
Abstract
Alpha-1 antitrypsin (AAT) is the most abundant serine protease inhibitor circulating in the blood. AAT deficiency (AATD) is an autosomal codominant condition affecting an estimated 3.4 million individuals worldwide. The clinical disease associated with AATD can present in a number of ways including COPD, liver disease, panniculitis and antineutrophil cytoplasmic antibody vasculitis. AATD is the only proven genetic risk factor for the development of COPD, and deficient individuals who smoke are disposed to more aggressive disease. Principally, AAT is a serine protease inhibitor; however, over the past number of years, the assessment of AAT as simply an antiprotease has evolved, and it is now recognized that AAT has significant anti-inflammatory properties affecting a wide range of cells, including the circulating neutrophil.
Introduction
Alpha-1 antitrypsin (AAT) is a member of the serpin family which also includes plasminogen activator inhibitor-1, alpha-1 antichymotrypsin, antithrombin and C1-inhibitor. These serpins play vital roles in the regulation of proteases involved in fibrinolytic, complement and coagulation pathways.Citation1 AAT is a 394-amino acid polypeptide chain encoded by the SERPINA1 gene located at the chromosomal region 14q32.1.Citation2 Aside from hepatocytes where it is mostly synthesized, AAT is also produced to a lesser degree by other cell types such as neutrophils,Citation4 macrophages,Citation3 monocytes,Citation5 intestinal epithelial cells,Citation6 pancreatic isletsCitation7 and cancer cells.Citation8 However, from these cellular sources, the AAT protein is unlikely to contribute to circulating plasma levels but rather to local AAT concentrations.Citation9 Within the circulation, the concentration of AAT is 1.21–2.17 g/L, making it one of the most abundant plasma proteins with a half-life of 4.6 days.Citation10 AAT is part of the acute-phase response, which means that a rapid rise in plasma levels of AAT is observed during acute inflammation,Citation11 with plasma levels increasing three- to four fold.Citation12 The aim of this review is to first introduce AAT deficiency (AATD) and then to consider the described anti-inflammatory activities of AAT in controlling key neutrophil functions, outline recognized signaling pathways and specifically recognize the features of neutrophil-driven airways disease in which AAT augmentation therapy has been demonstrated to be effective. Review of the literature was carried out using the MEDLINE (from 1986 to 2017), Google Scholar and The Cochrane Library databases.
The antiprotease AAT
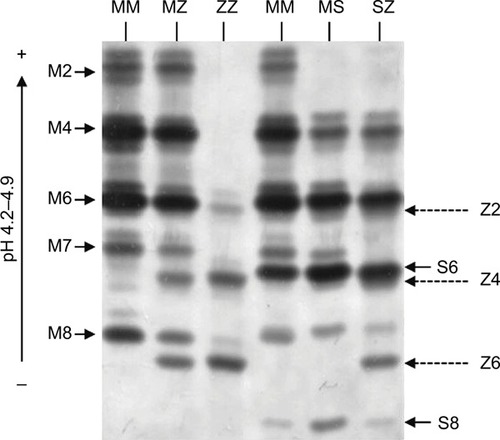
The predominate role of AAT is as a serine protease inhibitor, chiefly inhibiting neutrophil elastase (NE),Citation13 but also other proteases including chymotrypsin, cathepsin G (CathG), proteinase 3 (PR3) and thrombin. The structure of the AAT is critical for its antiprotease activity and comprises 3 beta sheets (A, B and C), 9 alpha helices and a reactive center loop (RCL) at the C-terminal end.Citation14 Furthermore, during AAT production, posttranslational modifications occur, and the protein undergoes addition of N-linked oligosaccharides at asparagines 70, 107 and 271. The three N-glycosylation sites on the AAT molecule contain mostly biantennary structures but also triantennery and traces of tetraantennary N-glycans.Citation15 Multiple glycoforms of AAT have been identified (M0–M8), and these can be visualized by isoelectric focusing (IEF) and separated by the charge of the N-glycans (). Adding to this field, we have recently published that during the acute inflammatory process of community-acquired pneumonia (CAP), the circulating AAT molecule differs due to variations in its glycosylation pattern and that AAT glycans containing 4 sialic acids appeared during the resolution phase of CAP.Citation16 Moreover, data highlight the role of sialylation in the anti-inflammatory activity of AAT, as during the resolving phase of infection there was a significant increase in circulating levels of interleukin (IL)-8 complexed to sialylated negative glycoforms of AAT. This binding event led to enhanced inhibition of C-X-C motif chemokine receptor (CXCR) 1 engagement on neutrophil plasma membranes,Citation16 which may serve to prevent further migration of cells to epithelial surfaces and decrease the potential for neutrophil-mediated damage.
Figure 1 Isoelectric focusing gel illustrating AAT phenotype mutations. The glycan numbers for the phenotypes are labeled.
Abbreviation: AAT, alpha-1 antitrypsin.
The antiprotease inhibitor activity of the molecule lies within the 9-amino acid RCL. AAT, unlike most proteins, folds into a metastable state which has a considerably lower conformational stability.Citation17 Fundamentally, the AAT molecule acts as a trap with the RCL as its bait. NE cleavage between amino acids 358 and 359 of the RCL results in the creation of an AAT:NE complex between the cleaved AAT molecule and NE. The process results in irreversible inactivation of both molecules, and thus, in the ideal scenario, AAT exists in the lungs surplus to the amount of protease in order to protect the lung parenchyma from degradation. Moreover, the structural rearrangement that enables the AAT:NE complex to form exposes a binding site that can engage with a receptor known as SERPIN:enzyme complex receptor. The interaction of this AAT:NE complex with the SERPIN:enzyme complex receptor on cell surfaces such as hepatocytes causes a positive feedback loop leading to increased expression of the SERPINA1 gene.Citation18
AAT deficiency
AATD is an autosomal, codominant, genetic disorder that is characterized by low circulating levels of AAT as a result of a mutation of the SERPINA1 gene. The worldwide frequency of AATD varies according to population and is particularly prevalent in Europe with multiple studies reporting a high prevalence of the deficiency alleles in Poland,Citation19,Citation20 France,Citation21 ItalyCitation22 and Ireland.Citation23 AATD is characterized by circulating levels <11 µmol/L, which is the putative protective threshold level.Citation24 The AAT phenotype is determined by codominant expression of parental alleles with the majority of individuals carrying 2 copies of non-mutated M allele. The M allele in homozygous individuals leads to AAT plasma levels >1.04 g/L or 20 µM. However, the SERPINA1 gene is highly pleomorphic, and at least 120 genetic variants have been reported to date,Citation25 with the Z (Glu342Lys) and S (Glu264Val) mutations () being the most common. The substitution of glutamic acid for lysine at position 342 leads to the Z mutation, and that of glutamic acid for valine at position 264 gives rise to the S mutation.Citation26,Citation27
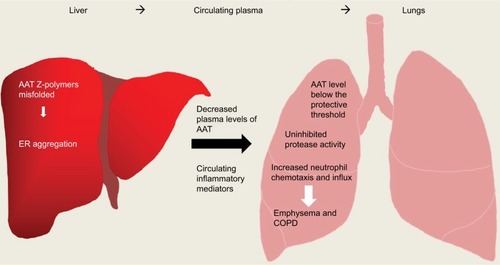
In individuals heterozygous for the S mutation, AAT levels typically remain above the protective threshold of 11 µM. This mutation is thus considered to carry a negligible risk of AATD-associated disease unless co-inherited with another deficiency allele such as the Z allele. The Z mutation gives rise to the most severe plasma deficiency and occurs in more than 95% of individuals with AATD.Citation28 Z-AAT protein polymerizes and becomes trapped within the endoplasmic reticulum (ER), thus accumulating in hepatocytes. This results in impaired secretion of the protein,Citation29 leading to plasma deficient in AAT, with individuals homozygous for the Z mutation having 10%–15% of normal circulating levels. It is because of these low circulating levels of AAT that patients with this condition are at a high risk of developing emphysema.Citation30 The low level of AAT in ZZ individuals results in an imbalance of proteases and antiproteases in the AATD lung, resulting in unchecked levels of active serine proteases that damage alveolar tissue leading to lung disease.Citation28,Citation31
Another class of mutations in the SERPINA1 gene are termed silent or “null” mutations. The plasma levels of this class of variants are undetectable by conventional techniques such as nephelometry and IEF, and as such, these mutations are classically thought to result in a complete absence of AAT production and therefore a high risk of developing emphysema.Citation32 While the consequence of the null alleles is unified, that is, undetectable plasma levels of AAT, the mutational events from which they arise vary from gene deletions, premature stop codon insertions, to mRNA degradation. For example, the Q0granite falls genotype arises from a single base pair deletion, resulting in a premature stop codon and a lack of mRNA production.Citation33
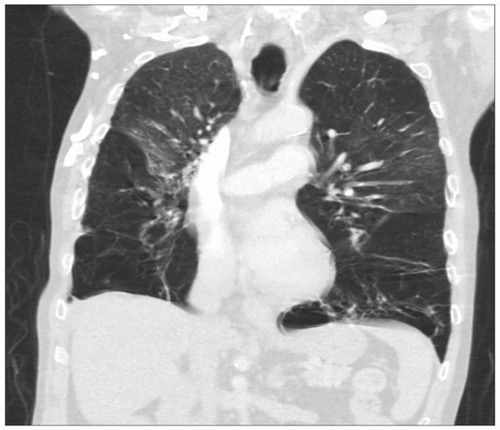
The classical manifestation of AATD in the lungs is panacinarCitation34 and lower lobe-predominant emphysema, involving the distal airway structures and resulting in uniform enlargement of the bronchioles and alveoli (). The clinical presentation of COPD in AATD is similar regardless of AAT phenotype, with patients commonly complaining of dyspnea and cough with frequent respiratory infections. Onset occurs at a significantly younger age in AATD patients however, often in the 3rd to the 4th decade of life. This is in contrast to typical COPD, which usually involves upper lobe-predominant centrilobular emphysemaCitation35 that commonly manifests in the late 6th or 7th decade.Citation30
Figure 2 A computed tomography scan showing severe bilateral, lower lobe-predominant panacinar emphysema in a patient with AATD homozygous for the Z mutation.
Abbreviation: AATD, alpha-1 antitrypsin deficiency.
The risk for emphysema is increased in individuals heterozygous for the Z mutation who are smokers.Citation29 Smoking has been demonstrated to further exacerbate the imbalance between proteases and antiproteases by rendering AAT inactive.Citation36,Citation37 Increased release of NE in bronchoalveolar lavage fluid (BALF)Citation38 in patients who smoke has been reported, and a direct correlation has been made between the NE burden in the BALF and the degree of emphysema seen on computed tomography scans.Citation40 Moreover, the inverse relationship between the degree of emphysema and the antielastase activity in BALF of COPD patients further supports the protease/antiprotease theory of emphysema.Citation41 In addition to the increased release of proteases, another process that can occur which further contributes to airway inflammation is oxidative stress. The conversion of hydrogen peroxidase to its product hypochlorous acid by myeloperoxidase, along with other reactive oxygen species, can render AAT inactive through oxidation and chlorination.Citation42 Hydrogen peroxidase, a component of cigarette smoke, oxidizes 2 methionine residues, 351 and 358, located on the RCL of AAT. The outcome of AAT oxidation is loss of anti-NE capacity.
The traditional view behind the cause of AATD-related emphysema is that it is a result of the imbalance of proteases and antiproteases in the lung. However, it has recently come to light that AAT is more than just an antiprotease. AAT has been shown to have anti-inflammatory capacities outside of its antiprotease activity. The loss of this AAT function is apparent as the manifestations of AATD are not confined to lung disease but extend to systemic inflammatory conditions such as vasculitisCitation43 and panniculitis,Citation44 which will be discussed below. Therefore, disease manifestations seen in AATD are due to loss of AAT function as both a protease inhibitor and an anti-inflammatory molecule.
The neutrophils in the pathogenesis of AATD disease
Neutrophils are the source of NE, CathG and PR3, the key drivers of inflammation that destroy alveolar tissue in AATD. Published data have demonstrated an increased number of these polymorphonuclear leukocytes in the BALF of AATD patients.Citation45 These cells are produced from myeloid precursors in the bone marrow and have a short circulating half-life of ~8 hours.Citation46 Neutrophils are typically the first leukocytes that migrate to the site of inflammation, exiting the bloodstream where they transmigrate through the endothelium and travel to the site of infection. Neutrophil localization to the infected site is crucial for the clearance of infection, as a defective neutrophil response has been shown to lead to bacterial colonization.Citation47
Upon arrival at the site, the activated neutrophil has an increased life span to ensure microbial clearance.Citation48 The neutrophil’s arsenal of antimicrobial serine proteases is stored within primary granules. Upon neutrophil activation, there is a rapid translocation of primary granules to the plasma or phagocytic membrane, thereby releasing serine proteases including NE, CathG and PR3 into the extracellular or phagocytic space where they play an important role in host defence.Citation49,Citation50 For example, NE is essential in the killing of Gram-negative bacteria, as evidenced by the finding that NE-knockout mice are more susceptible to these microbes.Citation51 Conversely, besides playing a protective role against invading microorganisms, these proteases have been associated in the pathogenesis of emphysema and COPD.Citation52,Citation53 Indeed, NE is referred to as a double-edged sword as unchecked levels can degrade a wide variety of host substrates. In AATD, NE is considered to be the major protease involved in the destruction of lung tissue as it possesses the ability to damage every component of the extracellular matrix including cross-linked fibrin, collagen and proteoglycans.Citation54
Additionally, NE amplifies the inflammatory burden by stimulating mucin secretion yet decreasing the beat frequency of cilia of bronchial epithelial cells, thereby interrupting mucociliary clearance.Citation55,Citation56 Studies in mice have shown that NE augments the activity of further destructive proteases, including matrix metalloprotease (MMP) 9.Citation52 Furthermore, NE also retains the capability to inactivate the innate inhibitors of these proteases including tissue inhibitors of metalloproteinases 1 and 2, inhibitors of MMP-9Citation57 and MMP-2Citation58 and other relevant protease inhibitors such as elafinCitation59 and secretory leukocyte protease inhibitor.Citation60
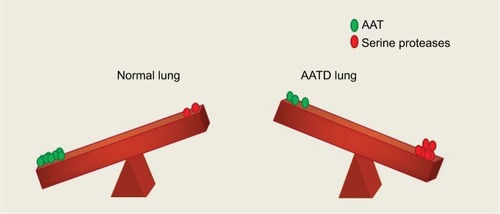
By proteolysis of complement receptorsCitation61,Citation62 and CXCR1Citation63 on neutrophils, NE quickly impairs the capacity of neutrophils to kill invading microbes. NE also weakens innate immunity by cleaving TIM-3 (T-cell Ig and mucin domain-containing molecule-3) from epithelial and neutrophil cell surfaces,Citation64,Citation65 and humoral immunity by cleaving immunoglobulins.Citation66 NE destroys signaling cytokines including the interferon-gamma (IFN-γ)-inducing factor IL-18,Citation67 thus potentiating the airway inflammatory environment. Adding to the proteolyic burden, CathG digests host substrates and is seen at higher levels in patients with emphysema.Citation52 There is thus a tremendous need to curtail the excessive activity of both NE and CathG, and by binding to them, AAT inhibits the destructive nature of these proteases. Indeed, AAT is a regulator of NE,Citation68 CathGCitation69 and PR3,Citation70 and it is for this reason that the balance created by AAT is essential for protection of the lung matrix (). During pulmonary exacerbations, the sputum of AATD patients has been found to have increased levels of PR3. As the elastolytic ability of PR3 is less than NE, PR3 likely plays a lesser role in the manifestation of emphysema.Citation71 In contrast, however, anti-PR3 autoantibodies have been proposed to exacerbate the degranulation process by binding PR3 expressed on surface membranes of monocytes and neutrophils, resulting in excess protease release and contributing to both vascular and endothelial injuries.Citation71,Citation72
Figure 3 Protease/antiprotease balance shown in the lung of a healthy individual and a person with AATD. In a normal individual, the lung parenchyma is protected from serine protease activity of NE, CathG and PR3 by AAT. In an AATD individual, unchecked levels of proteases damage lung tissue due to low levels of AAT.
Abbreviations: AAT, alpha-1 antitrypsin; AATD, AAT deficiency; CathG, cathepsin G; NE, neutrophil elastase; PR3, proteinase 3.
Studies aimed at understanding the increased neutrophil burden seen in AATD patients have demonstrated that individuals either homozygous or heterozygous for the Z allele have an increased influx of neutrophils into their lungs.Citation73 A number of models have been proposed to help explain this increase, and one suggestion is the deposition of polymers of mutated Z-AAT protein. Polymerization of the Z-AAT protein occurs mainly within hepatocytes but is also thought to occur spontaneously in the lungs of AATD individuals. In ZZ-AATD individuals, these polymers have been detected in BALFCitation74 and additionally in the alveolar walls of individuals with emphysema.Citation75 The presence of polymerized Z-AAT induces a pro-inflammatory response as these polymers can act as a chemoattractant, inducing chemotaxis levels comparable to IL-8Citation76 and complement component C5a ().Citation77 Z-AAT polymers have also been reported to stimulate neutrophil adhesion and induce neutrophil degranulation.Citation77 Extending on this concept, while many investigators agree that mutations of the AAT protein can lead to the formation of Z-AAT polymers that are retained within the ER of hepatocytes in ZZ-AATD,Citation78,Citation79 the characteristics of Z-AAT polymers and ER stress in immune cells from ZZ-AATD patients are less well studied. In neutrophils, an intrinsic defect due to misfolded AAT protein within the ER of circulating ZZ-AATD cells results in increased expression of the proapoptotic transcription factor CHOP, with accelerated apoptosis of ZZ-AATD neutrophils associated with decreased bacterial killing.Citation80
Figure 4 Overview of inflammation caused in AATD liver and lung disease. AAT Z-polymers misfold in the liver leading to retention/aggregation in the ER, resulting in low levels of plasma AAT. Low levels of AAT and active uninhibited serine proteases can cause damage to lung parenchyma ultimately leading to emphysema and COPD.
Abbreviations: AAT, alpha-1 antitrypsin; AATD, AAT deficiency; ER, endoplasmic reticulum.
When describing neutrophil-dominated inflammation with regard to AATD, it is important to discuss panniculitis. This rare condition (occurring in 0.1% of Z homozygotes)Citation81 is characterized by intense neutrophil infiltrates in the skin, presenting as a painful skin rash.Citation81 It has been described most commonly in ZZ patients but has also been found in MZ and SZ individuals.Citation81 The pathophysiology of AATD-related panniculitis has not been definitively described, but skin biopsies demonstrate neutrophil infiltration into the subcutaneous tissues and resultant tissue destruction due to the low levels of antiprotease and high levels of protease. The presence of polymers of Z-AAT in the skin is significant,Citation82 as these have been shown to be a powerful neutrophil attractant, as mentioned earlier.Citation75
The effect of AAT on neutrophil function
IL-8 is a commanding neutrophil chemoattractant produced by airway epithelial cells and alveolar macrophages in response to inflammationCitation83 and has been shown to be increased in the sputum of MZ heterozygous individuals.Citation84 IL-8 engages with CXCR1 on the neutrophil membrane, resulting in amplified neutrophil adhesion due to increased membrane expression of CD11b and CD18.Citation85 The engagement of IL-8:CXCR1 also causes a rise in intracellular calcium levels, which facilitates neutrophil cytoskeletal rearrangements ultimately enabling chemotaxis.Citation86 It has been shown that NE can induce the expression of this chemokine in bronchial epithelial cells via Toll-like receptor 4, thus adding to the inflammatory burden in the lung.Citation87 In a similar fashion, unopposed protease activity results in the activation of protease-activated receptors (PARs). PARs 1, 2, 3 and 4 have all been found in the lung,Citation88,Citation89 and in the absence of AAT, these PARs are overactivated by neutrophil serine proteases, thus amplifying inflammation.
In recent years, it has emerged that AAT possesses a variety of anti-inflammatory properties. In this regard, it has been found that AAT can modulate IL-8-induced chemotaxis by binding this chemokine. At physiological pH, AAT protein has an overall negative charge that enables electrostatic interaction between AAT and positively charged IL-8. The oligosaccharides on AAT are vital for this anti-inflammatory function as it has been shown that non-glycosylated AAT fails to bind IL-8.Citation90 This binding event between AAT and IL-8 impedes docking of IL-8 with CXCR1,Citation90 impacting negatively upon the downstream signaling events involved in cytoskeleton rearrangement, F-actin formation and calcium flux, ultimately resulting in decreased neutrophil migration.
AAT has also been shown to influence neutrophil chemotaxis in response to soluble immune complexes (sICs). Neutrophil engagement of sICs results in increased tumor necrosis factor-alpha (TNF-α)-converting enzyme (TACE) activity, causing release of the glycosylphosphatidylinositol-anchored Fc receptor (FcγRIIIB), which is a prerequisite for chemotaxis. AAT was shown to modulate TACE activity, thereby preventing the release of membrane FcγRIIIB.Citation90 Moreover, the ability of AAT to reduce neutrophil chemotaxis in response to a third stimuli, namely, formyl-methionyl-leucyl-phenylalanine, with subsequent reduced adherence to lung-derived endothelial cells has been demonstrated.Citation91 Furthermore, leukotriene B4 (LTB4) is another stimulant that can affect neutrophil function, increasing cell adhesion, degranulation and chemotaxis.Citation92–Citation94 In a vicious circle of inflammation, released NE can signal back to the neutrophil causing increased production of LTB4 and upregulation of its receptor BLT1 on the neutrophil membrane. In turn, in vitro AAT has been reported to exercise strong anti-inflammatory effects against LTB4, binding this lipid mediator via a central hydrophobic pocket on the protein surface. This binding event inhibits the engagement of LTB4 with BLT1 on the neutrophil plasma membrane, thereby preventing neutrophil activation ().Citation94
With regard to degranulation, in the neutrophil-burdened airways, there is an exuberant release of cytotoxic granule proteins to the outside of the cell. In landmark studies, degranulation of all granule subtypes in response to either TNF-α or LTB4 was significantly reduced by AAT. In this regard, Bergin et al reported a substantial rise in TNF-α membrane expression in circulating neutrophils isolated from AATD individuals compared to healthy controls, on par with levels in patients with rheumatoid arthritis.Citation95 The authors suggested that AAT may in turn control TNF-α bioactivity and therefore could reduce neutrophil degranulation in response to TNF-α.Citation96 Ensuing in vitro results demonstrated that AAT modulates degranulation of neutrophil secondary and tertiary granules and that the inhibitory mechanism involves the ability of AAT to bind TNF receptors (TNFRs), thereby blocking TNF-α engagement with TNFR1 and TNFR2.Citation95 This blockade of receptor engagement led to the prevention of downstream signaling events including MAPK p38 phosphorylation, a key step in the neutrophil degranulation process.Citation97 This is further supported by in vivo and in vitro data on the ability of AAT to modulate TNF-α-induced apoptosis.Citation98,Citation99 AAT’s capacity to inhibit ligand–receptor binding with resultant reduced downstream signaling events has also been described for transferrin.Citation100 Collectively, these studies strongly support the vital role that AAT plays in reducing the neutrophil burden in the airways and protecting lung tissue architecture from destruction, as seen in AATD.
The implications of augmentation therapy for neutrophil function in AATD patients
It is evident that the protease/antiprotease balance in the lung is of great importance in maintaining healthy tissue. Therefore, a logical treatment of AATD-related lung disease is to reestablish physiological concentrations of AAT. The current gold standard treatment for AATD is augmentation therapy utilizing AAT sourced from pooled human plasma at a dose of 60 mg/kg, administered by slow intravenous infusion once weekly. In the early 1980s, it was shown that intravenous infusion of purified human AAT corrected the concentration of AAT in plasma and on the lung epithelial surface which was used as a surrogate for lung parenchyma.Citation101,Citation102 It has been subsequently proven that AAT augmentation therapy ameliorates AATD-related lung disease, with the RAPID (Randomized, Placebo-controlled Trial of Augmentation Therapy in Alpha-1 Proteinase Inhibitor Deficiency) trial being the first to conclusively demonstrate the benefit of AAT augmentation therapy as compared to placebo.Citation103 The extension to this trial, RAPID-OLE (RAPID open-label extension), further supported the continued efficacy of AAT in decelerating the progression of AATD lung disease over 4 years.Citation104
Perhaps surprisingly, the RAPID trial did not demonstrate a significant reduction in frequency of exacerbations with AAT augmentation therapy.Citation103 Exacerbations are commonly neutrophil-driven events characterized by worsening of dyspnea and cough with increased sputum production. There is some evidence for a reduction in exacerbation frequency with augmentation therapy,Citation105 but this is a notoriously difficult outcome to capture, and thus, inadequate power could be an explanation for the lack of benefit observed. Further anecdotal evidence for a benefit comes from surveys amongst AATD patients, reporting a decrease in both chest infections and hospitalizations in association with augmentation therapy.Citation106
These results lend weight to the clinical use of AAT augmentation therapy for airways disease, which is currently available in the US, Canada and several European countries including Spain, Italy and Germany. Intravenous augmentation with AAT is also used as a treatment for panniculitis since the first report in 1987 in 2 ZZ patients whose skin condition had proven refractory to conventional therapy.Citation107 Both demonstrated remarkable improvement with augmentation therapy. Subsequently, numerous case reports support the use of this therapy, commonly at a higher dose of 120 mg/kg weekly.Citation81 Occasionally, panniculitis may coincide with a marked neutrophilic serositis manifesting as pleural effusions or arthritis and also characterized by dramatic improvement following AAT augmentation therapy.Citation108 Successful treatment of nonspecific vasculitis in an AATD patient with intravenous augmentation therapy has also been described.109
A number of studies have specifically investigated the effect of AAT augmentation therapy on neutrophil dysfunction seen in AATD. Bergin et al found that in clinically stable ZZ patients, the neutrophils have increased levels of TACE activity on their membranes, leading to a higher chemotactic index.Citation90 Post AAT augmentation therapy, the increase in plasma concentration of AAT resulted in normalized ZZ-AATD neutrophil chemotactic responses by inhibiting TACE activity and thereby preventing FcγRIIIB from being shed from the cell membrane, reducing it to that of healthy control levels. Further work demonstrated that augmentation therapy restored AAT plasma levels and normalized TNF-α signaling, thereby preventing TNF-α-induced neutrophil release of secondary and tertiary granules and resultant production of autoantibodies.Citation80 Additionally, it was recently reported that AATD is associated with increased neutrophil membrane-bound NE, which can trigger an inflammatory cycle inducing secretion of LTB4 that further stimulates primary granule release. Overall, these findings highlight a novel interplay between LTB4 and NE released from neutrophils. In vivo plasma levels of both LTB4 and neutrophil membrane-bound NE were reduced in AATD patients receiving AAT augmentation therapy, compared with untreated patients matched by forced expiratory volume in 1 second and in fact normalized to healthy control MM levels. Relevant to the latter findings, a reduction in airway levels of IL-8 and LTB4 in individuals with cystic fibrosis (CF) treated with aerosolized AAT has been reported,Citation93 suggesting the use of AAT outside of the context of AATD.
Moving to the end of the neutrophil’s life cycle, AAT augmentation therapy has been reported to impact upon neutrophil apoptosis. In AATD patients receiving AAT augmentation therapy, reduced TACE activity and TNF-α signaling and normalized neutrophil apoptosis were reported.Citation80 AAT also binds to and inhibits caspase-3, thereby preventing lung endothelial cell apoptosis.Citation111 Additionally, although not specific to neutrophils, AAT prolonged allograft survival and modulated cellular immunity in treatment of mice that had undergone pancreatic islet allograft.Citation99,Citation112–Citation114 An investigation into how AAT protects islets cells revealed that AAT potentiated insulin secretion and the effects of glucagon-like peptide-1 and forskolin.Citation115 Furthermore, AAT was shown to protect a diabetic cell line from TNF-α-mediated apoptosis and to significantly reduce apoptosis caused by a combination of TNF-α, IL-1β and IFN-γ.Citation115
Positive effects of AAT as treatment in other lung diseases
The increasing recognition of the immune-modulatory effects of AAT, alongside its broad antiprotease activity, has led to its consideration as a therapeutic agent in a wide range of conditions characterized by injurious neutrophilic inflammation,Citation116 most prominently in CF. CF is the most commonly inherited fatal disease in Caucasians. It results from mutations in the CFTR gene that codes for an ion channel present on the lung epithelial membrane (amongst other tissues). Mutation results in viscous respiratory mucus that cannot be cleared from the lung. Chronic respiratory infection ensues, ultimately resulting in bronchiectasis or permanent enlargement of the proximal airways with destruction of their walls. Neutrophils are centrally implicated in this condition, with increased levels of NE suggesting a relative deficiency of AAT in the lung,Citation117 leading to the evaluation of AAT as a potential therapy in CF. A clinical trial of aerosolized AAT at a dose of 1.5–3 mg/kg twice daily for 1 week demonstrated safety and tolerability as well as inhibition of airway NE and improved clearance of bacteria.Citation118 A subsequent study extended therapy to 4 weeks, demonstrating a reduction in airway neutrophils.Citation110 In contrast, however, a Phase II trial examining the effect of non-glycosylated recombinant AAT demonstrated safety and tolerability but showed a limited effect on NE activity and other markers of inflammation.Citation119 A similar result showed decreased taurine, a surrogate for neutrophils, with no change in NE activity.Citation120 These discrepancies may be caused by a number of reasons including but not limited to the glycosylation state of the AAT protein, different aerosol devices and methods of sampling CF airways. This remains an active area of research with randomized placebo-controlled trials in progress.
Another condition in which AAT is being investigated as a therapy in humans is bronchiolitis obliterans syndrome (BOS), a subtype of chronic rejection following lung transplant that is characterized by neutrophilic and lymphocytic inflammation and fibrosis of small airways.Citation121 There is a theoretical basis and some supporting data for the potential benefit of AAT in other conditions characterized by excessive neutrophilic inflammation such as inflammatory bowel disease, rheumatoid arthritis and postoperative systemic inflammatory response syndrome (SIRS).Citation122 For example, Daemen et al have demonstrated that AAT mitigates renal reperfusion injury in mice via reduced TNF-α and neutrophil influx.Citation123 Kaner et al have shown that transgenic mice expressing the human AAT gene have reduced levels of liver and pancreatic dysfunction compared to wild-type mice in an SIRS model, as well as improved survival at 24 hours.Citation124 Studies on human pancreatic beta cells in vitro and in mice have suggested a protective and regenerative effect of AAT on these insulin-producing cells, with potential implications in the treatment of autoimmune diabetes mellitus.Citation99,Citation125 Likewise, AAT has been shown to prolong survival of transplanted beta cells in miceCitation112 and monkeysCitation126 and ameliorate graft-vs-host disease in a mouse model.Citation127 These results are the basis for several ongoing clinical trials in humans. In summary, AAT also has numerous effects on a range of cell types including monocytes, B cells, T cells, dendritic cells and macrophages, both directly and indirectly. These effects have been reviewed in detail elsewhereCitation116,Citation128–Citation129 and further strengthen the concept of AAT as an important modifier of immune and inflammatory responses. Further understanding of how AAT interacts with the neutrophil, as well as other immune cells, may expand its use as a therapeutic agent outside of the setting of genetic AATD.
Conclusion
The lungs of an AATD individual are burdened with high levels of proteolytic agents including NE, CathG and PR3 as a result of neutrophilic inflammation (). The primary function of AAT as a protease inhibitor protecting the lung parenchyma from these destructive proteases is apparent in AATD as the diminished AAT levels in these individuals enable these proteases to go unchecked. However, recent work has shown that this multifaceted protein exerts more than just an antiprotease function in the circulation and lung. It has been found that AAT possesses anti-inflammatory properties independent of its antiprotease activity, bringing us away from the classical view of AAT. These anti-inflammatory properties are fundamental when trying to understand the manifestation of both the lung and systemic inflammation that is seen in AATD. The AATD neutrophils have dysregulated neutrophil adhesion, chemotaxis, degranulation and apoptosis. The ability of AAT to correct this dysregulation is seen following augmentation therapy, as AAT binds a number of pro-inflammatory mediators via hydrophobic and electrostatic interactions resulting in normalized neutrophil responses. This demonstrates that AAT exerts a wide array of immune-modulating effects, illustrating the wider role it plays, rather than just the protease inhibitor it was once thought. Nevertheless, a number of challenges remain in the development of AAT as an anti-inflammatory therapy such as expanding our knowledge of its mode of action and the development of new sources of glycosylated AAT with equivalent anti-inflammatory capacity to that of plasma purified protein. Despite these challenges, AAT holds incredible potential as a novel anti-inflammatory molecule, which has already been established as a safe and well-tolerated therapeutic agent.
Acknowledgments
The authors would like to thank the Medical Research Charities Group and the Health Research Board (MRCG-2014-1) and the US Alpha-1 Foundation for support.
Disclosure
The authors report no conflicts of interest in this work.
References
- KhanMSSinghPAzharASerpin inhibition mechanism: a delicate balance between native metastable state and polymerizationJ Amino Acids2011201160679722312466
- CoxDWMarkovicVDTeshimaIEGenes for immunoglobulin heavy chains and for alpha 1-antitrypsin are localized to specific regions of chromosome 14qNature198229758654284306804874
- PerlmutterDHColeFSKilbridgePRossingTHColtenHRExpression of the alpha 1-proteinase inhibitor gene in human monocytes and macrophagesProceedings of the National Academy of Sciences1985823795799
- du BoisRMBernaudinJFPaakkoPHuman neutrophils express the alpha 1-antitrypsin gene and produce alpha 1-antitrypsinBlood19917712272427302043769
- PerlmutterDHKayRMColeFSRossingTHVan ThielDColtenHRThe cellular defect in alpha 1-proteinase inhibitor (alpha 1-PI) deficiency is expressed in human monocytes and in Xenopus oocytes injected with human liver mRNAProc Natl Acad Sci U S A19858220691869213876562
- MolmentiEPPerlmutterDHRubinDCCell-specific expression of alpha 1-antitrypsin in human intestinal epitheliumJ Clin Invest1993924202220348408656
- BoscoDMedaPMorelPExpression and secretion of alpha1-proteinase inhibitor are regulated by proinflammatory cytokines in human pancreatic islet cellsDiabetologia20054881523153316001235
- ChenXLZhouLYangJShenFKZhaoSPWangYLHepatocellular carcinoma-associated protein markers investigated by MALDI-TOF MSMol Med Rep20103458959621472284
- LewisECExpanding the clinical indications for α(1)-antitrypsin therapyMol Med20121895797022634722
- JonesEAVergallaJSteerCJBradley-MoorePRVierlingJMMetabolism of intact and desialylated alpha 1-antitrypsinClin Sci Mol Med1978552139148307998
- PerlmutterDHAlpha-1-antitrypsin deficiency: diagnosis and treatmentClin Liver Dis200484839859viiiix15464658
- JanciauskieneSMBalsRKoczullaRVogelmeierCKöhnleinTWelteTThe discovery of α1-antitrypsin and its role in health and diseaseRespir Med201110581129113921367592
- GreeneCMMcElvaneyNGProteases and antiproteases in chronic neutrophilic lung disease – relevance to drug discoveryBr J Pharmacol200915841048105819845686
- ElliottPRPeiXYDaffornTRLomasDATopography of a 2.0 A structure of alpha1-antitrypsin reveals targets for rational drug design to prevent conformational diseaseProtein Sci2000971274128110933492
- KolarichDTurecekPLWeberABiochemical, molecular characterization, and glycoproteomic analyses of alpha(1)-proteinase inhibitor products used for replacement therapyTransfusion200646111959197717076852
- McCarthyCDunleaDMSaldovaRGlycosylation repurposes alpha-1 antitrypsin for resolution of community-acquired-pneumoniaAm J Respir Crit Care Med Epub20171116
- PaceCNConformational stability of globular proteinsTrends Biochem Sci199015114172107612
- PerlmutterDHJoslinGNelsonPSchasteenCAdamsSPFallonRJEndocytosis and degradation of alpha 1-antitrypsin-protease complexes is mediated by the serpin-enzyme complex (SEC) receptorJ Biol Chem19902652816713167162211587
- Chorostowska-WynimkoJStruniawskiRPopławskaBBorszewska-KornackaMThe incidence of alpha-1-antitrypsin (A1AT) deficiency alleles in population of Central Poland–preliminary results from newborn screeningPneumonol Alergol Pol2012805450453 Polish [with English abstract]22926906
- PopławskaBJanciauskieneSChorostowska-WynimkoJGenetic variants of alpha-1 antitrypsin: classification and clinical implicationsPneumonol Alergol Pol20138114554 Polish [with English abstract]23258471
- SesboüéRMartinJPAlpha-1-antitrypsin (PI) polymorphism in France, with special regard to the PI*Z alleleHum Hered19914153403461778610
- MassiGFabianoARagusaDAlpha 1-antitrypsin phenotypes and pi m subtypes in ItalyBull Eur Physiopathol Respir198016Suppl3013066971674
- CarrollTPO’ConnorCAFloydOThe prevalence of alpha-1 antitrypsin deficiency in IrelandRespir Res2011129121752289
- CrystalRGAlpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapyJ Clin Invest1990855134313522185272
- DeMeoDLSilvermanEKAlpha1-antitrypsin deficiency. 2: genetic aspects of alpha(1)-antitrypsin deficiency: phenotypes and genetic modifiers of emphysema riskThorax200459325926414985567
- CurielDChytilACourtneyMCrystalRGSerum alpha 1-antitrypsin deficiency associated with the common S-type (Glu264—Val) mutation results from intracellular degradation of alpha 1-antitrypsin prior to secretionJ Biol Chem19892641810477104862567291
- LomasDAEvansDLFinchJTCarrellRWThe mechanism of Z alpha 1-antitrypsin accumulation in the liverNature199235763796056071608473
- BrantlyMNukiwaTCrystalRGMolecular basis of alpha-1-antitrypsin deficiencyAm J Med1988846A13313289385
- MolloyKHershCPMorrisVBClarification of the risk of chronic obstructive pulmonary disease in α1-antitrypsin deficiency PiMZ heterozygotesAm J Respir Crit Care Med2014189441942724428606
- KellyEGreeneCMCarrollTPMcElvaneyNGO’NeillSJAlpha-1 antitrypsin deficiencyRespir Med2010104676377220303723
- JanoffAElastase in tissue injuryAnnu Rev Med1985362072163888051
- CoxDWLevisonHEmphysema of early onset associated with a complete deficiency of alpha-1-antitrypsin (null homozygotes)Am Rev Respir Dis198813723713753257661
- NukiwaTTakahashiHBrantlyMCourtneyMCrystalRGalpha 1-Antitrypsin nullGranite Falls, a nonexpressing alpha 1-antitrypsin gene associated with a frameshift to stop mutation in a coding exonJ Biol Chem19872622511999120043040726
- ParrDGStoelBCStolkJStockleyRAPattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairmentAm J Respir Crit Care Med2004170111172117815306534
- TomashefskiJFJrCrystalRGWiedemannHPMaschaEStollerJKAlpha 1-Antitrypsin Deficiency Registry Study GroupThe bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the national registry for individuals with Severe Deficiency of Alpha-1 AntitrypsinHum Pathol200435121452146115619203
- KorkmazBAttucciSEpinetteCMeasurement of neutrophil elastase, proteinase 3, and cathepsin G activities using intramolecularly quenched fluorogenic substratesMethods Mol Biol201284412513822262439
- TaggartCCervantes-LaureanDKimGOxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activityJ Biol Chem200027535272582726510867014
- McCarthyCSaldovaRWormaldMRRuddPMMcElvaneyNGReevesEPThe role and importance of glycosylation of acute phase proteins with focus on alpha-1 antitrypsin in acute and chronic inflammatory conditionsJ Proteome Res20141373131314324892502
- KasturiLEshlemanJRWunnerWHShakin-EshlemanSHThe hydroxy amino acid in an Asn-X-Ser/Thr sequon can influence N-linked core glycosylation efficiency and the level of expression of a cell surface glycoproteinJ Biol Chem19952702414756147617782341
- BetsuyakuTNishimuraMTakeyabuKTaninoMMiyamotoKKawakamiYDecline in FEV(1) in community-based older volunteers with higher levels of neutrophil elastase in bronchoalveolar lavage fluidRespiration200067326126710867593
- SilbersteinSGilmoreRBiochemistry, molecular biology, and genetics of the oligosaccharyltransferaseFASEB J19961088498588666161
- SchönbergMReibetanzURathmannSLessigJMaintenance of α(1)-antitrypsin activity by means of co-application of hypochlorous acid-scavengers in vitro and in the supernatant of polymorphonuclear leukocytes: as a basis for a new drug delivery approachBiomatter201221243623507783
- SegelmarkMElzoukiANWieslanderJErikssonSThe PiZ gene of alpha 1-antitrypsin as a determinant of outcome in PR3-ANCA-positive vasculitisKidney Int19954838448507474674
- PittelkowMRSmithKCSuWPAlpha-1-antitrypsin deficiency and panniculitis. Perspectives on disease relationship and replacement therapyAm J Med1988846A80863260076
- HubbardRCFellsGGadekJPacholokSHumesJCrystalRGNeutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophagesJ Clin Invest19918838918971653278
- GalliSJBorregaardNWynnTAPhenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophilsNat Immunol201112111035104422012443
- HaraokaMHangLFrendéusBNeutrophil recruitment and resistance to urinary tract infectionJ Infect Dis199918041220122910479151
- SummersCRankinSMCondliffeAMSinghNPetersAMChilversERNeutrophil kinetics in health and diseaseTrends Immunol201031831832420620114
- OwenCACampbellMABoukedesSSCampbellEJCytokines regulate membrane-bound leukocyte elastase on neutrophils: a novel mechanism for effector activityAm J Physiol19972723 Pt 1L385L3939124593
- WeissSJRegianiSNeutrophils degrade subendothelial matrices in the presence of alpha-1-proteinase inhibitor. Cooperative use of lysosomal proteinases and oxygen metabolitesJ Clin Invest1984735129713036325501
- BelaaouajAMcCarthyRBaumannMMice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsisNat Med1998456156189585238
- GuyotNWartelleJMalleretLUnopposed cathepsin G, neutrophil elastase, and proteinase 3 cause severe lung damage and emphysemaAm J Pathol201418482197221024929239
- SmedlyLATonnesenMGSandhausRANeutrophil-mediated injury to endothelial cells. Enhancement by endotoxin and essential role of neutrophil elastaseJ Clin Invest1986774123312433485659
- TravisJStructure, function, and control of neutrophil proteinasesAm J Med1988846A3742
- LundgrenJDRievesRDMullolJLogunCShelhamerJHThe effect of neutrophil proteinase enzymes on the release of mucus from feline and human airway culturesRespir Med19948875115187972975
- KimKCWasanoKNilesRMSchusterJEStonePJBrodyJSHuman neutrophil elastase releases cell surface mucins from primary cultures of hamster tracheal epithelial cellsProc Natl Acad Sci U S A19878424930493083480544
- GaggarALiYWeathingtonNMatrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patientsAm J Physiol Lung Cell Mol Physiol20072931L96L10417384080
- ZhuYKLiuXDSköldCMSynergistic neutrophil elastase-cytokine interaction degrades collagen in three-dimensional cultureAm J Physiol Lung Cell Mol Physiol20012814L868L87811557590
- GuyotNButlerMWMcNallyPElafin, an elastase-specific inhibitor, is cleaved by its cognate enzyme neutrophil elastase in sputum from individuals with cystic fibrosisJ Biol Chem200828347323773238518799464
- WeldonSMcNallyPMcElvaneyNGDecreased levels of secretory leucoprotease inhibitor in the Pseudomonas-infected cystic fibrosis lung are due to neutrophil elastase degradationJ Immunol2009183128148815620007580
- BergerMSorensenRUTosiMFDearbornDGDöringGComplement receptor expression on neutrophils at an inflammatory site, the Pseudomonas-infected lung in cystic fibrosisJ Clin Invest1989844130213132507578
- TosiMFZakemHBergerMNeutrophil elastase cleaves C3bi on opsonized pseudomonas as well as CR1 on neutrophils to create a functionally important opsonin receptor mismatchJ Clin Invest19908613003082164045
- HartlDLatzinPHordijkPCleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung diseaseNat Med200713121423143018059279
- Vega-CarrascalIBerginDAMcElvaneyOJGalectin-9 signaling through TIM-3 is involved in neutrophil-mediated Gram-negative bacterial killing: an effect abrogated within the cystic fibrosis lungJ Immunol201419252418243124477913
- Vega-CarrascalIReevesEPNikiTDysregulation of TIM-3-galectin-9 pathway in the cystic fibrosis airwaysJ Immunol201118652897290921263071
- FickRBJrNaegelGPSquierSUWoodREGeeJBReynoldsHYProteins of the cystic fibrosis respiratory tract. Fragmented immunoglobulin G opsonic antibody causing defective opsonophagocytosisJ Clin Invest19847412362486429195
- ReevesEPWilliamsonMByrneBIL-8 dictates glycosaminoglycan binding and stability of IL-18 in cystic fibrosisJ Immunol201018431642165220026745
- CarrellRWJeppssonJOLaurellCBStructure and variation of human alpha 1-antitrypsinNature198229858723293347045697
- Vercaigne-MarkoDDavrilMLaineAHayemAInteraction of human alpha1-proteinase inhibitor with human leukocyte cathepsin GBiol Chem Hoppe Seyler198536676556613876103
- DurantonJBiethJGInhibition of proteinase 3 by [alpha]1-antitrypsin in vitro predicts very fast inhibition in vivoAm J Respir Cell Mol Biol2003291576112600819
- YingQLSimonSRElastolysis by proteinase 3 and its inhibition by alpha(1)-proteinase inhibitor: a mechanism for the incomplete inhibition of ongoing elastolysisAm J Respir Cell Mol Biol200226335636111867344
- FalkRJTerrellRSCharlesLAJennetteJCAnti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitroProc Natl Acad Sci U S A19908711411541192161532
- RouhaniFPaoneGSmithNKKreinPBarnesPBrantlyMLLung neutrophil burden correlates with increased pro-inflammatory cytokines and decreased lung function in individuals with alpha(1)-antitrypsin deficiencyChest20001175 Suppl 1250S251S10843938
- ElliottPRBiltonDLomasDALung polymers in Z alpha1-antitrypsin deficiency-related emphysemaAm J Respir Cell Mol Biol19981856706749569237
- MahadevaRAtkinsonCLiZPolymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivoAm J Pathol2005166237738615681822
- MulgrewATTaggartCCLawlessMWZ alpha1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractantChest200412551952195715136414
- ParmarJSMahadevaRReedBJPolymers of alpha(1)-anti-trypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysemaAm J Respir Cell Mol Biol200226672373012034572
- LomasDACarrellRWSerpinopathies and the conformational dementiasNat Rev Genet200231075976812360234
- WuYWhitmanIMolmentiEMooreKHippenmeyerPPerlmutterDHA lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiencyProc Natl Acad Sci U S A19949119901490188090762
- HurleyKLaceyNO’DwyerCAAlpha-1 antitrypsin augmentation therapy corrects accelerated neutrophil apoptosis in deficient individualsJ Immunol201419383978399125217166
- BlancoILipskerDLaraBJanciauskieneSNeutrophilic panniculitis associated with alpha-1-antitrypsin deficiency: an updateBr J Dermatol2016174475376226595240
- GrossBGrebeMWenckerMStollerJKBjurstenLMJanciauskieneSNew findings in PiZZ alpha1-antitrypsin deficiency-related panniculitis. Demonstration of skin polymers and high dosing requirements of intravenous augmentation therapyDermatology2009218437037519218787
- KunkelSLStandifordTKasaharaKStrieterRMInterleukin-8 (IL-8): the major neutrophil chemotactic factor in the lungExp Lung Res199117117232013270
- MalerbaMRicciardoloFRadaeliANeutrophilic inflammation and IL-8 levels in induced sputum of alpha-1-antitrypsin PiMZ subjectsThorax200661212913316284217
- DetmersPALoSKOlsen-EgbertEWalzABaggioliniMCohnZANeutrophil-activating protein 1/interleukin 8 stimulates the binding activity of the leukocyte adhesion receptor CD11b/CD18 on human neutrophilsJ Exp Med19901714115511621969919
- ZeilhoferHUSchorrWRole of interleukin-8 in neutrophil signalingCurr Opin Hematol20007317818210786656
- DevaneyJMGreeneCMTaggartCCCarrollTPO’NeillSJMcElvaneyNGNeutrophil elastase up-regulates interleukin-8 via toll-like receptor 4FEBS Lett20035441–312913212782302
- OstrowskaESokolovaEReiserGPAR-2 activation and LPS synergistically enhance inflammatory signaling in airway epithelial cells by raising PAR expression level and interleukin-8 releaseAm J Physiol Lung Cell Mol Physiol20072935L1208L121817766588
- RamachandranRHollenbergMDProteinases and signalling: pathophysiological and therapeutic implications via PARs and moreBr J Pharmacol2008153Suppl 1S263S28218059329
- BerginDAReevesEPMeleadyPα-1 antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8J Clin Invest2010120124236425021060150
- JonigkDAl-OmariMMaegelLAnti-inflammatory and immunomodulatory properties of α1-antitrypsin without inhibition of elastaseProc Natl Acad Sci U S A201311037150071501223975926
- Ford-HutchinsonAWBrayMADoigMVShipleyMESmithMJLeukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytesNature198028657702642656250050
- HooverRLKarnovskyMJAustenKFCoreyEJLewisRALeukotriene B4 action on endothelium mediates augmented neutrophil/endothelial adhesionProc Natl Acad Sci U S A1984817219121936326110
- O’DwyerCAO’BrienMEWormaldMRThe BLT1 inhibitory function of alpha-1 antitrypsin augmentation therapy disrupts leukotriene B4 neutrophil signalingJ Immunol201519583628364126371243
- BerginDAReevesEPHurleyKThe circulating proteinase inhibitor α-1 antitrypsin regulates neutrophil degranulation and autoimmunitySci Transl Med20146217217ra1
- BrandtEPetersenFFladHDRecombinant tumor necrosis factor-alpha potentiates neutrophil degranulation in response to host defense cytokines neutrophil-activating peptide 2 and IL-8 by modulating intracellular cyclic AMP levelsJ Immunol19921494135613641323612
- MócsaiAJakusZVántusTBertonGLowellCALigetiEKinase pathways in chemoattractant-induced degranulation of neutrophils: the role of p38 mitogen-activated protein kinase activated by Src family kinasesJ Immunol200016484321433110754332
- Van MolleWLibertCFiersWBrouckaertPAlpha 1-acid glycoprotein and alpha 1-antitrypsin inhibit TNF-induced but not anti-Fas-induced apoptosis of hepatocytes in miceJ Immunol19971597355535649317155
- ZhangBLuYCampbell-ThompsonMAlpha1-antitrypsin protects beta-cells from apoptosisDiabetes20075651316132317360983
- GraziadeiIKaserbacherRBraunsteinerHVogelWThe hepatic acute-phase proteins alpha 1-antitrypsin and alpha 2-macroglobulin inhibit binding of transferrin to its receptorBiochem J1993290Pt 11091137679893
- SchmidtEWRascheBUlmerWTReplacement therapy for alpha-1-protease inhibitor deficiency in PiZ subjects with chronic obstructive lung diseaseAm J Med1988846A63693289388
- WewersMDCasolaroMASellersSEReplacement therapy for alpha 1-antitrypsin deficiency associated with emphysemaN Engl J Med198731617105510623494198
- ChapmanKRBurdonJGPiitulainenERAPID Trial Study GroupIntravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trialLancet2015386999136036826026936
- McElvaneyNGBurdonJHolmesMRAPID Extension Trial GroupLong-term efficacy and safety of α1 proteinase inhibitor treatment for emphysema caused by severe α1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE)Lancet Respir Med201751516027916480
- LiebermanJAugmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting dataChest200011851480148511083705
- MolloyMO’ConnorCFeeLCarrollTPMcElvaneyNGReal life treatment benefit of intravenous augmentation therapy for severe alpha-1 antitrypsinIr J Med Sci20161859S439S508
- SmithKCPittelkowMRSuWPPanniculitis associated with severe alpha 1-antitrypsin deficiency. Treatment and review of the literatureArch Dermatol198712312165516613318708
- FranciosiANMcCarthyCCarrollTPMcElvaneyNGUnusual acute sequelae of α1-antitrypsin deficiency: a myriad of symptoms with one common cureChest20151485e136e13826527439
- DowdSKRodgersGCCallenJPEffective treatment with alpha 1-protease inhibitor of chronic cutaneous vasculitis associated with alpha 1-antitrypsin deficiencyJ Am Acad Dermatol1995335 Pt 29139167593810
- GrieseMLatzinPKapplerMAlpha1-antitrypsin inhalation reduces airway inflammation in cystic fibrosis patientsEur Respir J200729224025017050563
- PetracheIFijalkowskaIMedlerTRAlpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosisAm J Pathol200616941155116617003475
- LewisECShapiroLBowersOJDinarelloCAAlpha1-antitrypsin monotherapy prolongs islet allograft survival in miceProc Natl Acad Sci U S A200510234121531215816093309
- MolanoRDPileggiASongSProlonged islet allograft survival by alpha-1 antitrypsin: the role of humoral immunityTransplant Proc200840245545618374099
- PileggiAMolanoRDSongSAlpha-1 antitrypsin treatment of spontaneously diabetic nonobese diabetic mice receiving islet allograftsTransplant Proc200840245745818374100
- KalisMKumarRJanciauskieneSSalehiACilioCMα 1-Antitrypsin enhances insulin secretion and prevents cytokine-mediated apoptosis in pancreatic β-cellsIslets20102318518921099312
- BerginDAHurleyKMcElvaneyNGReevesEPAlpha-1 antitrypsin: a potent anti-inflammatory and potential novel therapeutic agentArch Immunol Ther Exp (Warsz)2012602819722349104
- McElvaneyNGAlpha-1 antitrypsin therapy in cystic fibrosis and the lung disease associated with alpha-1 antitrypsin deficiencyAnn Am Thorac Soc201613Suppl 2S191S19627115956
- McElvaneyNGHubbardRCBirrerPAerosol alpha 1-antitrypsin treatment for cystic fibrosisLancet199133787383923941671425
- MartinSLDowneyDBiltonDKeoganMTEdgarJElbornJSRecombinant AAT CF Study TeamSafety and efficacy of recombinant alpha(1)-antitrypsin therapy in cystic fibrosisPediatr Pulmonol200641217718316372352
- CantinAMBerthiaumeYCloutierDMartelMProlastin aerosol therapy and sputum taurine in cystic fibrosisClin Invest Med200629420120716986483
- VerledenGMRaghuGMeyerKCGlanvilleARCorrisPA new classification system for chronic lung allograft dysfunctionJ Heart Lung Transplant201433212713324374027
- LiorYGeyraALewisECTherapeutic compositions and uses of alpha1-antitrypsin: a patent review (2012–2015)Expert Opin Ther Pat201626558158926966859
- DaemenMAHeemskerkVHvan’t VeerCFunctional protection by acute phase proteins alpha(1)-acid glycoprotein and alpha(1)-antitrypsin against ischemia/reperfusion injury by preventing apoptosis and inflammationCirculation2000102121420142610993862
- KanerZOchayonDEShahafGAcute phase protein α1-antitrypsin reduces the bacterial burden in mice by selective modulation of innate cell responsesJ Infect Dis201521191489149825389308
- KoulmandaMBhasinMHoffmanLCurative and beta cell regenerative effects of alpha1-antitrypsin treatment in autoimmune diabetic NOD miceProc Natl Acad Sci U S A200810542162421624718852471
- KoulmandaMSampathkumarRSBhasinMPrevention of nonimmunologic loss of transplanted islets in monkeysAm J Transplant20141471543155124913821
- TawaraISunYLewisECAlpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantationProc Natl Acad Sci U S A2012109256456922203983
- GuttmanOBaranovskiBMSchusterRAcute-phase protein α1-anti-trypsin: diverting injurious innate and adaptive immune responses from non-authentic threatsClin Exp Immunol2015179216117225351931
- de SerresFBlancoIRole of alpha-1 antitrypsin in human health and diseaseJ Intern Med2014276431133524661570