we manage the haematological aspects of major obstetric haemorrhageBr J Haematol2014164217718824383841
- JohanssonPStensballeJOstrowskiSShock induced endotheliopathy (SHINE) in acute critical illness - a unifying pathophysiologic mechanismCrit Care20172112528179016
- WhiteNJWardKRPatiSStrandenesGCapAPHemorrhagic blood failure: oxygen debt, coagulopathy, and endothelial damageJ Trauma Acute Care Surg2017826S Suppl 1S41S4928328671
- OstrowskiSRJohanssonPIEndothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathyJ Trauma Acute Care Surg2012731606622743373
- JohanssonPIOstrowskiSRAcute coagulopathy of trauma: balancing progressive catecholamine induced endothelial activation and damage by fluid phase anticoagulationMed Hypotheses201075656456720708846
- HayakawaMGandoSOnoYWadaTYanagidaYSawamuraAFibrinogen level deteriorates before other routine coagulation parameters and massive transfusion in the early phase of severe trauma: a retrospective observational studySemin Thromb Hemost2015411354225590522
- HayakawaMSawamuraAGandoSDisseminated intravascular coagulation at an early phase of trauma is associated with consumption coagulopathy and excessive fibrinolysis both by plasmin and neutrophil elastaseSurgery2011149222123020655560
- YanagidaYGandoSSawamuraANormal prothrombinase activity, increased systemic thrombin activity, and lower antithrombin levels in patients with disseminated intravascular coagulation at an early phase of trauma: comparison with acute coagulopathy of trauma-shockSurgery20131541485723684364
- CardenasJCMatijevicNBaerLAHolcombJBCottonBAWadeCEElevated tissue plasminogen activator and reduced plasminogen activator inhibitor promote hyperfibrinolysis in trauma patientsShock201441651452124667610
- CardenasJCWadeCEHolcombJBMechanisms of trauma-induced coagulopathyCurr Opin Hematol201421540440925010798
- ChapmanMPMooreEERamosCRFibrinolysis greater than 3% is the critical value for initiation of antifibrinolytic therapyJ Trauma Acute Care Surg201375696196724256667
- MooreHBMooreEEHuebnerBRFibrinolysis shutdown is associated with a fivefold increase in mortality in trauma patients lacking hypersensitivity to tissue plasminogen activatorJ Trauma Acute Care Surg20178361014102229190254
- DavenportRPathogenesis of acute traumatic coagulopathyTransfusion201353Suppl 123S27S23301969
- RazaIDavenportRRourkeCThe incidence and magnitude of fibrinolytic activation in trauma patientsJ Thromb Haemost201311230731423176206
- ShakurHRobertsIBautistaREffects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trialLancet20103769734233220554319
- MooreHBMooreEEGonzalezEHyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapyJ Trauma Acute Care Surg201477681181725051384
- AlamAQUse of TXA to Prevent Postpartum Hemorrhage (TAPPH-1) Available from: https://clinicaltrials.gov/ct2/show/NCT03069859?term=NCT03069859&rank=1Accessed May 18, 2018
- ShakurHHaemorrhage Alleviation With Tranexamic Acid-Intestinal System (HALT-IT) NCT016581242018 Available from: https://clinicaltrials.gov/ct2/show/NCT01658124?term=HALT-IT&rank=1Accessed May 5, 2018
- PanteliMPountosIGiannoudisPVPharmacological adjuncts to stop bleeding: options and effectivenessEur J Trauma Emerg Surg201642330331026660675
- RourkeCCurryNKhanSFibrinogen levels during trauma hemorrhage, response to replacement therapy, and association with patient outcomesJ Thromb Haemost20121071342135122519961
- BesserMWMacDonaldSGAcquired hypofibrinogenemia: current perspectivesJ Blood Med2016721722527713652
- HagemoJSStanworthSJuffermansNPPrevalence, predictors and outcome of hypofibrinogenaemia in trauma: a multicentre observational studyCrit Care2014182R5224666991
- vanDeWaterLCarrJMAronsonDMcDonaghJAnalysis of elevated fibrin(ogen) degradation product levels in patients with liver diseaseBlood1986675146814732938648
- MartiniJMaischSPilshoferLStreifWMartiniWFriesDFibrinogen concentrate in dilutional coagulopathy: a dose study in pigsTransfusion201454114915723672536
- MartiniWZCoagulopathy by hypothermia and acidosis: mechanisms of thrombin generation and fibrinogen availabilityJ Trauma200967120220819590336
- HuntBJSegalHHyperfibrinolysisJ Clin Pathol199649129589038728
- StachurskaJLatalloZKopecMInhibition of platelet aggregation by dialysable fibrinogen degradation products (FDP)Thromb Diath Haemorrh197023191985420436
- LevyJHGreenbergCBiology of Factor XIII and clinical manifestations of Factor XIII deficiencyTransfusion20135351120113122928875
- AndreasonCLPohlmanTHDamage control resuscitation for catastrophic bleedingOral Maxillofac Surg Clin North Am201628455356827745621
- PapadopoulosDSiempisTTheodorakouETsoulfasGHepatic ischemia and reperfusion injury and trauma: current conceptsArch Trauma Res201322637024396796
- SatterlySAMartinMWingerdMHempelJHofferZStallingsJDFlutamide fails to reduce resuscitation requirements in a porcine ischemia-reperfusion modelJ Surg Res2013184147247923791438
- SingerMCritical illness and flat batteriesCrit Care201721Suppl 330929297363
- WaxmanKShock: ischemia, reperfusion, and inflammationNew Horiz1996421531608774791
- JorchSKKubesPAn emerging role for neutrophil extracellular traps in noninfectious diseaseNat Med201723327928728267716
- RaymondSLHoldenDCMiraJCMicrobial recognition and danger signals in sepsis and traumaBiochim Biophys Acta2017186310 Pt B2564257328115287
- WagnerNDieterenSFranzNEthyl pyruvate ameliorates hepatic injury following blunt chest trauma and hemorrhagic shock by reducing local inflammation, NF-kappaB activation and HMGB1 releasePLoS One2018132e019217129420582
- EppensteinerJDavisRPBarbasASKwunJLeeJImmunothrombotic activity of damage-associated molecular patterns and extracellular vesicles in secondary organ failure induced by trauma and sterile insultsFront Immunol2018919029472928
- LevyRMMollenKPPrinceJMSystemic inflammation and remote organ injury following trauma require HMGB1Am J Physiol Regul Integr Comp Physiol20072934R1538R154417652366
- EskiciZMAcikgozSPiskinNHigh mobility group B1 levels in sepsis and disseminated intravascular coagulationActa Biochim Pol201259456156623094260
- HatadaTWadaHNoboriTPlasma concentrations and importance of high mobility group box protein in the prognosis of organ failure in patients with disseminated intravascular coagulationThromb Haemost200594597597916363239
- GandoSMicrovascular thrombosis and multiple organ dysfunction syndromeCrit Care Med2010382 SupplS35S4220083912
- ItoTKawaharaKNakamuraTHigh-mobility group box 1 protein promotes development of microvascular thrombosis in ratsJ Thromb Haemost20075110911617239166
- ZettelKRDyerMRavalJSAged human stored red blood cell supernatant inhibits macrophage phagocytosis in an HMGB1 dependent manner after trauma in a murine modelShock201747221722427488090
- ThurairajahKBriggsGDBaloghZJThe source of cell-free mitochondrial DNA in trauma and potential therapeutic strategiesEur J Trauma Emerg Surg Epub201849
- NakahiraKKyungSYRogersAJCirculating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validationPLoS Med20131012e100157724391478
- BlackGESokolKKMoeDMImpact of a novel phosphoinositol-3 kinase inhibitor in preventing mitochondrial DNA damage and damage-associated molecular pattern accumulation: results from the Biochronicity ProjectJ Trauma Acute Care Surg201783468368928930961
- McNabABurnsBBhullarIChesireDKerwinAA prehospital shock index for trauma correlates with measures of hospital resource use and mortalitySurgery2012152347347622938906
- MitraBFitzgeraldMChanJThe utility of a shock index ≥1 as an indication for pre-hospital oxygen carrier administration in major traumaInjury2014451616523391451
- MutschlerMNienaberUMunzbergMThe Shock Index revisited - a fast guide to transfusion requirement? A retrospective analysis on 21,853 patients derived from the TraumaRegister DGU(R)Crit Care2013174R17223938104
- CamazineMNHemmilaMRLeonardJCMassive transfusion policies at trauma centers participating in the American College of Surgeons Trauma Quality Improvement ProgramJ Trauma Acute Care Surg2015786 Suppl 1S485326002263
- TongletMLEarly Prediction of Ongoing Hemorrhage in Severe Trauma: Presentation of the Existing Scoring SystemsArch Trauma Res201654e3337728144603
- ArakakiLSLBulgerEMCiesielskiWAMuscle oxygenation as an early predictor of shock severity in trauma patientsShock201747559960527820776
- SchenkmanKACarlbomDJBulgerEMMuscle oxygenation as an indicator of shock severity in patients with suspected severe sepsis or septic shockPLoS One2017128e018235128771567
- SchenkmanKAHawkinsDSCiesielskiWADelaneyMArakakiLSNon-invasive assessment of muscle oxygenation may aid in optimising transfusion threshold decisions in ambulatory paediatric patientsTransfus Med2017271252928070916
- ArakakiLSSchenkmanKACiesielskiWAShaverJMMuscle oxygenation measurement in humans by noninvasive optical spectroscopy and locally weighted regressionAnal Chim Acta2013785273323764440
- HolcombJBTilleyBCBaraniukSTransfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trialJAMA2015313547148225647203
- FahrendorffMOliveriRSJohanssonPIThe use of viscoelastic haemostatic assays in goal-directing treatment with allogeneic blood products – a systematic review and meta-analysisScand J Trauma Resusc Emerg Med20172513928403868
- LopezESrivastavaAKPatiSHolcombJBWadeCEPlatelet-derived microvesicles: a potential therapy for trauma-induced coagulopathyShock201849324324828858139
- HickmanDAPawlowskiCLSekhonUDSMarksJGuptaASBiomaterials and advanced technologies for hemostatic management of bleedingAdv Mater20183041700859
- Centers for Disease C, PreventionFatal bacterial infections associated with platelet transfusions -- United States, 2004MMWR Morb Mortal Wkly Rep200554716817015729217
- EstcourtLJMaloufRHopewellSPathogen-reduced platelets for the prevention of bleedingCochrane Database Syst Rev20177CD00907228756627
- VamvakasECRelative safety of pooled whole blood-derived versus single-donor (apheresis) platelets in the United States: a systematic review of disparate risksTransfusion200949122743275819682339
- AckerJPMarksDCSheffieldWPQuality assessment of established and emerging blood components for transfusionJ Blood Transfus20162016486028428070448
- Neisser-SvaeATrawnicekLHegerAMehtaTTriulziDFive-day stability of thawed plasma: solvent/detergent-treated plasma comparable with fresh-frozen plasma and plasma frozen within 24 hoursTransfusion201656240440926419404
- HessJRHolcombJBResuscitating PROPPRlyTransfusion20155561362136425858048
- MayrWRHaemovigilance: are there significant differences among plasma products?Transfus Apher Sci201043340740920934384
- PitkanenHJouppilaAMowinckelMCEnhanced thrombin generation and reduced intact protein S in processed solvent detergent plasmaThromb Res2015135116717425466844
- HegerANeisser-SvaeATrawnicekLTriulziDThrombin generation potential and clot-forming capacity of thawed fresh-frozen plasma, plasma frozen within 24 h and solvent/detergent-treated plasma (octaplasLG((R))), during 5-day storage at 1–6 degrees CVox Sang Epub2018423
- USFDA2018 Available from: https://www.fda.gov/downloads/biologicsbloodvaccines/bloodbloodproducts/approvedproducts/licensedproductsblas/ucm336161.pdfAccessed May 18, 2018
- ChhibberVGreeneMVauthrinMBaileyJWeinsteinRIs group A thawed plasma suitable as the first option for emergency release transfusion? (CME)Transfusion20145471751175524400951
- CoolingLGoing from A to B: the safety of incompatible group A plasma for emergency release in trauma and massive transfusion patientsTransfusion20145471695169725041147
- MehrCRGuptaRvon RecklinghausenFMSzczepiorkowskiZMDunbarNMBalancing risk and benefit: maintenance of a thawed Group A plasma inventory for trauma patients requiring massive transfusionJ Trauma Acute Care Surg20137461425143123694868
- WinearlsJCampbellDHurnCFibrinogen in traumatic haemorrhage: a narrative reviewInjury201748223024228088374
- WinearlsJWullschlegerMWakeEFibrinogen Early In Severe Trauma studY (FEISTY): study protocol for a randomised controlled trialTrials201718124128549445
- OkerbergCKWilliamsLA3rdKilgoreMLCryoprecipitate AHF vs. fibrinogen concentrates for fibrinogen replacement in acquired bleeding patients - an economic evaluationVox Sang2016111329229827248502
- KriebardisAGAntonelouMHGeorgatzakouHTTzounakasVLStamoulisKEPapassideriISMicroparticles variability in fresh frozen plasma: preparation protocol and storage time effectsBlood Transfus201614222823727136430
- FrancisCWMarderVJIncreased resistance to plasmic degradation of fibrin with highly crosslinked alpha-polymer chains formed at high factor XIII concentrationsBlood1988715136113652965927
- GerlachRTolleFRaabeAZimmermannMSiegemundASeifertVIncreased risk for postoperative hemorrhage after intracranial surgery in patients with decreased factor XIII activity: implications of a prospective studyStroke20023361618162312053001
- KarkoutiKvon HeymannCJespersenCMEfficacy and safety of recombinant factor XIII on reducing blood transfusions in cardiac surgery: a randomized, placebo-controlled, multicenter clinical trialJ Thorac Cardiovasc Surg2013146492793923820174
- NascimentoBGoodnoughLTLevyJHCryoprecipitate therapyBr J Anaesth2014113692293424972790
- KeyNSNegrierCCoagulation factor concentrates: past, present, and futureLancet2007370958543944817679021
- HauserCJBoffardKDuttonRResults of the CONTROL trial: efficacy and safety of recombinant activated Factor VII in the management of refractory traumatic hemorrhageJ Trauma201069348950020838118
- MurthiSBStansburyLGDuttonRPEdelmanBBScaleaTMHessJRTransfusion medicine in trauma patients: an updateExpert Rev Hematol20114552753721939420
- KrautJAMadiasNESerum anion gap: its uses and limitations in clinical medicineClin J Am Soc Nephrol20072116217417699401
- McCarronDAElliottWCRoseJSBennettWMSevere mixed metabolic acidosis secondary to rhabdomyolysisAm J Med1979675905908507101
- KrautJAMadiasNEMetabolic acidosis: pathophysiology, diagnosis and managementNat Rev Nephrol20106527428520308999
- WolbergASMengZHMonroeDM3rdHoffmanMA systematic evaluation of the effect of temperature on coagulation enzyme activity and platelet functionJ Trauma20045661221122815211129
- KermodeJCZhengQMilnerEPMarked temperature dependence of the platelet calcium signal induced by human von Willebrand factorBlood199994119920710381514
- KramerLBauerEJoukhadarCCitrate pharmacokinetics and metabolism in cirrhotic and noncirrhotic critically ill patientsCrit Care Med200331102450245514530750
- KornblithLZKutcherMERedickBJCalfeeCSVilardiRFCohenMJFibrinogen and platelet contributions to clot formation: implications for trauma resuscitation and thromboprophylaxisJ Trauma Acute Care Surg201476225526324458031
- AsehnouneKBaloghZCiterioGThe research agenda for trauma critical careIntensive Care Med20174391340135128756471
- ShuklaMSekhonUDBetapudiVIn vitro characterization of SynthoPlate (synthetic platelet) technology and its in vivo evaluation in severely thrombocytopenic miceJ Thromb Haemost201715237538727925685
- HarrJNMooreEEGhasabyanAFunctional fibrinogen assay indicates that fibrinogen is critical in correcting abnormal clot strength following traumaShock2013391454923247121
- JegerVWilliSLiuTThe Rapid TEG a-Angle may be a sensitive predictor of transfusion in moderately injured blunt trauma patientsScientific World Journal2012201282179422547997
- JegerVZimmermannHExadaktylosAKCan RapidTEG accelerate the search for coagulopathies in the patient with multiple injuries?J Trauma20096641253125719359945
- SchlimpCJSolomonCRanucciMHochleitnerGRedlHSchochlHThe effectiveness of different functional fibrinogen polymerization assays in eliminating platelet contribution to clot strength in thromboelastometryAnesth Analg2014118226927624445628
- KashukJLMooreEESawyerMPostinjury coagulopathy management: goal directed resuscitation via POC thrombelastographyAnn Surg2010251460461420224372
- KashukJLMooreEEWohlauerMInitial experiences with point-of-care rapid thrombelastography for management of life-threatening postinjury coagulopathyTransfusion2012521233321790635
- PommereningMJGoodmanMDFarleyDLEarly diagnosis of clinically significant hyperfibrinolysis using thrombelastography velocity curvesJ Am Coll Surg201421961157116625458237
- SheenJHHeegerPSEffects of complement activation on allograft injuryCurr Opin Organ Transplant201520446847526132735
- DavisAE3rdMejiaPLuFBiological activities of C1 inhibitorMol Immunol200845164057406318674818