
Abstract
Acquired hemophilia A is a rare autoimmune disorder caused by an autoantibody (inhibitor) to factor VIII (FVIII) that interferes with its coagulant function and predisposes to severe, potentially life-threatening hemorrhage. Disease management focuses on controlling bleeding, primarily with the use of bypassing therapy and recombinant porcine FVIII, and permanently eradicating the autoantibody using various immunosuppressants. Treatment challenges include delayed diagnosis, difficulty achieving hemostasis and durable remissions, and complications associated with the use of hemostatic and immunosuppressive therapy in a primarily older patient population.
Introduction
Acquired hemophilia A (AHA) is a rare autoimmune disease caused by immunoglobulin G antibodies that bind to specific domains on the factor (F) VIII molecule, partially or completely neutralizing its coagulant function.Citation1–Citation3 Because FVIII serves as a cofactor to activated FIX in the tenase complex, its deficiency reduces thrombin generation on the surface of activated platelets,Citation4 predisposing to bleeding that may be life-threatening.Citation1,Citation2 The incidence of AHA increases with age, ranging from an estimated 0.045 per million per year in children <16 years to 14.7 per million annually in people >85 years.Citation5 Approximately 10% of persons with AHA are younger women diagnosed during or after pregnancy, while more than 80% of those affected are men and women 65 years and older (median age: 78 years).Citation5–Citation9 Roughly, half of all AHA cases are attributable to an underlying medical condition, typically another autoimmune disorder, malignancy, or a drug/allergic reaction (); the remainder are idiopathic.Citation5–Citation8
Table 1 Underlying conditions associated with AHA
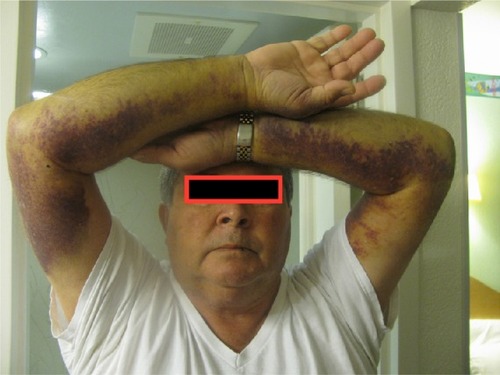
The usual clinical presentation of AHA is spontaneous or provoked bleeding and an unexplained, prolonged activated partial thromboplastin time (aPTT) in a person with a negative personal or family history of a coagulopathy.Citation10,Citation11 In contrast to the joint bleeds that characterize congenital hemophilia A,Citation12 bleeding associated with AHA usually manifests as spontaneous subcutaneous hematomas and extensive bruising (), although muscle bleeding, hematuria, epistaxis, gastrointestinal bleeding, and even intracranial hemorrhage may occur.Citation8,Citation10,Citation11 A substantial number of individuals – nearly 7% of the 501 patients enrolled in the European Acquired Haemophilia Registry (EACH2), the largest reported AHA observational datasetCitation8 – had no bleeding symptoms, and the diathesis was detected incidentally after routine blood testing.Citation10,Citation11
Figure 1 Bleeding associated with acquired hemophilia A.
The management of AHA requires a two-pronged, parallel approach: 1) control bleeding and 2) eradicate the inhibitor.Citation1,Citation7,Citation13,Citation14 Additionally, an underlying cause should be investigated. Here we review the strategies used to treat patients with AHA and discuss newer treatments and emerging therapies that may surmount some of the challenges posed by this complex bleeding disorder.
Hemostatic therapy
The need for hemostatic therapy in patients with AHA is determined by bleeding site and severity.Citation13–Citation16 The inhibitor titer provides little guidance for clinical decision making, as it does not correlate with bleeding phenotype. Unlike the alloantibodies associated with congenital hemophilia A, which inactivate FVIII in direct proportion to their concentration (type 1 kinetics), acquired inhibitors exhibit type 2/nonlinear kinetics, whereby rapid initial inactivation is followed by a slower phase of equilibrium.Citation4,Citation11,Citation17 Some measurable FVIII (usually <10% of normal) is often detected in patients with high-titer anti-FVIII antibodies (>5 Bethesda units [BU]), but this small amount of residual circulating FVIII offers no protection from bleeding. In fact, residual FVIII activity levels as high as 10% – consistent with a diagnosis of mild congenital hemophilia A in which bleeding seldom occurs – have been observed in the presence of severe hemorrhage.Citation11
Minor bleeding
In persons with low-titer acquired inhibitors (≤5 BU) who have no/minimal bleeding and do not require surgery, observation and laboratory monitoring may be sufficient.Citation18 Even extensive subcutaneous hemorrhage can usually be managed without treatment, although patients may be hospitalized to facilitate close monitoring.Citation19 Inhibitors spontaneously disappear within a few months in approximately 25% of cases, primarily those associated with pregnancy or antibiotic therapy.Citation2,Citation20
When minor bleeding requires treatment, initial interventions include routine hemostatic techniques; avoidance of invasive procedures; and discontinuation of any therapies that can exacerbate bleeding (ie, antiplatelet or anticoagulant drugs), if at all possible. For patients with an FVIII level >5% and a very low-titer inhibitor (<2 BU), desmopressin (DDAVP) may cause a transient rise in FVIII levels capable of controlling minor bleeding.Citation1,Citation4,Citation14 Human FVIII is more likely than DDAVP to achieve hemostasis in patients with low-titer inhibitors, but large, even massive doses may be required to saturate the autoantibody. Human FVIII is almost never effective in patients with high-titer inhibitors.
Major bleeding
Severe bleeding symptoms on presentation have been reported in up to 72% of patients and require the prompt initiation of antihemorrhagic treatment.Citation21 Human FVIII concentrates or DDAVP may have some value as first-line treatment if the inhibitor titer is ≤5 BU but will only control bleeding to the degree that hemostatic circulating levels of FVIII can be achieved. Thus, these interventions are likely to be completely ineffective in patients with high-titer autoantibodies.
Bypassing agents
Bypassing therapy with agents that circumvent the need for FVIII is the current treatment of choice for major bleeding in patients with high-titer (>5 BU) acquired inhibitors.Citation13,Citation15 Two bypassing agents are available: plasma-derived activated prothrombin complex concentrate (aPCC) (FEIBA; Baxter Healthcare Corporation, Deerfield, IL, USA), which contains exogenously activated FII, FVII, FIX, and FX, and recombinant activated FVII (rFVIIa) (Novo Seven; Novo Nordisk A/S, Bagsværd, Denmark). Both achieve hemostasis by generating thrombin (in the absence of FVIII) at the site of bleeding,Citation22,Citation23 but they differ with respect to their biochemical properties and pharmacokinetics. In particular, aPCC has a longer half-life of 4–7 hoursCitation24 compared with approximately 2 hours for rFVIIa.Citation25
Although good and equivalent success rates have been reported for aPCC and rFVIIa used for hemostatic management in AHA,Citation26 neither is predictably effective. In the EACH2, bleeding was controlled in 60 of 64 patients (93%) treated with aPCC first-line and 159 of 174 (91%) of those treated with rFVIIa first-line.Citation26 Additionally, patients may exhibit a differential response to treatment. A randomized crossover study comparing aPCC and rFVIIa in the management of joint bleeding associated with congenital hemophilia A and inhibitors found that some patients reported better bleed resolution with one bypassing agent than the other,Citation27 and in our clinical practices, we have also observed this phenomenon in patients with AHA.
Either bypassing agent can be used first-line for bleeding in AHA, with choice determined by a patient’s previous response (if any), dosing convenience, possible need for early redosing, preference for a plasma-derived or recombinant product, and availability.Citation14 Mucosal bleeds may benefit from the addition of an antifibrinolytic drug (ie, epsilon-aminocaproic acid or tranexamic acid), with accumulating data supporting the safety of concomitant treatment, even with aPCC.Citation28 If first-line therapy fails, treatment with the alternative bypassing agent should be quickly initiated and may prove successful. The use of higher than recommended doses of aPCC (100 U/kg per dose or 200 U/kg/day) or rFVIIa (90 μg/kg every 2 hours) should be considered only under exceptional circumstances because of the increased risk for arterial and venous thrombosis.Citation14,Citation29 Such risk may be heightened in patients with AHA, who are frequently older and have comorbidities, such as cardiovascular disease, cancer, and pregnancy, that increase thrombotic risk.Citation13,Citation30,Citation31 The combined use of both bypassing agents should be avoided except in the setting of life-threatening bleeding uncontrolled by either agent used alone.Citation32
No laboratory tests have been validated for monitoring the efficacy of bypassing therapy. Treatment is assessed clinically, which is often difficult due to the location of bleeding. Although global hemostatic assays have been evaluated as a strategy for measuring thrombin generation after the application of bypassing therapy, these measurements do not correlate with clinical efficacy or bleed resolution.Citation33
Once hemostasis has been achieved, ongoing treatment at reduced dose (as prophylaxis) may be needed to prevent recurrence of bleeding.
Case study 1: challenges to achieving hemostasis – part 1
A 74-year-old woman with hypertension, type 2 diabetes mellitus, hyperlipidemia, and chronic obstructive pulmonary disease presents to the emergency room with right leg swelling. Deep vein thrombosis is suspected, but a venous Doppler ultrasound is negative, and she is released home. She returns 2 days later with worsening leg swelling, tingling in her foot, cold and bluish toes, and absent pedal pulses. Ultrasound shows a large intramuscular hematoma; compartment syndrome is diagnosed, and she is taken to the operating room for emergent release. Significant intraoperative bleeding occurs, and active bleeding from the fasciotomy incision continues in the recovery room. Laboratory studies show a hemoglobin of 5.6 g/dL, a prolonged aPTT of 72 seconds, a FVIII level of <1%, and an inhibitor titer of 134 BU. She is diagnosed with AHA.
The patient is given 4 units of packed red blood cells (RBCs), and hemostatic therapy is started with rFVIIa 90 μg/kg every 2 hours. After 6 hours, the patient is still actively bleeding, and the hemostatic regimen is changed to aPCC 75 U/kg every 6 hours. Active bleeding persists 24 hours after switching products, and another RBC transfusion is given. Combined sequential bypassing therapy is initiated using alternating doses of aPCC and rFVIIa (rFVIIa dosing 6 hours after aPCC and then aPCC dosing 2 hours after rFVIIa) ().
Figure 2 Regimen for combined sequential bypassing therapy.

Bleeding continues unabated after 24 hours of combined sequential therapy. Another RBC transfusion is given and intravenous tranexamic acid 10 mg/kg every 6 hours is added. Bleeding persists despite maximum doses of hemostatic agents, and there is growing concern about the potential for thrombotic complications, given the patient’s age and comorbidities.
Porcine FVIII
Despite the prompt initiation of appropriate bypassing therapy, fatal hemorrhage occurs in 3%–8% of patients with AHA.Citation5,Citation8 In the past, a highly purified plasma-derived porcine FVIII (Hyate:C; Speywood Pharmaceuticals Ltd, Maidenhead, Berks, UK) was an option for the management of bleeding caused by acquired inhibitors. In clinical practice, the hemostatic efficacy of porcine FVIII was 90%, and inhibitory antibodies against hFVIII showed low cross-reactivity with porcine FVIII (~15%).Citation34,Citation35 A major advantage to the use of porcine FVIII compared with bypassing agents was the ability to measure circulating FVIII levels using a standard FVIII assay, thus allowing dose adjustment. Manufacture of this product ceased in 2004 because of concerns about possible residual viral contamination, primarily by porcine parvovirus,Citation36 leaving a void in the AHA treatment armamentarium.
A recombinant porcine sequence FVIII (rpFVIII) (Obizur; Baxter) formulated without animal-derived products has recently received US Food and Drug Administration approval for use in AHA. The pivotal Phase II/III clinical trial that led to product licensure enrolled 28 adults with AHA (age range: 40–≥65 years) experiencing serious bleeding episodes that required hospitalization.Citation37 Following administration of an initial rpFVIII dose of 200 U/kg, therapeutic FVIII activity levels were achieved and maintained with intermittent rpFVIII administration until bleeding resolved. Response was determined by clinical assessment and FVIII activity levels. A positive response at 24 hours, defined as effective or partially effective bleeding control as determined by clinical assessment and FVIII activity levels, was reported for all 28 patients, and treatment of the initial bleeding episode was successful in 24 of 28 (86%). (The remaining four patients were taken off study before a final assessment could be made.) No thrombotic or other serious adverse events occurred that were related to the study drug. At baseline and before exposure to rpFVIII, ten of 28 patients (36%) demonstrated anti-rpFVIII antibody activity (0.8–29 BU) due to cross-reactivity with the patients’ existing FVIII autoantibodies. Antibody status was not known before initiation of treatment with rpFVIII, however, and all study patients achieved FVIII levels >100%, although higher doses were required in those with anti-rpFVIII antibodies at baseline. Four patients without baseline antibodies eventually developed anti-porcine inhibitors (range: 8–166 BU), and treatment was subsequently discontinued in two of these individuals.
Case study 1: challenges to achieving hemostasis – part 2
The patient is given rpFVIII at 200 U/kg. Her FVIII level rises from <1% to 76% 1 hour after infusion, and she receives additional doses every 2–3 hours for the first 24 hours to maintain FVIII levels >80%. Overall, she has good clinical bleeding control at 24 hours. Bolus doses of rpFVIII are continued every 4–6 hours, with a target trough level of >70%. The fasciotomy incision begins to heal by secondary intention, and bleeding continues to be well controlled. rpFVIII dosing continues, with the goal of maintaining FVIII levels >25% to ensure wound healing and prevent rebleeding.
After 100 rpFVIII doses, FVIII recovery is less than expected, and an anti-rpFVIII antibody measuring 20 BU is detected. At that point, bleeding is controlled, and the patient is transitioned to aPCC (75 IU/kg/day). The wound continues to heal by primary intention with no further bleeding.
Emerging hemostatic therapies
A recombinant humanized anti-FIXa/FX bispecific antibody has been studied in a primate model of severe AHA.Citation38 The investigational agent brings FIX and FX into spatially appropriate position, mimicking the cofactor function of FVIII without the need for activation. Weekly subcutaneous doses of the experimental agent (ACE910) significantly prevented spontaneous bleeding in all four treated cynomolgus monkeys (one animal experienced traumatic bleeding), and no hematuria or organ bleeding was seen at necropsy. Shortening of the prolonged aPTT observed immediately after the initial injection gradually disappeared in three of four monkeys in the middle of the dosing period, suggesting the development of alloantibodies to ACE910. In another primate model of acute bleeding, the hemostatic efficacy of a single 1–3 mg/kg bolus dose of ACE910 was equivalent to twice-daily doses of rpFVIII 10 U/kg. Because ACE910 has a 3-week half-life and nearly 100% subcutaneous bioavailability, once-weekly dosing to maintain hemostatic trough levels is feasible. Clinical investigation of ACE910 is ongoing.
Augmenting the activity of activated FV (FVa) is another novel approach for bypassing inhibitors in congenital hemophilia A. As a cofactor in the prothrombinase complex, FVa enhances the rate of thrombin generation by approximately 10,000-fold; however, this clotting factor is rapidly inactivated by protein C.Citation39,Citation40 SuperFVa is a molecularly engineered FVa variant in which an interdomain disulfide bond covalently anchors the A2 to the A3 domain, thus providing increased cofactor activity, while protein C cleavage site mutations enhance biologic stability.Citation41,Citation42 When evaluated in in vitro studies in hemophilia plasma with and without inhibitors and in in vivo mouse models, SuperFVa had excellent hemostatic properties.Citation41 All prohemostatic effects, including clot stability and prolongation of clot lysis, were synergistically enhanced when SuperFVa was used in combination with trace amounts of FVIIa in hemophilic or normal plasma with inhibitors.Citation43
Inhibitor eradication
Immunosuppression to eradicate an acquired inhibitor should begin immediately upon diagnosis in all patients.Citation13 Even those who are not actively bleeding remain at risk for life-threatening hemorrhage until the autoantibody has been eliminated,Citation5 and overall survival is improved in patients who achieve a complete response to inhibitor eradication.Citation7
Steroids and cyclophosphamide
Although the optimal immunosuppressive regimen and duration of treatment remains to be determined, prednisone (1 mg/kg/day) alone or in combination with oral cyclophosphamide (50–100 mg/day) is currently recommended as first-line treatment for autoantibody eradication.Citation13–Citation15 (Azathioprine, 6-mercaptopurine, and vincristine may be considered as alternatives to cyclophosphamide for cytotoxic therapy).Citation2,Citation44–Citation46 In the EACH2 that included data on first-line immunosuppression for 276 patients, those given steroids were compared with patients receiving steroids plus cyclophosphamide.Citation47 The groups were matched by logistic regression and a propensity score for age, sex, inhibitor titer, FVIII level, and underlying etiology. Among patients treated with steroids alone, 48% (68 of 142) achieved a stable complete remission (CR), as compared with 70% (58 of 83) given combination therapy (median time to CR for both regimens: ~5 weeks). These findings are in agreement with several other reports showing that first-line treatment with steroids plus cyclophosphamide is more likely to achieve a CR than steroids alone.Citation5,Citation7,Citation48,Citation49 Nonetheless, the final outcome of treatment, as measured by survival and sustained remission, was the same for both regimens in all large studies.Citation5,Citation7,Citation47
Successful eradication is defined as an undetectable inhibitor (<0.6 BU) and normal FVIII levels (>50%).Citation15 If the FVIII level is not increasing and the inhibitor titer is not dropping after 3–5 weeks of treatment, second-line immunosuppression should begin.Citation14
Case study 2: challenges to inhibitor eradication – part 1
A 62-year-old woman presents to the emergency room with multiple large bruises on her lower and upper extremities and a 2-day history of severe left hip pain that radiates into her back and lower abdomen, worsens with walking and leg extension, and improves with hip and knee flexion. Her medical history includes a diagnosis of systemic lupus erythematosus that was intermittently treated with corticosteroids (the last time approximately 2 years ago). She has experienced periodic rashes over most of her body for the last 5 years and reports increased bruising in the past 2 months. Computed tomography reveals a large retroperitoneal hematoma and bleeding into the iliopsoas plane. The potential for femoral nerve compression from an expanding iliopsoas bleed is of major concern. Hemoglobin is 5.5 g/dL, and she is given 6 units of RBCs. The prothrombin time is normal, but the aPTT is markedly elevated at 89 seconds and is not corrected by incubation of patient’s plasma with normal plasma at 37°C for 2 hours (mixing study). The diagnosis of AHA is confirmed by an FVIII level <1% and a Bethesda assay showing an inhibitor titer of 1,200 BU.
Hemostatic therapy is initiated with rFVIIa 90 μg/kg every 3 hours, and bleeding symptoms stabilize within 24 hours. Over the next 2 days, dosing frequency is gradually reduced to every 8 hours, but bleeding recurs when the dosing interval is further extended. Daily immunosuppressive therapy with oral prednisone 1 mg/kg and cyclophosphamide 1 mg/kg is started immediately upon diagnosis. During the first 5 days of treatment, the inhibitor titer decreases to 829 BU and holds at this level for the next 2 weeks.
The patient remains hospitalized because of the need for ongoing hemostatic therapy.
Rituximab
The anti-CD20 monoclonal antibody rituximab is increasingly used first- and second-line for inhibitor eradication in AHA. No data support the contention that rituximab used alone or in combination with other immunosuppressants results in higher remission rates or more rapid remissions.Citation14 In the EACH2, a stable CR was achieved by 59% of patients (30 of 51) treated with any rituximab – a success rate halfway between that achieved with steroids alone (48%) and steroids plus cyclophosphamide (70%).Citation47 Nevertheless, some patients resistant to standard first-line immunosuppression respond to second-line rituximab. In the EACH2, seven of 14 patients (50%) achieved a stable CR with rituximab-based regimens used after the failure of first-line drug(s) or relapse.
Other immunosuppressants
Calcineurin inhibitors (eg, cyclosporine, tacrolimus) and mycophenolate mofetil are alternatives to rituximab in patients who do not respond to first-line treatment.Citation14 The addition of immune tolerance with FVIII may be considered, although the benefits are unclear, and such treatment significantly increases the cost of inhibitor eradication. Available evidence does not support the use of intravenous immunoglobulin for the elimination of acquired inhibitors.Citation5,Citation7,Citation47
On the basis of data from EACH2, approximately 50% of patients can be expected to achieve a stable remission with second-line therapy using a variety of immunosuppressive agents.Citation47
Case study 2: challenges to inhibitor eradication – part 2
The patient receives rituximab 375 mg/m2 weekly for 4 weeks, and prednisone and cyclophosphamide are slowly tapered. Her inhibitor titer ranges between 700 to 800 BU for the next 4 weeks, after which it slowly decreases and becomes undetectable over another 4-week period. At 1-year follow-up, her B-cells are reconstituted, FVIII remains in the normal range, and the autoantibody is undetectable.
Follow-up
Long-term laboratory follow-up is needed to demonstrate a stable CR and rule out relapse, which occurs in up to 20% of patients.Citation13,Citation14,Citation19 Monthly monitoring of aPTT and FVIII activity (FVIII:C) for the first 6 months, then every 2 to 3 months up to 12 months, and every 6 months during the second year – and possibly beyond – is recommended.Citation15 Patients should be educated about the signs and symptoms of recurrence and urged to promptly report any bleeding or bruising.
Remission of AHA is often associated with high FVIII levels. Because many patients have thromboembolic risk factors associated with comorbidities, discontinuation of antiplatelet or antithrombotic therapy during bleeding, and/or reduced mobilization due to hospitalization, pharmacologic thromboprophylaxis is recommended after a stable CR is achieved.Citation15
Summary, future directions, and perspective commentary
The rarity of AHA means that patients usually present to clinicians with no previous experience in its management, often resulting in delays in diagnosis and treatment. Owing to the high morbidity and mortality associated with the condition and the complexities of management, immediate consultation with a hemophilia treatment center experienced in the care of inhibitor patients is recommended whenever AHA is suspected, whether or not bleeding is present.
Achieving hemostasis and eliminating the inhibitor represent the core of AHA management. The return of porcine FVIII has been eagerly awaited by clinicians and is now available as a recombinant concentrate. Promising new investigational agents may soon provide additional options for successfully controlling serious bleeding episodes in patients with FVIII autoantibodies. Although inhibitor eradication is successful in most individuals with AHA, immunosuppression in this primarily older patient population is associated with significant side effects, including sepsis, neutropenia, diabetes, and psychiatric illness.Citation47 Development of less toxic and more targeted immunosuppressive regimens capable of achieving faster, more durable responses remains a critical unmet need in AHA therapy.
Acknowledgments
Michele Grygotis, an independent consultant, provided medical writing services that were funded by Tulane University School of Medicine.
Disclosure
RKJ has received research support from Baxter Bioscience and Biogen Idec and received honoraria for advisory board participation and consulting fees from Baxter Bioscience, Bayer Healthcare, Grifols, and Novo Nordisk. MJ received honoraria for advisory board participation from Bayer and CSL Behring. CAL has received research funding from Baxter, Bayer, CSL Behring, and Novo Nordisk and received honoraria for advisory board participation from Baxter, Bayer, Biogen Idec, CSL Behring, Kedrion, Novo Nordisk, and Pfizer. The authors report no other conflicts of interest in this work.
References
- CollinsPWManagement of acquired haemophilia AJ Thromb Haemost20119Suppl 122623521781259
- CohenAJKesslerCMAcquired inhibitorsBaillieres Clin Haematol1996923313548800509
- ToschiVBaudoFDiagnosis, laboratory aspects and management of acquired hemophilia AIntern Emerg Med20105432533320407848
- MaADCarrizosaDAcquired factor VIII inhibitors: pathophysiology and treatmentHematology Am Soc Hematol Educ Program200643243717124095
- CollinsPWHirschSBaglinTPUK Haemophilia Centre Doctors’ OrganisationAcquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ OrganisationBlood200710951870187717047148
- GreenDLechnerKA survey of 215 non-hemophilic patients with inhibitors to Factor VIIIThromb Haemost19814532002036792737
- DelgadoJJimenez-YusteVHernandez-NavarroFVillarAAcquired haemophilia: review and meta-analysis focused on therapy and prognostic factorsBr J Haematol20031211213512670328
- KnoeblPMarcoPBaudoFEACH2 Registry ContributorsDemographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2)J Thromb Haemost201210462263122321904
- BorgJYGuilletBLe Cam-DuchezVGoudemandJLévesqueHSACHA Study GroupOutcome of acquired haemophilia in France: the prospective SACHA (Surveillance des Auto antiCorps au cours de l’Hémophilie Acquise) registryHaemophilia201319456457023574453
- WebertKEAcquired hemophilia ASemin Thromb Hemost201238773574122941793
- CoppolaAFavaloroEJTufanoADi MinnoMNCerboneAMFranchiniMAcquired inhibitors of coagulation factors: part I-acquired hemophilia ASemin Thromb Hemost201238543344622740182
- SrivastavaABrewerAKMauser-BunschotenEPTreatment Guidelines Working Group on Behalf of The World Federation of HemophiliaGuidelines for the management of hemophiliaHaemophilia2013191e1e4722776238
- CollinsPBaudoFHuth-KühneAConsensus recommendations for the diagnosis and treatment of acquired hemophilia ABMC Res Notes2010316120529258
- W CollinsPChalmersEHartDUnited Kingdom Haemophilia Centre Doctors’ OrganizationDiagnosis and management of acquired coagulation inhibitors: a guideline from UKHCDOBr J Haematol2013162675877323889317
- Huth-KühneABaudoFCollinsPInternational recommendations on the diagnosis and treatment of patients with acquired hemophilia AHaematologica200994456657519336751
- CollinsPWTreatment of acquired hemophilia AJ Thromb Haemost20075589390017461924
- ShettySBhaveMGhoshKAcquired hemophilia a: diagnosis, aetiology, clinical spectrum and treatment optionsAutoimmun Rev201110631131621115138
- SborovDWRodgersGMAcquired hemophilia a: a current review of autoantibody diseaseClin Adv Hematol Oncol2012101192722398803
- CollinsPWTherapeutic challenges in acquired factor VIII deficiencyHematology Am Soc Hematol Educ Program2012201236937423233606
- FranchiniMPostpartum acquired factor VIII inhibitorsAm J Hematol2006811076877316868941
- ZeitlerHGoldmannGMarquardtNUlrich-MerzenichGLong term outcome of patients with acquired haemophilia – a monocentre interim analysis of 82 patientsAtheroscler Suppl201314122322823357169
- TurecekPLVáradiKGritschHSchwarzHPFEIBA: mode of actionHaemophilia200410Suppl 23915385040
- HoffmanMMonroeDM3rdThe action of high-dose factor VIIa (FVIIa) in a cell-based model of hemostasisDis Mon2003491142112525825
- VáradiKNegrierCBerntorpEMonitoring the bioavailability of FEIBA with a thrombin generation assayJ Thromb Haemost20031112374238014629472
- VillarAAronisSMorfiniMPharmacokinetics of activated recombinant coagulation factor VII (NovoSeven) in children vs adults with haemophilia AHaemophilia200410435235915230949
- BaudoFCollinsPHuth-KühneAEACH2 registry contributorsManagement of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) RegistryBlood20121201394622618709
- AstermarkJDonfieldSMDiMicheleDMFENOC Study GroupA randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: the FEIBA NovoSeven Comparative (FENOC) StudyBlood2007109254655116990605
- HolmströmMTranHTHolmePACombined treatment with APCC (FEIBA®) and tranexamic acid in patients with haemophilia A with inhibitors and in patients with acquired haemophilia A – a two-centre experienceHaemophilia201218454454922348384
- AledortLMComparative thrombotic event incidence after infusion of recombinant factor VIIa versus factor VIII inhibitor bypass activityJ Thromb Haemost20042101700170815456478
- EhrlichHJHenzlMJGompertsEDSafety of factor VIII inhibitor bypass activity (FEIBA): 10-year compilation of thrombotic adverse eventsHaemophilia200282839011952842
- O’ConnellKAWoodJJWiseRPLozierJNBraunMMThromboembolic adverse events after use of recombinant human coagulation factor VIIaJAMA2006295329329816418464
- SchneidermanJRubinENugentDJYoungGSequential therapy with activated prothrombin complex concentrates and recombinant FVIIa in patients with severe haemophilia and inhibitors: update of our previous experienceHaemophilia20071324424817498072
- DehmelHWerwitzkeSTrummerAGanserATiedeAThrombelastographic monitoring of recombinant factor VIIa in acquired haemophiliaHaemophilia200814473674218445011
- HayCRPorcine factor VIII: past, present and futureHaematologica20008510 Suppl2124 discussion 24–2511187865
- MorrisonAELudlamCAKesslerCUse of porcine factor VIII in the treatment of patients with acquired hemophiliaBlood1993816151315208453098
- GiangrandePLPorcine factor VIIIHaemophilia201218330530922531020
- OBIZUR [prescribing information]Westlake Village, CABaxter Healthcare Corporation2014
- MutoAYoshihashiKTakedaMAnti-factor IXa/X bispecific antibody ACE910 prevents spontaneous joint bleeds in a long-term primate model of acquired hemophilia ABlood2014124203165317125274508
- MannKGJennyRJKrishnaswamySCofactor proteins in the assembly and expression of blood clotting enzyme complexesAnnu Rev Biochem1988579159563052293
- NesheimMETaswellJBMannKGThe contribution of bovine Factor V and Factor Va to the activity of prothrombinaseJ Biol Chem1979254211095210962500617
- von DrygalskiACramerTJBhatVGriffinJHGaleAJMosnierLOImproved hemostasis in hemophilia mice by means of an engineered factor Va mutantJ Thromb Haemost201412336337224818532
- GaleAJXuXPellequerJLGetzoffEDGriffinJHInterdomain engineered disulfide bond permitting elucidation of mechanisms of inactivation of coagulation factor Va by activated protein CProtein Sci20021192091210112192065
- von DrygalskiABhatVGaleAJGriffinJHMosnierLOSynergism of augmented FVa activity and recombinant human FVIIa represent a novel bypassing strategy in hemophilic patients with inhibitorsHaemophilia201420Suppl 34142
- HayCRAcquired haemophiliaBaillieres Clin Haematol199811228730310097808
- LianECLarcadaAFChiuAYCombination immunosuppressive therapy after factor VIII infusion for acquired factor VIII inhibitorAnn Intern Med1989110107747782496636
- BaudoFCaimiTde CataldoFDiagnosis and treatment of acquired haemophiliaHaemophilia20101610210210620536992
- CollinsPBaudoFKnoeblPEACH2 registry collaboratorsImmunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2)Blood20121201475522517903
- GreenDRademakerAWBriëtEA prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodiesThromb Haemost19937057537578128430
- BittingRLBentSLiYKohlwesJThe prognosis and treatment of acquired hemophilia: a systematic review and meta-analysisBlood Coagul Fibrinolysis200920751752319644360