
Abstract
Liver cancer is highly malignant, has a low sensitivity to chemotherapy, and is associated with poor patient prognosis. The last 3 years have seen the emergence of promising targeted therapies for the treatment of hepatocellular carcinoma (HCC). For over 10 years, before the discovery of lenvatinib, sorafenib was only first-line therapeutic agent available for the treatment of advanced HCC. However, several clinical studies have shown that a considerable proportion liver cancer patients are insensitive to sorafenib. Very few patients actually substantially benefit from treatment with sorafenib, and the overall efficacy of the drug has not been satisfactory; therefore, sorafenib has attracted considerable research attention. This study, which is based on previous studies and reports, reviews the potential mechanisms underlying sorafenib resistance and summarizes combination therapies and potential drugs that can be used to sensitize HCC cells to sorafenib.
Introduction
Hepatocellular carcinoma (HCC) is insidious in nature and has a low early diagnosis rate. Surgical resection, local ablation, and liver transplantation are potential curative treatment options for early-stage HCC. Sorafenib has long been the main targeted drug used for the systemic treatment of advanced liver cancer. It is an oral multi-kinase inhibitor and inhibits intracellular serine/threonine kinases (including Raf-1, wild-type B-Raf, and mutant B-Raf) and receptor tyrosine kinases (RTKs) (including the vascular endothelial growth factor receptors 1, 2, and 3, platelet-derived growth factor receptors β and c-KIT, FMS-like tyrosine kinase 3, and RET), thereby inhibiting tumor proliferation and angiogenesis.Citation1,Citation2 However, only 35–43% of patients respond to sorafenib, and most of them relapse within 6 months.Citation3,Citation4 Therefore, elucidating the mechanisms underlying sorafenib resistance (SR) is important for the improvement of survival in HCC patients. Because of the existence of complicated signaling cross-talk within HCC cells, which leads to the development of multiple compensatory mechanisms and alternative pathways, HCC patients respond differentially to sorafenib. Elucidating the mechanisms underlying SR and enhancing drug sensitivity are key strategies for improving the clinical efficacy of sorafenib.
Alleviation of SR Through the Hypoxia-Related Pathway in HCC Cells
Mechanism of Hypoxia-Related SR
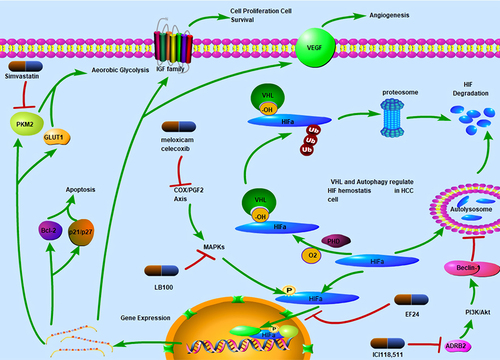
Hypoxia is commonly occurs in solid tumors such as HCC.Citation5 Since the speed of vasculogenesis is not sufficient to meet up with rapid tumor growth, oxygen and nutrient supply is usually limited. Hypoxia inducible factors (HIFs) are the main transcriptional regulators of the adaptive response to hypoxia in HCC cells.Citation6 Oxygen content is a critical determinant of cellular HIF homeostasis. HIFs contain an oxygen-dependent degradation domain (ODDD) of approximately 200 residues that regulates their degradation. The ODDD contains two oxygen-dependent prolyl hydroxylation sites, both of which can be hydroxylated by prolyl hydroxylases.Citation7 Under normal oxygen supply conditions (normoxia), hydroxylated HIF1α is recruited by the von Hippel-Lindau ubiquitination complex (VHL) and degraded in a ubiquitination-dependent manner (). In addition, HIF1α can degraded through the autophagy pathway.Citation8 Under hypoxia-induced stress conditions, HIF cannot be hydroxylated or stabilized for the activation the transcription of over 40 genes that facilitate angiogenesis and the shift in cell energy metabolism to anaerobic glycolysis.Citation9 HIF1α has been identified to be relevant in chemoresistance, tumor aggressiveness, and poor prognosis in HCC patients.Citation10 Interestingly, HIFs tend to exhibit dual characteristics under sorafenib-induced stress conditions. As a drug with anti-angiogenesis activity, sorafenib has VEGF as one of its main targets.Citation11 In vivo studies have shown that sorafenib downregulates the synthesis of both HIF1α and VEGF.Citation12 Of note, VEGF is also a key downstream factor that affects HIF expression. However, long-term treatment with sorafenib paradoxically induces an increase in HIF transcription and translation.Citation13 This could be because the hypoxia microenvironment, which develops due to the anti-angiogenesis effects of sorafenib, selects for more resistant and invasive HCC clones.Citation13,Citation14
Figure 1 Mechanism of HIF-dependent sorafenib sensitization. HIFα is hydroxylated by prolyl hydroxylases under normoxia conditions and binds with VHL. Following ubiquitination by VHL, HIFα is degraded by 26S proteosomes. HIFα can be packaged by autophagosomes and transported to lysosomes for hydrolysis. ICI118.511 can inhibit autophagy-dependent HIF degradation by inhibiting ADRB2. HIFα can be translocated to the nucleus where it functions as a transcription factor. This process is pharmacologically inhibited by EF24. HIF translocation is also dependent on MAPKs located in the cytosol. LB100 inhibits HIF function through MAPK inhibition. Meloxicam and celecoxib can also inhibit MAPK through the COX-PGF2 axis, thereby inhibiting HIF translocation and function. HIFs transcriptionally activate multiple genes, including Bcl-2 and p21/p27, which mediate cell apoptosis, PKM2 and GLUT1, induce metabolic reprogramming and shift cell energy metabolism to aerobic glycolysis. Simvastatin inhibits PKM2 and sensitizes HCC cells to sorafenib. HIFs also regulate cell proliferation and angiogenesis by upregulating IGF and VEGF levels.
HIF1α
As HIF1α contributes to SR in HCC cells, its inhibition could be a potential strategy for overcoming SR (). Liang et al found that EF24 suppresses HIF translocation to the nucleus and upregulates VHL expression, thereby inducing an increase in HIF1α degradation through proteasome activity.Citation13 This enhances the antitumor effects of sorafenib. In addition, curcumin, which has a similar structure to EF24, was found to potentiate the effects of sorafenib.Citation15 Wu et al found that ICI118,551, an ADRB2 antagonist, in combination with sorafenib, destabilized HIF1α, thereby inhibiting tumor growth.Citation16 Mechanistically, ADRB2 signaling inhibits autophagy by facilitating Beclin1 homodimer formation in an Akt-dependent manner. Inhibition of autophagy further stabilizes HIF1α, thereby inducing SR.Citation16 Thus, the pharmacological inhibition of ADRB2 promotes autophagy and HIF1α degradation. Of note, ADRB2 is also degraded in a VHL-dependent manner.Citation17 Since VHL is the main regulatory factor for HIF, it is hypothesized that targeting the HIF/VHL axis might serve as a potential therapeutic strategy for reversing SR. However, the role of VHL in HCC chemoresistance still needs to be further investigated. Liu et al found VHL instability to enhance HCC metastasis and adaption to the hypoxia microenvironment.Citation18 Furthermore, Feng et al found sorafenib-resistant HCC cells to be more sensitive to simvastatin than non-resistant cells in LM3 cells.Citation19 Mechanistically, HCC cells evade the effects of sorafenib by upregulating the expression levels of both PKM2 and HIF1α and by shifting energy metabolism from OXPHOS to aerobic glycolysis. Simvastatin inhibits PKM2 and HIF1α, thereby reversing SR.Citation19
HIF2α
The HIF family is constituted of three members (HIF1α, HIF2α, and HIF3α). HIF2α is closely related to HIF1α homologues.Citation20 HIF1α and HIF2α jointly control hypoxia response in HCC cells.Citation21 Clinical data have shown that the overexpression levels of both HIF1α and HIF2α are reliable poor prognostic markers for HCC.Citation22 Interestingly, sorafenib was found to exert downregulatory bioeffects against HIF1α expression, and to shift hypoxic response from an HIF1α-dependent pathway to an HIF2α-dependent pathway.Citation23 This consequently leads to the upregulation of VEGF and cyclin D1 expression, and SR. Thus, treatment strategies targeting HIF1α alone may not be sufficient to overcome SR; HIF2α targeting may equally be necessary for sensitizing HCC cells to sorafenib (). Dong et al found that the activation of COX-2/PGE2 axis effectively induced a decrease in VHL levels and stabilized HIF2α. In addition, it was found to enhance HIF2α activity by promoting HIF2α nuclear translocation via the p38 mitogen-activated protein kinase (MAPK) pathway. Consequently, VEGF, cyclin D, and TGFα/EGFR were all activated, and this led to SR. The COX-2 inhibitors, meloxicam and celecoxib, were found to enhance response to sorafenib both in an in vivo CDX model and in vitro.Citation24 Furthermore, hypoxic conditions were shown to activate the MAPK signaling pathway, consequently leading to SR through the enhancement of p-Smad3-dependent B‑cell lymphoma (Bcl)-2 inhibition. Interestingly, LB-100, a serine/threonine protein phosphatase 2A inhibitor, which was found to be functional only in hypoxic environments, was shown to sensitize cells to sorafenib. LB-100 sensitizes cells to sorafenib by upregulating p-Smad3 expression, thereby decreasing Bcl-2 expression and increasing HCC cell apoptosis.Citation25 Furthermore, Ma et al found that the antitumor drug, 2-methoxyestradiol, which acts by dysregulating HIF-1α expression, showed synergistic effects with sorafenib by inhibiting both the expression and translocation of HIF-1α and HIF-2α.Citation23 As HIF-2α is upregulated in HCC cells and contributes to chemoresistance, its downregulation using the antidiabetic drug, metformin, was also found to sensitize HCC cells to sorafenib both in vitro and in vivo.Citation26,Citation27 Moreover, regorafenib was found exhibit synergistic effects with metformin in HCC cells through a similar mechanism.Citation28
Hypoxia and the Nuclear Factor κ-Light-Chain-Enhancer of Activated B Cells (NF-κB) Pathway
The NF-kB pathway, which is a critical downstream pathway for HIF regulation, plays a critical role in HCC cell resistance to sorafenib.Citation29 Studies have shown that NF-κB is significantly activated in sorafenib-resistant HCC cells.Citation29 Cheng et al demonstrated that the inhibition of interleukin-1 receptor-associated kinase 1 (IRAK1) axis, which is an upstream regulator of the NF-kB pathway, using a specific IRAK1/4 kinase inhibitor, induces effects that are synergistic to those of sorafenib.Citation30 In line with these findings, Alsaied et al found that triptolide, which decreases NF-κB activity, in combination with sorafenib, can significantly control mouse tumor growth. Of note, in this study, 10 mg/kg of sorafenib, which is lower that 10% of the dose currently prescribed for HCC patients, was found to exert significant effects in combination with triptolide in an in vivo HCC model.Citation31 CD47 is a downstream NF-κB regulatory factor; its inhibition using anti-CD47 antibodies was found to induce an increase in cell sensitization to sorafenib.Citation29 Furthermore, as an antiangiogenic drug, sorafenib was shown to decrease vessel density and induce the development of a hypoxic microenvironment for tumor cell destruction.Citation32 Vorinostat, a traditional and well-discussed HDACi was shown to exhibit synergistic effects with sorafenib and to induce apoptosis and cell cycle stagnation in HCC cells.Citation33 Mechanistically, vorinostat promotes NF-κB activation and induces cancer cell progression and chemoresistance (). However, sorafenib inhibits NF-κB and its downstream signaling pathway, thereby further inhibiting vorinostat-induced HCC progression and drug resistance.Citation34 It has been shown that the strategy of using sorafenib and HDACi in combination has yielded unsatisfactory results in patients with HCC expressing low CD95.Citation35 Hence, Hamed et al found that TRAIL, a death receptor agonist, in combination with sorafenib and HDACi induced apoptosis more significantly than TRAIL or [sorafenib + HDACi] in CD95 null Huh7 HCC cells.Citation35 A Phase I/II study on the sorafenib, HDACi, and resminostat combination is ongoing.Citation36 This triple combination therapy offers a novel potential strategy for the reduction of SR in HCC cells ().
Trans-Arterial Chemoembolization (TACE) in Combination with Sorafenib
TACE is recognized as the most common nonsurgical treatment method for HCC.Citation37 Trans-arterial chemoembolization involves the infusion of cytotoxic agents mixed with lipiodol into hepatic arteries.Citation38 This induces cell apoptosis, and subsequently, cancer tissue necrosis as a result of local angiogenesis inhibition in HCC, which is a well-vascularized tumor type. Consequently, several growth factors, including HIF-1α and VEGF, are activated. Several studies have reported the efficacy of the TACE plus sorafenib strategy. A randomized, multi-center, prospective clinical trial (NCT01217034) compared the efficacy and safety of TACE plus sorafenib with those of TACE alone in patients with unresectable HCC. Median progression free survival (PFS) significantly increased in patients treated with the combination therapy than in those treated with TACE alone (25.2 vs 13.5 months; p=0.006).Citation39 Furthermore, a meta-analysis carried out by Zhang et al also confirmed the clinical efficacy of the combination therapy to be better than that of TACE alone.Citation40 Li et al confirmed that serum VEGF concentrations were significantly elevated following TACE in rabbit model.Citation41 This suggests that VEGF is a key factor that enables HCC cells to survive TACE treatment. As a VEGF antagonist, sorafenib specifically inhibits cell clones that survive TACE. However, the biological mechanism underlying the action of TACE plus sorafenib, as well as the reason why the combination therapy shows better clinical efficacy than TACE alone, still need to be determined.
Energy Metabolism
Glutamine Metabolism
Metabolic reprogramming contributes significantly to tumor metastasis and drug resistance.Citation42 Recently, it was discovered that cancer cells reprogram nutrient metabolism in the tumor microenvironment.Citation43 Cancer cells shift their energy metabolism to glutamine and lipids but not to glucose as initially believed. In line with this, Kim et al observed glutamine metabolism and reductive glutamine carboxylation in sorafenib-resistant HCC cells.Citation44 Based on this, targeting glutamine metabolism could be a potential strategy for reversing SR. The glutaminase inhibitor, bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl) ethyl sulfide, also known as BPTES, was found be effective in sensitizing HCC cells to sorafenib. Besides, oxoglutarate dehydrogenase-like (OGDHL), one of rate-limiting components of the mitochondrial multi-enzyme OGDH complex and plays a critical role in down-regulating reductive glutamine metabolism.Citation45 Dai et al found that lower expression of OGDHL was associate with advanced tumor stage, significantly worse survival and more frequent tumor recurrence in 681 patients.Citation45 Over expression of OGDHL enhanced sorafenib chemo-sensitization to HCC CDX models.Citation45 Kim et al attribute this glutamine-dependent energetic shift to peroxisome proliferator-activated receptor-δ (PPARδ), as its inhibition by GSK0660 was found to effectively reverse SR.Citation44
Lipid Metabolism
Lally et al highlighted the importance of de novo lipogenesis (DNL) in HCC energy metabolism. Acetyl-CoA carboxylases (ACC) 1 and 2 are both rate-limiting enzymes in the DNL process, which is activated by AMPK. ND-654, a liver-specific ACC inhibitor, was found to improve survival in HCC-bearing rats in combination with sorafenib through the inhibition of hepatic DNL.Citation46
ROS
Increasing evidence shows that sorafenib enhances oxidative stress in HCC cells.Citation47,Citation48 However, HCC cells upregulate the expression of antioxidant genes through different mechanisms to avoid sorafenib-induced oxidative damage. For instance, the facilitates chromatin transcription (FACT) complex, a histone chaperone,Citation49 was found to be upregulated in HCC cells. The FACT complex is a key regulatory factor for NRF2 expression and its downstream factors include NQO1, TXNRD1, and TKT, which are essential for HCC evasion of oxidative damage.Citation49,Citation50 Thus, by inhibiting the FACT complex and inducing oxidative stress, curaxin showed promising synergistic effects with sorafenib in vivo and in vitro.Citation50 In addition, auranofin (AUR)-induced inhibition of TXNRD1 transcription, which is mediated by NRF2 and downregulated in HCC cells,Citation51 also induces an increase in ROS levels and leads to sorafenib sensitization in these cells.Citation52 Targeting the FACT/NRF2/TXNRD1 axis and inducing ROS production could be a promising strategy for sensitizing HCC cells to sorafenib. The curaxin and sorafenib combination also exhibits acceptable safety profiles in mice. However, these studies are mostly experimental and there is need for further clinical trials to be carried out to validate the safety and effectiveness of these agents.
Signaling Pathways
Upstream RTKs
Transforming growth factor β (TGFβ) is a key upstream RTK regulatory factor. TGFβ was found to be upregulated in HCC patients,Citation53 and over-activated especially in sorafenib-resistant HCC patients.Citation54 Matsuda et al found that TGF enhanced SR in HCC.Citation6 Valproic acid (VPA) is a histone deacetylase inhibitor that is used as an anti-epileptic agent. Interestingly, VPA was found to be effective in reversing TGFβ-induced SR and exhibited synergistic effects with sorafenib in HCC cells.Citation55,Citation56 Mechanistically, VPA modulates the Jagged 2-mediated Notch1 signaling pathway and reverses the EMT phenotype, which are significantly correlated with chemoresistance, especially resistance to sorafenib, in HCC cells.Citation55 The TGFβRI kinase inhibitor, LY2157399, was found to effectively enhance sorafenib-induced apoptosis in HCC cells.Citation57 Studies have shown that TGFβ can upregulate receptor tyrosine kinase (RTK) levels. Furthermore, RTKs, including IGF1R and EGFR, have been shown to be responsible for chemoresistance to sorafenib.Citation58,Citation59 Thus, LY2157399 can improve sensitivity to sorafenib by suppressing RTK-induced SR via the inhibition of TGFβ. In consonance with these findings, the liver X receptor (LXR) agonist, T0901317, was also found to be effective in inhibiting RTK function and sensitizing HCC cells to sorafenib. LXR is a key factor involved in liver lipid metabolismCitation60 and its activation induces cholesterol efflux and interferes with the stability of the cytomembrane, thereby suppressing the sorafenib-dependent recruitment of multiple RTKs, including MET and EGFR, in lipid rafts; this leads to an enhancement of the antitumor effects of sorafenib.Citation61
Downstream RTKs
Sorafenib is a well-known tyrosine kinase inhibitor, and it inhibits several different targets.Citation62 However, compensatory pathways are usually activated in HCC cells to evade the cytotoxic effects of sorafenib. Thus, targeting compensatory signaling pathways is a potential strategy for alleviating chemoresistance to sorafenib.
Increasing evidence has shown that drug resistance can be acquired by HCC cells through RTK-mediated activation of several signaling pathways. SRC homology 2 domain-containing phosphatase 2 (SHP2) is an important downstream RTK regulatory factor. It has been shown that SHP2 is activated by sorafenib in HCC cells and that further activation of SHP2 induces the suppression of STAT3, an important transcription factor that has been shown to be involved in chemoresistance in different cancer types, including HCC.Citation63–67 Sorafenib has been shown to suppress STAT3 function through SHP2 activation,Citation68,Citation69 thereby further inducing apoptosis in HCC cells. In addition, by directly activating SHP-1, dovitinib, another multiple kinase inhibitor, inhibits STAT3 and exhibits synergistic effects with sorafenib.Citation70 Moreover, SC-2001, which has a similar structure to obatoclax, has been shown to exhibit synergistic effects with sorafenib by inducing RFX-1/SHP-1 expression, and consequently, STAT3 suppression.Citation71 Furthermore, Leung et al found that SHP2 inhibition using SHP099 also sensitizes HCC cells to sorafenib. It has been reported that RTKs can induce the activation of the MEK/ERK and Akt signaling pathways, thereby inducing SR.Citation72 In summary, SHP2 could be an important downstream effector of RTKs, which are key inducers of SR, and induces sorafenib sensitization through dephosphorylation and STAT3 inhibition. The dual nature of the interaction between SHP and sorafenib still needs to be further investigated.
The Akt and c-Met Pathways
The Akt pathway is an important compensatory pathway in HCC cells under sorafenib-induced stress.Citation73 Several Akt inhibitors are under investigation in clinical trials to determine their efficacy in HCC treatment.Citation73 Increasing evidence has shown that the IGF/FGF axis, which is an upstream Akt regulator, plays an important role in acquired SR.Citation74 The inhibition of these pathways may contribute to the restoration of HCC cell sensitivity to sorafenib and further increase this sensitivity.Citation74 For example, Wang et al found that ceritinib, which is an IGFR inhibitor initially used for the treatment of non-small cell lung cancer, could sensitize HCC cells to sorafenib by inhibiting the IGF1R/Akt pathway both in vitro and in xenograft and HCC mouse models.Citation75 Interestingly, despite the significant IGF1R inhibition induced by shRNA and ceritinib, they do not exhibit significant suppressive effects against HCC cells when used alone. Ceritinib exhibits significant inhibitory effects against HCC cell proliferation when used in combination with sorafenib as compared to when it is used alone. In addition, the IGFR inhibitor, linsitinib, and the FGFR inhibitor, brigatinib, were found to be effective in decreasing sorafenib-resistant HCC cell viability through the Akt pathway.Citation74 In line with this, Xu et al found that liver-specific microRNA-122, which targets IGF1R and inhibits its expression, was significantly downregulated in sorafenib-resistant cells. The IGF-1R inhibitors, PPP and NVP-AEW541, in combination with sorafenib, significantly induced cell apoptosis and decreased drug tolerance in vitro.Citation58 In addition, Zhai et al found that bufalin, a natural compound extracted from bufonid, reverses acquired SR by downregulating Akt phosphorylation. Moreover, Lu et al found that 20(S)-Ginsenoside Rg3 sensitizes HCC cells to sorafenib through the PI3K/Akt pathway. The PI3K inhibitor, SF1126, was also found to be effective in reversing SR in vivo.Citation76
However, targeting the Akt pathway may not be sufficient to reverse sorafenib chemoresistance due to the activation of the c-Met pathway, which can function as a compensatory pathway to enhance HCC cell progression and proliferation.Citation73 Thus, Han et al developed a dual inhibition therapeutic strategy that combined the Akt inhibitor, MK2206, and the c-Met inhibitor, capmatinib, with sorafenib, and this combination showed significant effectiveness in inducing apoptosis and cell cycle arrest. These studies provide new evidence to support further clinical trials.
Natural Compounds and Nano-Particles
Several natural compounds have been found to show synergistic effects with sorafenib. A study compared the sensitization effect of different natural phenolic compounds, including curcumin (Cur), quercetin (Que), and kaempferol (Kmf), with that of sorafenib. Cur and Kmf were found to be effective in sensitizing both Hep3b and HepG2 cells to sorafenib by decreasing cyclins A, B2, and D1 levels, and this led to S-phase and G2/M-phase cell arrest. Furthermore, concomitant treatment with sorafenib and Cur also induced an increase in apoptosis by decreasing Bcl extra‑large protein levels and increasing Bcl-2-associated X protein, caspase-3, and caspase-9 levels.Citation15 In this study, Que was found to be ineffective in sensitizing cells to sorafenib. However, in another study,Citation77 it was found to be effective in sensitizing cells to sorafenib when packaged into dual-targeted lactobionic acid (LA)/ lactoferrin (LF)-NCs. These nanocapsules, which were modified by LF and LA/glycyrrhetinic acid (GA), exhibited increased selectivity and specificity due to the presence of asialoglycoprotein and GA receptors on liver cancer cells and LF receptor overexpression.
Conclusion
HCC has the second lowest cancer survival rate (20%) worldwide.Citation78 Over the last decade, sorafenib has been the only first-line agent available for the treatment of advanced liver cancer. In this study, we review key factors involved in SR, including hypoxia, metabolic factors, and the corresponding signaling pathways. Through the elaboration of the mechanisms involving these resistance factors, we listed their related pharmacological treatments, which have shown potential in alleviating SR. Recently, Lenvatinib was approved as a first-line agent for the treatment of HCC, and showed almost the same efficiency as sorafenib. However, the occurrence of resistance to lenvatinib has also become a major obstacle to its use in improving prognosis in HCC patients.Citation79 For a long time, HCC has been has been known to be insensitive to chemotherapy, including TKIs treatment.Citation80 There is an urgent need to understand the mechanisms underlying this resistance to potentiate the antitumor effects of sorafenib and identify novel chemotherapeutic agents. Therefore, through the description of the mechanisms underlying SR and the summarizing of corresponding SR inhibitors, this review attempts to provide potential solutions to this challenge and provide a significant reference for treating TKI resistance. Given the high degree of heterogeneity and the complexity of SR in HCC, in the future, we will focus on the exploration of the molecular, genetic, and cellular mechanisms underlying drug resistance in HCC cells. Some new tools, including CRISPR/Cas9 technology, 3-dimensional culturing, spheroids, and organoids can be used to identify specific molecular targets and investigate the tumor microenvironment during HCC chemoresistance. In vivo models such as patients, cell-line derived xenografts, and transgenic animals are still relevant for the confirmation of the synergistic effects of different drugs with sorafenib.
Abbreviations
HCC, hepatocellular carcinoma; LF, lactoferrin; RTK, receptor tyrosine kinase; SR, sorafenib resistance; HIF, hypoxia inducible factor; ODDD, oxygen-dependent degradation domain; VHL, von Hippel-Lindau ubiquitination complex; MAPK, mitogen-activated protein kinase; SHP2, SRC homology 2 domain-containing phosphatase 2; GA, glycyrrhetinic acid; Bcl, B‑cell lymphoma; LXR, liver X receptor; IRAK, interleukin-1 receptor-associated kinase; TACE, trans-arterial chemoembolization.
Disclosure
Dr Zhonghao Jiang and Prof. Dr. Chaoliu Dai report grants from Education Department of Liaoning Province, during the conduct of the study. The authors declare that they have no competing interests.
Additional information
Funding
References
- Zhu AX, Kang Y-K, Yen C-J, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, Phase 3 trial. Lancet Oncol. 2019;20(2):282–296. doi:10.1016/S1470-2045(18)30937-9
- Nishida N, Kitano M, Sakurai T, Kudo M. Molecular mechanism and prediction of sorafenib chemoresistance in human hepatocellular carcinoma. Dig Dis. 2015;33(6):771–779. doi:10.1159/000439102
- Sonbol MB, Riaz IB, Naqvi SAA, et al. Systemic therapy and sequencing options in advanced hepatocellular carcinoma. JAMA Oncol. 2020;6(12):e204930. doi:10.1001/jamaoncol.2020.4930
- Wu L-W, Zhou D-M, Zhang Z-Y, et al. Suppression of LSD1 enhances the cytotoxic and apoptotic effects of regorafenib in hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2019;512(4):852–858. doi:10.1016/j.bbrc.2019.03.154
- Li Y-L, Zhang N-Y, Hu X, et al. Evodiamine induces apoptosis and promotes hepatocellular carcinoma cell death induced by vorinostat via downregulating HIF-1α under hypoxia. Biochem Biophys Res Commun. 2018;498(3):481–486. doi:10.1016/j.bbrc.2018.03.004
- Bao MH-R, Wong CC-L. Hypoxia, metabolic reprogramming, and drug resistance in liver cancer. Cells. 2021;10(7):1715. doi:10.3390/cells10071715
- Chowdhury R, Hardy A, Schofield CJ. The human oxygen sensing machinery and its manipulation. Chem Soc Rev. 2008;37(7):1308. doi:10.1039/b701676j
- Maxwell PH, Wiesener MS, Chang G-W, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. doi:10.1038/20459
- Méndez-Blanco C, Fondevila F, García-Palomo A, González-Gallego J, Mauriz JL. Sorafenib resistance in hepatocarcinoma: role of hypoxia-inducible factors. Exp Mol Med. 2018;50(10):1–9. doi:10.1038/s12276-018-0159-1
- Mu H, Yu G, Li H, et al. Mild chronic hypoxia-induced HIF-2α interacts with c-MYC through competition with HIF-1α to induce hepatocellular carcinoma cell proliferation. Cell Oncol. 2021;44(5):1151–1166. doi:10.1007/s13402-021-00625-w
- Chen JC-H, Chuang H-Y, Liao Y-J, et al. Enhanced cytotoxicity of human hepatocellular carcinoma cells following pretreatment with sorafenib combined with trichostatin A. Oncol Lett. 2019;17(1):638–645. doi:10.3892/ol.2018.9582
- Liu L-P, Ho RLK, Chen GG, Lai PBS. Sorafenib inhibits hypoxia-inducible factor-1α synthesis: implications for antiangiogenic activity in hepatocellular carcinoma. Clin Cancer Res. 2012;18(20):5662–5671. doi:10.1158/1078-0432.CCR-12-0552
- Liang Y, Zheng T, Song R, et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1α inhibition in hepatocellular carcinoma. Hepatology. 2013;57(5):1847–1857. doi:10.1002/hep.26224
- Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99(19):1441–1454. doi:10.1093/jnci/djm135
- Bahman AA, Abaza MSI, Khoushiash SI, Al-Attiyah RJ. Sequence‑dependent effect of sorafenib in combination with natural phenolic compounds on hepatic cancer cells and the possible mechanism of action. Int J Mol Med. 2018;42(3):1695–1715. doi:10.3892/ijmm.2018.3725
- Wu F-Q, Fang T, Yu L-X, et al. ADRB2 signaling promotes HCC progression and sorafenib resistance by inhibiting autophagic degradation of HIF1α. J Hepatol. 2016;65(2):314–324. doi:10.1016/j.jhep.2016.04.019
- Xie L, Xiao K, Whalen EJ, et al. Oxygen-regulated β 2 -adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by pVHL. Sci Signal. 2009;2(78):ra33–ra33. doi:10.1126/scisignal.2000444
- Liu X, Zhang X, Peng Z, et al. Deubiquitylase OTUD6B governs pVHL stability in an Enzyme-independent manner and suppresses hepatocellular carcinoma metastasis. Adv Sci. 2020;7(8):1902040. doi:10.1002/advs.201902040
- Feng J, Dai W, Mao Y, et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1α/PPAR-γ/PKM2-mediated glycolysis. J Exp Clin Cancer Res. 2020;39(1):24. doi:10.1186/s13046-020-1528-x
- Albadari N, Deng S, Li W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin Drug Discov. 2019;14(7):667–682. doi:10.1080/17460441.2019.1613370
- Gonzalez FJ, Xie C, Jiang C. The role of hypoxia-inducible factors in metabolic diseases. Nat Rev Endocrinol. 2019;15(1):21–32. doi:10.1038/s41574-018-0096-z
- Wilson GK, Tennant DA, McKeating JA. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: current understanding and future directions. J Hepatol. 2014;61(6):1397–1406. doi:10.1016/j.jhep.2014.08.025
- Ma L, Li G, Zhu H, et al. 2-Methoxyestradiol synergizes with sorafenib to suppress hepatocellular carcinoma by simultaneously dysregulating hypoxia-inducible factor-1 and −2. Cancer Lett. 2014;355(1):96–105. doi:10.1016/j.canlet.2014.09.011
- Dong X-F, Liu T-Q, Zhi X-T, et al. COX-2/PGE2 axis regulates HIF2α activity to promote hepatocellular carcinoma hypoxic response and reduce the sensitivity of sorafenib treatment. Clin Cancer Res. 2018;24(13):3204–3216. doi:10.1158/1078-0432.CCR-17-2725
- Fu Q-H, Zhang Q, Zhang J-Y, et al. LB-100 sensitizes hepatocellular carcinoma cells to the effects of sorafenib during hypoxia by activation of Smad3 phosphorylation. Tumour Biol. 2016;37(6):7277–7286. doi:10.1007/s13277-015-4560-2
- You A, Cao M, Guo Z, et al. Metformin sensitizes sorafenib to inhibit postoperative recurrence and metastasis of hepatocellular carcinoma in orthotopic mouse models. J Hematol Oncol. 2016;9:20. doi:10.1186/s13045-016-0253-6
- Ling S, Song L, Fan N, et al. Combination of metformin and sorafenib suppresses proliferation and induces autophagy of hepatocellular carcinoma via targeting the mTOR pathway. Int J Oncol. 2017;50(1):297–309. doi:10.3892/ijo.2016.3799
- Yang Q, Guo X, Yang L. Metformin enhances the effect of regorafenib and inhibits recurrence and metastasis of hepatic carcinoma after liver resection via regulating expression of hypoxia inducible factors 2α (HIF-2α) and 30 kDa HIV Tat-Interacting Protein (TIP30). Med Sci Monit. 2018;24:2225–2234. doi:10.12659/msm.906687
- Lo J, Lau EY, Ching RH, et al. Nuclear factor kappa B-mediated CD47 up-regulation promotes sorafenib resistance and its blockade synergizes the effect of sorafenib in hepatocellular carcinoma in mice. Hepatology. 2015;62(2):534–545. doi:10.1002/hep.27859
- Cheng BY, Lau EY, Leung H-W, et al. IRAK1 augments cancer stemness and drug resistance via the AP-1/AKR1B10 signaling cascade in hepatocellular carcinoma. Cancer Res. 2018;78(9):2332–2342. doi:10.1158/0008-5472.CAN-17-2445
- Alsaied OA, Sangwan V, Banerjee S, et al. Sorafenib and triptolide as combination therapy for hepatocellular carcinoma. Surgery. 2014;156(2):270–279. doi:10.1016/j.surg.2014.04.055
- Ma L, Liu W, Xu A, et al. Activator of thyroid and retinoid receptor increases sorafenib resistance in hepatocellular carcinoma by facilitating the Warburg effect. Cancer Sci. 2020;111(6):2028–2040. doi:10.1111/cas.14412
- Yuan H, Li A-J, Ma S-L, et al. Inhibition of autophagy significantly enhances combination therapy with sorafenib and HDAC inhibitors for human hepatoma cells. World J Gastroenterol. 2014;20(17):4953–4962. doi:10.3748/wjg.v20.i17.4953
- Hsu F-T, Liu Y-C, Chiang IT, et al. Sorafenib increases efficacy of vorinostat against human hepatocellular carcinoma through transduction inhibition of vorinostat-induced ERK/NF-κB signaling. Int J Oncol. 2014;45(1):177–188. doi:10.3892/ijo.2014.2423
- Hamed HA, Yamaguchi Y, Fisher PB, Grant S, Dent P. Sorafenib and HDAC inhibitors synergize with TRAIL to kill tumor cells. J Cell Physiol. 2013;228(10):1996–2005. doi:10.1002/jcp.24362
- Tak WY, Ryoo B-Y, Lim HY, et al. Phase I/II study of first-line combination therapy with sorafenib plus resminostat, an oral HDAC inhibitor, versus sorafenib monotherapy for advanced hepatocellular carcinoma in east Asian patients. Invest New Drugs. 2018;36(6):1072–1084. doi:10.1007/s10637-018-0658-x
- Raoul J-L, Forner A, Bolondi L, Cheung TT, Kloeckner R, de Baere T. Updated use of TACE for hepatocellular carcinoma treatment: how and when to use it based on clinical evidence. Cancer Treat Rev. 2019;72:28–36. doi:10.1016/j.ctrv.2018.11.002
- Omyła-Staszewska J, Deptała A. Effective therapeutic management of hepatocellular carcinoma – on the basis of a clinical case [Polish version: skuteczne postępowanie terapeutyczne w nieresekcyjnym raku wątrobowokomórkowym – na przykładzie przypadku klinicznego p. 64]. Contemp Oncol. 2012;1:60–67. doi:10.5114/wo.2012.27339
- Kudo M, Ueshima K, Ikeda M, et al. Randomised, multicentre prospective trial of transarterial chemoembolisation (TACE) plus sorafenib as compared with TACE alone in patients with hepatocellular carcinoma: TACTICS trial. Gut. 2020;69(8):1492–1501. doi:10.1136/gutjnl-2019-318934
- Zhang T, Huang W, Dong H, Chen Y. Trans-catheter arterial chemoembolization plus Sorafenib, an unsuccessful therapy in the treatment of hepatocellular carcinoma? Medicine. 2020;99(29):e20962. doi:10.1097/MD.0000000000020962
- Li Z. Cell apoptosis and regeneration of hepatocellular carcinoma after transarterial chemoembolization. WJG. 2004;10(13):1876. doi:10.3748/wjg.v10.i13.1876
- Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368:6487. doi:10.1126/science.aaw5473
- Reinfeld BI, Madden MZ, Wolf MM, et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature. 2021. doi:10.1038/s41586-021-03442-1
- Kim M-J, Choi Y-K, Park SY, et al. PPARδ reprograms glutamine metabolism in sorafenib-resistant HCC. Mol Cancer Res. 2017;15(9):1230–1242. doi:10.1158/1541-7786.MCR-17-0061
- Dai W, Xu L, Yu X, et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J Hepatol. 2020;72(5):909–923. doi:10.1016/j.jhep.2019.12.015
- Lally JSV, Ghoshal S, DePeralta DK, et al. Inhibition of Acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab. 2019;29(1):174–182.e5. doi:10.1016/j.cmet.2018.08.020
- Xu IM-J, Lai RK-H, Lin S-H, et al. Transketolase counteracts oxidative stress to drive cancer development. Proc Natl Acad Sci U S A. 2016;113(6):E725–E734. doi:10.1073/pnas.1508779113
- Lee D, Xu IM-J, Chiu DK-C, et al. Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma. J Clin Invest. 2017;127(5):1856–1872. doi:10.1172/JCI90253
- Zhou K, Liu Y, Luger K. Histone chaperone FACT facilitates chromatin transcription: mechanistic and structural insights. Curr Opin Struct Biol. 2020;65:26–32. doi:10.1016/j.sbi.2020.05.019
- Shen J, Chen M, Lee D, et al. Histone chaperone FACT complex mediates oxidative stress response to promote liver cancer progression. Gut. 2020;69(2):329–342. doi:10.1136/gutjnl-2019-318668
- Tuo L, Xiang J, Pan X, et al. PCK1 downregulation promotes TXNRD1 expression and hepatoma cell growth via the Nrf2/Keap1 pathway. Front Oncol. 2018;8:611. doi:10.3389/fonc.2018.00611
- Lee D, Xu IM-J, Chiu DK-C, et al. Induction of oxidative stress through inhibition of thioredoxin reductase 1 is an effective therapeutic approach for hepatocellular carcinoma. Hepatology. 2019;69(4):1768–1786. doi:10.1002/hep.30467
- Sacco R, Leuci D, Tortorella C, et al. Transforming growth factor beta1 and soluble Fas serum levels in hepatocellular carcinoma. Cytokine. 2000;12(6):811–814.
- Lin T-H, Shao -Y-Y, Chan S-Y, Huang C-Y, Hsu C-H, Cheng A-L. High serum transforming growth factor-β1 levels predict outcome in hepatocellular carcinoma patients treated with sorafenib. Clin Cancer Res. 2015;21(16):3678–3684. doi:10.1158/1078-0432.CCR-14-1954
- Matsuda Y, Wakai T, Kubota M, et al. Valproic acid overcomes transforming growth factor-β-mediated sorafenib resistance in hepatocellular carcinoma. Int J Clin Exp Pathol. 2014;7(4):1299–1313.
- Liu J, Yang X, Liang Q, Yu Y, Shen X, Sun G. Valproic acid overcomes sorafenib resistance by reducing the migration of Jagged2-mediated Notch1 signaling pathway in hepatocellular carcinoma cells. Int J Biochem Cell Biol. 2020;126:105820. doi:10.1016/j.biocel.2020.105820
- Ungerleider N, Han C, Zhang J, Yao L, Wu T. TGFβ signaling confers sorafenib resistance via induction of multiple RTKs in hepatocellular carcinoma cells. Mol Carcinog. 2017;56(4):1302–1311. doi:10.1002/mc.22592
- Xu Y, Huang J, Ma L, et al. MicroRNA-122 confers sorafenib resistance to hepatocellular carcinoma cells by targeting IGF-1R to regulate RAS/RAF/ERK signaling pathways. Cancer Lett. 2016;371(2):171–181. doi:10.1016/j.canlet.2015.11.034
- Ezzoukhry Z, Louandre C, Trécherel E, et al. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int J Cancer. 2012;131(12):2961–2969. doi:10.1002/ijc.27604
- Lin C-Y, Gustafsson J-Å. Targeting liver X receptors in cancer therapeutics. Nat Rev Cancer. 2015;15(4):216–224. doi:10.1038/nrc3912
- Shao W, Zhu W, Lin J, et al. Liver X receptor agonism sensitizes a subset of hepatocellular carcinoma to sorafenib by dual-inhibiting MET and EGFR. Neoplasia. 2020;22(1):1–9. doi:10.1016/j.neo.2019.08.002
- Xia S, Pan Y, Liang Y, Xu J, Cai X. The microenvironmental and metabolic aspects of sorafenib resistance in hepatocellular carcinoma. EBioMedicine. 2020;51. doi:10.1016/j.ebiom.2019.102610
- Tai W-T, Shiau C-W, Chen P-J, et al. Discovery of novel Src homology region 2 domain-containing phosphatase 1 agonists from sorafenib for the treatment of hepatocellular carcinoma. Hepatology. 2014;59(1):190–201. doi:10.1002/hep.26640
- Long J, Jiang C, Liu B, et al. Maintenance of stemness by miR-589-5p in hepatocellular carcinoma cells promotes chemoresistance via STAT3 signaling. Cancer Lett. 2018;423:113–126. doi:10.1016/j.canlet.2017.11.031
- Wang J, Zhou M, Jin X, et al. Glycochenodeoxycholate induces cell survival and chemoresistance via phosphorylation of STAT3 at Ser727 site in HCC. J Cell Physiol. 2020;235(3):2557–2568. doi:10.1002/jcp.29159
- Duan Z, Foster R, Bell DA, et al. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clin Cancer Res. 2006;12(17):5055–5063.
- Ji T, Gong D, Han Z, et al. Abrogation of constitutive Stat3 activity circumvents cisplatin resistant ovarian cancer. Cancer Lett. 2013;341(2):231–239. doi:10.1016/j.canlet.2013.08.022
- Hung M-H, Tai W-T, Shiau C-W, Chen K-F. Downregulation of signal transducer and activator of transcription 3 by sorafenib: a novel mechanism for hepatocellular carcinoma therapy. World J Gastroenterol. 2014;20(41):15269–15274. doi:10.3748/wjg.v20.i41.15269
- Blechacz BRA, Smoot RL, Bronk SF, Werneburg NW, Sirica AE, Gores GJ. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology. 2009;50(6):1861–1870. doi:10.1002/hep.23214
- Tai W-T, Cheng A-L, Shiau C-W, et al. Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP-1-mediated inhibition of STAT3. Mol Cancer Ther. 2012;11(2):452–463. doi:10.1158/1535-7163.MCT-11-0412
- Su J-C, Tseng P-H, Wu S-H, et al. SC-2001 overcomes STAT3-mediated sorafenib resistance through RFX-1/SHP-1 activation in hepatocellular carcinoma. Neoplasia. 2014;16(7):595–605. doi:10.1016/j.neo.2014.06.005
- Leung CON, Tong M, Chung KPS, et al. Overriding adaptive resistance to sorafenib through combination therapy with src homology 2 domain-containing phosphatase 2 blockade in hepatocellular carcinoma. Hepatology. 2020;72(1):155–168. doi:10.1002/hep.30989
- Han P, Li H, Jiang X, et al. Dual inhibition of Akt and c-Met as a second-line therapy following acquired resistance to sorafenib in hepatocellular carcinoma cells. Mol Oncol. 2017;11(3):320–334. doi:10.1002/1878-0261.12039
- Tovar V, Cornella H, Moeini A, et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut. 2017;66(3):530–540. doi:10.1136/gutjnl-2015-309501
- Wang F, Bank T, Malnassy G, et al. Inhibition of insulin-like growth factor 1 receptor enhances the efficacy of sorafenib in inhibiting hepatocellular carcinoma cell growth and survival. Hepatol Commun. 2018;2(6):732–746. doi:10.1002/hep4.1181
- Singh AR, Joshi S, Burgoyne AM, et al. Single agent and synergistic activity of the “First-in-Class” dual PI3K/BRD4 Inhibitor SF1126 with sorafenib in hepatocellular carcinoma. Mol Cancer Ther. 2016;15(11):2553–2562. doi:10.1158/1535-7163.MCT-15-0976
- Abdelmoneem MA, Elnaggar MA, Hammady RS, et al. Dual-targeted lactoferrin shell-oily core nanocapsules for synergistic targeted/herbal therapy of hepatocellular carcinoma. ACS Appl Mater Interfaces. 2019;11(30):26731–26744. doi:10.1021/acsami.9b10164
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA a Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
- Fu R, Jiang S, Li J, Chen H, Zhang X. Activation of the HGF/c-MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-MET expression. Med Oncol. 2020;37(4). doi:10.1007/s12032-020-01350-4
- Wei L, Lee D, Law C-T, et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat Commun. 2019;10(1). doi:10.1038/s41467-019-12606-7