
Abstract
Sepsis was known to ancient Greeks since the time of great physician Hippocrates (460–377 BC) without exact information regarding its pathogenesis. With time and medical advances, it is now considered as a condition associated with organ dysfunction occurring in the presence of systemic infection as a result of dysregulation of the immune response. Still with this advancement, we are struggling for the development of target-based therapeutic approach for the management of sepsis. The advancement in understanding the immune system and its working has led to novel discoveries in the last 50 years, including different pattern recognition receptors. Inflammasomes are also part of these novel discoveries in the field of immunology which are <20 years old in terms of their first identification. They serve as important cytosolic pattern recognition receptors required for recognizing cytosolic pathogens, and their pathogen-associated molecular patterns play an important role in the pathogenesis of sepsis. The activation of both canonical and non-canonical inflammasome signaling pathways is involved in mounting a proinflammatory immune response via regulating the generation of IL-1β, IL-18, IL-33 cytokines and pyroptosis. In addition to pathogens and their pathogen-associated molecular patterns, death/damage-associated molecular patterns and other proinflammatory molecules involved in the pathogenesis of sepsis affect inflammasomes and vice versa. Thus, the present review is mainly focused on the inflammasomes, their role in the regulation of immune response associated with sepsis, and their targeting as a novel therapeutic approach.
Video abstract
Point your SmartPhone at the code above. If you have a QR code reader the video abstract will appear. Or use:
Introduction
The word sepsis originated from the Greek word “σηψις” (pronounced as “sipsis”) that is originally used in Greek language for describing deterioration of the animal or vegetable organic matter due to the presence of microbes.Citation1 However, the word sepsis was used in medical literature for the first time by Homer in his poems almost 2,700 years ago and the word was directly derived from the word “sepo” (σηπω) meaning “I rot”.Citation2 The term sepsis was also used by the Greek physician Hippocrates in 430 BC to describe deterioration of the organic matter.Citation1 He is well known for his work in ancient Greek medicine. He introduced the concept of impairment in the body humors or fluids including blood, bile (yellow and black bile), and phlegm as a major cause of the disease. Thereafter, Ibna Sina (also known as Avicenna, AD 980–1037), a Persian philosopher and the father of modern medicine, described sepsis/septicemia as putrefaction of blood and tissues with fever.Citation3 The emergence of classic or modern concept of sepsis happened in the 19th century. Ignaz Semmelweis (a Vienna, Austria-based obstetrician, 1818–1865) is considered as the first physician to provide the modern view of sepsis while studying the cause of death in infants getting puerperal fever (also called childbed fever). He concluded that the childbed fever was caused by the decomposed animal matter that entered their blood system. He is also considered an early pioneer in developing antiseptic techniques.
It was the beginning toward understanding sepsis. In 1914, a German physician called Hugo Schottmüller established for the first time that the presence of infection is the basic requirement for the development of sepsis.Citation3,Citation4 In 1989, a USA-based intensive care unit specialist Roger C Bone (1941–1997) defined sepsis as the invasion of microorganisms and/or their toxins into the bloodstream of patients and induction of the host’s immune response against the microbial invasion called systemic inflammatory response syndrome (SIRS).Citation5,Citation6 This definition of sepsis remained valid for more than two decades. However, in the year 2016, the third consensus on sepsis, which is also called sepsis 3, defined sepsis as a condition of life-threatening organ dysfunction generated due to the dysregulation of the host’s immune response during infection.Citation7 Sepsis 3 also led to the development of quick Sequential Organ Failure Assessment (qSOFA) scoring system for the early identification of simultaneous organ dysfunction in sepsis and challenged the SIRS criteria for sepsis definition because sepsis involves an early activation of both pro- and anti-inflammatory immune responses.Citation8–Citation11 Hence, sepsis 3 emphasized on early diagnosis and quick initiation of treatment during the ongoing process of sepsis to prevent mortality. Sepsis accompanied with the incidence of hypoperfusion and hypotension along with organ dysfunction is considered as severe sepsis,Citation12,Citation13 while septic shock is defined as severe sepsis with profound hypo-tension that persists during volume resuscitation and needs the administration of vasopressors including epinephrine, norepinephrine, isoproterenol, dobutamine, and so on.Citation10,Citation13,Citation14 Septic shock predisposes the patient towards an increased risk of mortality as compared to sepsis/severe sepsis alone.
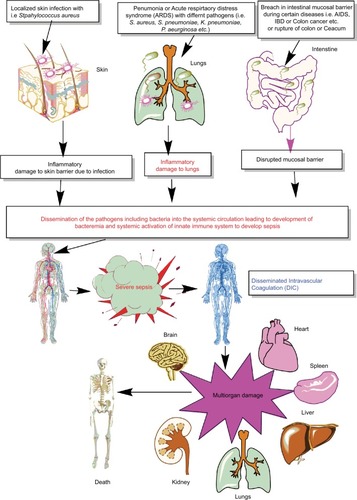
Thus, sepsis is a condition of severely dysregulated immune response against a pathogen, where both pronounced and overwhelming activation of innate immunity along with the suppression of conventional T-cell–mediated immune response and an upregulation of regulatory T cells (Tregs) causing sepsis-associated immunosuppression are observed.Citation10,Citation15–Citation22 It is now well established that sepsis is a condition of an aberrant immune response where an initial innate immune activation fails to clear the source of the infection or pathogen, causing the generation of an overwhelming proinflammatory reaction responsible for multiorgan damage or failure (). Furthermore, patients surviving the episode of sepsis suffer from long-term immunosuppression, making them prone to develop secondary infections.Citation8,Citation9,Citation23,Citation24
Figure 1 Schematic representation of breach in host innate immunity due to different bacterial infections and development of bacteremia and sepsis.
Notes: Once the bacteremia is developed in the host and the initial control of systemic infection fails, it leads to increased activation of the innate immune response systemically. This exaggerated innate immune response causes MOD or MODS. The development of MODS leads to the ultimate death of the patients.
Abbreviations: ARDS, acute respiratory distress syndrome; DIC, disseminated intravascular coagulation; IBD, inflammatory bowel disease; MOD, multiorgan damage; MODS, multiorgan dysfunction syndrome.
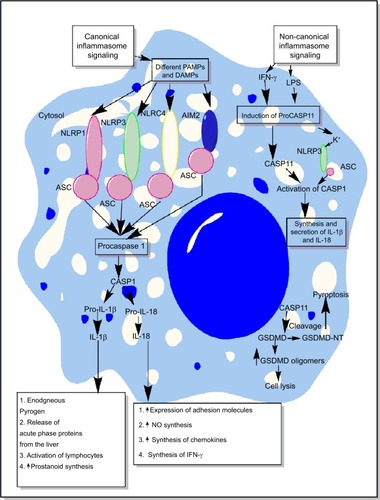
Inflammasomes are the essential components of host’s immune defense mechanisms that regulate the generation of proinflammatory immune response along with the recognition of intracellular pathogens.Citation25–Citation29 However, activation of NLRP3 inflammasome is also shown to exhibit an inhibitory action on protective gastrointestinal immune response against helminthic infections.Citation30 The activation of both the classical/canonical and non-canonical/non-classical inflammasome signaling pathways under diverse inflammatory conditions is shown in and the detailed mechanisms are described elsewhere in detail.Citation31–Citation38 The primary aim of the present review is to highlight the role of inflammasomes in sepsis-associated proinflammatory immune response and its targeting.
Figure 2 Schematic representation of canonical and non-canonical inflammasome signaling pathways.
Notes: The figure describes both the canonical and non-canonical signaling pathways involved in the activation of different inflammasome components including NLRP1, NLRP3, NLRC4, and AIM2. The canonical inflammasome signaling pathway causes activation of procaspase 1 into mature CASP1 that cleaves immature proIL-1β and proIL-18 into mature IL-1β and IL-18 involved in the generation of inflammatory immune response. The intracellular LPS and type-1 IFNs cause induction of non-canonical signaling in macrophages and DCs via the induction of procaspase 11 that further activates NLRP3 inflammasome by inducing a decrease in cytosolic K+ through different mechanisms described in the text. Additionally, CASP11 cleaves GSDMD into N- and C-terminal components that, upon their cytosolic accumulation, form GSDMD oligomer causing cell death, while GSDMD-NT causes pyroptosis, but it does not cause the death of adjacent cells upon its extracellular release. Detailed mechanism of inflammasome signaling pathway is mentioned elsewhere in the text.
Abbreviations: DAMP, death/damage-associated molecular pattern; DCs, dendritic cells; GSDMD, gasdermin D; IFN, interferon; LPS, lipopolysaccharide; PAMP, pathogen-associated molecular pattern.
Inflammasomes as a regulator of immune homeostasis during infection and inflammation
Inflammasomes were first discovered as a caspase-activating complex comprising mainly caspase-1 (CASP1), caspase-5 (CASP5), Pycard/Asc, and NALP1, a pyrin domain-containing protein sharing structural homology with NODs, in the year 2002 by the research team led by Jurg Tschopp.Citation39 After 16 years of their discovery, they have been studied extensively and are still the topic of research targeting infectious and inflammatory diseases including cancer and neurodegeneration.Citation40–Citation47 Thus, inflammasomes might be playing an important role in the homeostasis of the immune system to maintain a healthy stage within the host. For example, inflammasome-deficient mice harbor an aberrant microbial community that can be dominantly transmissible to healthy animals.Citation48 This alteration in the microbial community due to the deficiency of inflammasomes causes a profound defect in intestinal immunity along with that in distant organs systemically.Citation48 In addition, the AIM2 and NLRP3-deficient New Zealand Black mice exhibit autoimmune phenotype responsible for the development of anti-erythrocyte antibodies and autoimmune hemolytic anemia.Citation49,Citation50 These mice also develop antinuclear antibodies typical of systemic lupus erythematosus.Citation49
The recognition of PAMPs or DAMPs involves specific germline-encoded receptors called pattern recognition receptors (PRRs) expressed either on the outer surface of the plasma membrane or intracellular cytoplasmic vesicles including endosomes, lysosome, or endolysosomes.Citation51–Citation53 In comparison to a class of PRRs called Toll-like receptors (TLRs) that are present both on the cell surface and intracellular environment, certain receptors called Nod-like receptors (NLRs), AIM-2 like receptors (ALRs), pyrin and HIN domain-containing family proteins, and cytosolic nucleoside sensors are present only intracellularly and guard against potential pathogens or inflammogens.Citation25,Citation54 These NLRs and ALRs are distinct from other cytosolic PRRs mentioned, as activation of these receptors forms a specific complex via the assembly and activation of a complex called inflammasomes to activate CASP1 and CASP11 (also known as caspase-4).Citation31,Citation39 Only five PRRs including NLRP1, NLRP3, NLRC4, pyrin, and AIM2 are able to form inflammasomes.Citation55,Citation56 Under resting conditions, the inflammasomes are considered to exist in an inhibitory state.Citation57,Citation58 Once activated, they cause conversion of proinflammatory procaspases into their mature form. This inflammasome-mediated activation of CASP1 and CASP11 causes the release of proinflammatory cytokines called IL-1β and IL-18.Citation55,Citation59 Thus, the activation of inflammasomes under diverse conditions causes proinflammatory cell death via several mechanisms including pyroptosis (inflammatory cell death) and necroptosis (a form of lytic inflammatory cell death or a physiological suicide initiated by RIPK3-mediated phosphorylation of MLKL).Citation57,Citation60–Citation63 Necroptosis is involved in the development, inflammation, and disease pathogenesis.Citation64 Hence, via inducing necroptosis or regulating the release of IL-1β and IL-18, inflammasomes play an active role in the process of generation of inflammatory immune response and in the disease pathogenesis, depending on the condition/stimulus.Citation65,Citation66
Inflammasome-mediated release of IL-1β and IL-18 and the maintenance of immune homeostasis
IL-1β and IL-18 are the very first proinflammatory cytokines released by the macrophages and dendritic cells (DCs) upon an encounter with different pathogens.Citation59,Citation67 IL-1β acts as an endogenous pyrogen and is responsible for the induction of fever, release of hepatic acute-phase proteins, activation of lymphocytes (ie, release of IL-2 form T-cells), and an upregulation of prostanoid synthesis.Citation67–Citation69 However, IL-18 does not produce fever or induce prostaglandins (PGs) or prostanoids due to its inability to induce cyclooxygenase-2 (COX-2), but induces inflammation via other mechanisms.Citation70–Citation72 The inability of IL-18 to induce COX-2 activation and, thus, fever can be attributed to the fact that it acts by stimulating p38 mitogen-activated protein (MAP) kinase pathway, not via NF-κB activation.Citation71 In addition, IL-18 can attenuate IL-1beta (an endogenous pyrogen) induced fever.Citation73 Instead, it induces the overexpression of adhesion molecules required for neutrophil infiltration at the site of infection or inflammation and increases nitric oxide (NO) synthesis and chemokines required for neutrophil and T-cell chemotaxis.Citation74 IL-18 was found to induce the synthesis of interferon (IFN)-γ, and thus plays an important role in the polarization of T-cells toward proinflammatory Th1 cells.Citation67,Citation75 IL-18 controls energy homeostasis by suppressing appetite and feed efficiency in mice, as IL-18−/− mice exhibit hyperphagia leading to the development of obesity, and insulin resistance causing an induction of metabolic syndrome phenotype due to a defective STAT3 phosphorylation.Citation76,Citation77 Thus, inflammasome-mediated CASP1 and CASP11 activation controls several immunological mechanisms via mediating the release of these cytokines.
Additionally, inflammasomes have also been implicated in the maintenance of normal and healthy intestinal microbiota.Citation78,Citation79 For example, NLRP6 inflammasome affects the intestinal microbiota in mice via recognizing various low-molecular-weight metabolites including taurine, pinitol, sebacate, and undecanedioate derived from colonic bacteria.Citation37,Citation80,Citation81 Thus, any disruption in the function of inflammasomes in the intestinal immune environment has a potential to induce a dysfunctional immune response that may lead to the development of an environment supportive to the dysbiosis of the gut bacteria into peripheral circulation, leading to the development of bacteremia and sepsis.Citation82–Citation84 Thus, inflammasomes are one of the essential components of vertebrate immunity playing an important role in the defense against pathogens via regulating the generation of inflammatory immune response and other components of immunity required for the host defense.
Inflammasomes in the immunopathogenesis of sepsis
The induction of profound and dysregulated immune response during uncontrolled infections of different origins including bacterial, viral, parasitic, and fungal infections may lead to the development of sepsis. However, sepsis in most cases seems to be clinically associated with the occurrence of an uncontrolled bacterial infection caused either by gram-negative or gram-positive bacteria or both (polymicrobial sepsis). The induction of an effective innate immune response to clear the pathogen requires the identification of pathogens via different PRRs that recognize different PAMPs.Citation85 PAMPs are evolutionarily conserved molecular structures expressed by different pathogens, but are not synthesized or expressed by the host cells.Citation86 These PRRs can be classified into phagocytic PRRs and sensor PRRs. The phagocytic PRRs directly bind to the pathogens and prepare them for the process of phagocytosis via opsonization.Citation87,Citation88 On the other hand, sensor PRRs include different TLRs (TLR1–TLR13) that are present both extracellularly on the plasma membrane or intracellularly in endosomes or lysosomes, NLRs, and ALRs.Citation89–Citation98 Thus, any defect in any of these PRRs and associated signaling pathway during the recognition of the potential pathogen and their PAMPs proves detrimental to the host by supporting the development of sepsis.
The pathogen is recognized by the various innate immune cells (including neutrophils recruited at the site of infection) located in the vicinity of their route of exposure, that is, respiratory tract, gastrointestinal tract, genitourinary tract, and skin.Citation99–Citation101 Thus, the initial recognition of the pathogen by PRRs (ie, TLRs including TLR4 [present both on the cell membrane and intracellularly in endosomes and phagosomes], TLR2, TLR1, TLR6, and TLR5) located on the surface of immune cells initiates the signaling cascade responsible for the generation of proinflammatory immune response via releasing various proinflammatory molecules including various cytokines and chemokines (ie, tumor necrosis factor [TNF]-α, IL-1α, IL-6, IL-8 [CXCL8], monocyte chemoattractant protein 1 [MCP1 or CCL2], etc) along with the generation of type 1 IFNs.Citation102–Citation106 The intact pathogens or PAMPs that get into the cytosol are also recognized by intracellular PRRs. Inflammasomes act as a major class of intracellular PRRs, along with TLR3, TLR7, TLR8, and TLR9. However, inflammasomes are freely circulating in the cytosol, while intracellular TLRs are present in cellular organelles including endosomes, lysosomes, and phagosomes. Thus, the initial recognition of the pathogens and PAMPs by TLRs strengthens the inflammasome signaling required for the release of potent proinflammatory mediators and generates several other molecules that act as endogenous activators of inflammasomes. Both the canonical and non-canonical inflammasomes are activated under different infectious conditions, which may lead to the development of sepsis if not regulated/controlled efficiently.Citation31,Citation32,Citation34,Citation107–Citation111 All these factors and events are described subsequently under various subheadings.
Adhesion molecules, sepsis, and inflammasomes
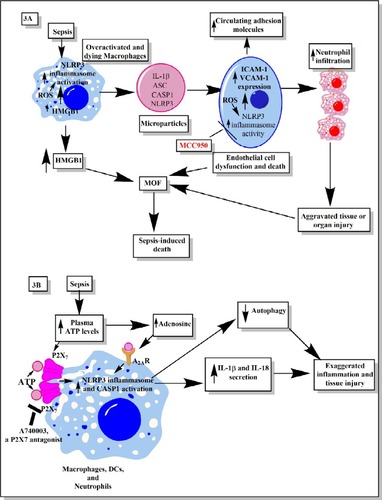
A higher level of circulating adhesion molecules called intracellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) are seen in the clinical cases of sepsis in patients of all age groupsCitation112–Citation114 (). The increased circulating levels of ICAM-1 and VCAM-1 are well correlated with the higher incidence of sepsis-associated multiorgan failure (MOF) and mortality in patients admitted to the hospitals.Citation115,Citation116 The elevated serum level of VCAM-1 is associated with septic encephalopathy.Citation117 Thus, an increase in the circulating adhesion molecules is used as a prognostic marker for sepsis and septic shock.Citation113,Citation116 The expression of adhesion molecules on endothelial cells is required for the migration of immune cells (neutrophils, monocytes, and T-cells) at the site of infection to clear the pathogens.Citation118 However, overexpression of these adhesion molecules during sepsis leads to the increased infiltration of proinflammatory immune cells at the site of infection, which also causes tissue and organ damage.Citation118–Citation121 One of the factors contributing to the increased serum levels of adhesion molecules and MOF during sepsis is endothelial dysfunction and their death due to apoptosis or inflammasome-mediated pyroptosisCitation122,Citation123 ().
Figure 3 Schematic representation of the role of inflammasomes and their regulation via different molecules or factors elevated during sepsis.
Notes: (A) This part figure shows the induction of NLRP3 inflammasomes during sepsis in macrophages, causing their death, release of high-mobility group box 1 protein (HMGB1), microparticles (consisting of IL-1β, NLRP3, ASC, and CASP1) binding to endothelial cells and stimulating the induction of higher expression of adhesion molecules (ICAM-1 and VCAM-1), causing profound infiltration of neutrophils at the site of infection, which leads to increased tissue or organ damage and death of endothelial cells, in turn leading to increased circulating levels of ICAM-1 and VCAM-1, MOF, and finally death of the septic patient. MCC950, an NLRP3 inflammasome inhibitor, caused inhibition of increased expression of ICAM-1 and VCAM-1 on the endothelial cells and prevented MOF. (B) This part figure shows increased circulating levels of ATP and its metabolite called adenosine act via different receptors called P2X7 and A2ARs expressed on the neutrophils, DCs, and macrophages. The binding then activates the NLRP3 inflammasomes and CASP1 to release IL-1β and IL-18. These inflammatory cytokines cause increased neutrophil infiltration and tissue/organ damage during sepsis. The P2X7 receptor antagonist is shown to inhibit the activation of NLRP3 inflammasome and, thus, the inflammatory tissue damage. Furthermore, activation of NLRP3 inflammasomes decreases the process of autophagy that contains the inflammation, as restoring autophagy has been used to target sepsis. See text for full details.
Abbreviations: ATP, adenosine 5′-triphosphate; DC, dendritic cell; ICAM, intracellular adhesion molecule; MOF, multiorgan failure; VCAM, vascular cell adhesion molecule.
NLRP3 inflammasome activation also increases the expression of chemotactic molecules (ie, α4β1 integrin, CCL7, CCL8, and CXCL16).Citation124 The overactivated and dying macrophages during sepsis also release microparticles (MPs) in the systemic circulation that consist of IL-1β and components of the inflammasome, including ASC, CASP-1, and NLRP3Citation125 (). These MPs bind to and are internalized by human endothelial cells, causing induction of phosphorylation of extracellular signal-related kinase (ERK)1/2, NF-κB activation, and expression of ICAM-1, VCAM-1, and E-selectin.Citation125 However, macrophages lacking NLRP3 inflammasome exhibit reduced production and activity of MPs.Citation125 The endothelial cells lacking IL-1 receptor (IL-1R) showed decreased expression of adhesion molecules in the presence of these MPs.Citation125 Thus, NLRP3 inflammasome-mediated release of MPs containing IL-1β is essential to upregulate the expression of adhesion molecules on endothelial cells. The expression of different adhesion molecules on endothelial cells plays an important role in neutrophil chemotaxis and their infiltration at the site of infection to clear the infection initially. However, in the case of sepsis, persistent infection along with overactivation of the immune response may cause increased activation of the NLRP3 inflammasome. This causes increased release of MPs from the dying macrophages, which leads to higher expression of various adhesion molecules on the endothelial cells required for neutrophil/monocyte adhesion and migration to the distant organs. This increased infiltration of neutrophils/monocytes along with clearing the pathogen also exerts collateral damage and, if not controlled, may cause severe organ damage, including lungs, kidneys, liver, and so on.Citation126,Citation127 On the other hand, this overactivation of endothelial cells also leads to their destruction and release of MPs which further enhances the pathogenic effect of endothelial cells.Citation127 Thus, a prolonged interaction between the neutrophils/monocytes and endothelial cells, where NLRP3 inflammasome activation induces increased expression of adhesion molecules on the endothelial cells, causes their overactivation and shedding of the adhesion molecules in the circulation and organ damage.
NLRP3 inflammasome activation in the endothelial cells during Lactobacillus casei infection causes dysfunction in endothelial cells and increases the upregulation of VCAM-1Citation128 (). ICAM-1 levels are also elevated on endothelial cells upon inflammasome activationCitation129 (). Thus, this increased expression of ICAM-1 and VCAM-1 upon inflammasome stimulation plays an important role in neutrophil adhesion, transcellular migration, and neutrophil-dependent and -independent organ (ie, lungs, liver, heart, etc) damage during sepsisCitation130–Citation134 (). Furthermore, an increased expression of ICAM-1 on the neutrophils is also observed during sepsis, which enhances their phagocytic potential and proinflammatory action.Citation135 However, it would be interesting to explore if the increased expression of ICAM-1 on the neutrophils is also a result of an inflammasome-based event or is independent of inflammasome activation. A potent NLRP3 inflammasome inhibitor called MCC950 was found to decrease the expression of ICAM-1 and VCAM-1 on the endothelial cellsCitation136 (). Thus, it would be novel to study the impact of MCC950 on these adhesion molecules’ expression on endothelial cells, circulating adhesion molecules, and MOF during sepsis. It would also be interesting to observe its action on the expression of ICAM-1 on neutrophils isolated from sepsis patients. Human neutrophils also express NLRP3, AIM2 inflammasomes, and CASP1.Citation137 Thus, activation of these inflammasomes on neutrophils during sepsis can provide defense against the invading cytosolic pathogens, along with an increased proinflammatory action via releasing IL-1β and IL-18.Citation137,Citation138 However, if this inflammasome-based protective action gets dysegulated, it may cause collateral organ damage as reactive oxygen species (ROS) generated by neutrophils exert a tissue destructive effect, and also, inflammasomes are further activated by ROS, which may further aggravate the tissue damage by releasing proinflammatory moleculesCitation139–Citation141
HMGB1, sepsis, and inflammasomes
HMGB1 is a late mediator of sepsis that is released either actively by activated monocytes/macrophages or passively by cells that died due to necrosis or necroptosisCitation142–Citation145 (). This release of HMGB1 in the circulation during sepsis increases the expression of ICAM-1 and VCAM-1 via binding to the receptor for advanced glycation end products (RAGE).Citation146 This HMGB1–RAGE interaction also enhances the release of TNF-α, IL-8, MCP-1 or CCL2, plasminogen activator inhibitor 1, and tissue plasminogen activator.Citation146–Citation148 The binding of HMGB1 to RAGE on endothelial cells causes transient phosphorylation of MAP kinases, ERK, JNK, and p38 and activation of NF-κB and Sp1 for its proinflammatory action.Citation146 It was found that as sepsis worsens, ROS generation and oxidation of HMGB1 further increase its proinflammatory potential, which aggravates the sepsis-associated inflammatory organ damage including acute kidney injury.Citation149
Therapeutic targeting of HMGB1 during sepsis modulated the proinflammatory cytokine profile in such a way that it improved several clinical features of sepsis and survival among experimental animals.Citation150 Furthermore, the mice that survived the episode of sepsis showed resistance toward developing secondary infections, but most of the patients surviving sepsis become prone to develop secondary infection later.Citation150 However, studies have shown the inflammasome-mediated regulation of HMGB1 release from activated immune cells including macrophages during sepsis.Citation151–Citation154 The deletion of inflammasomes including NLRP3 in mice during endotoxemia and bacterial sepsis decreases HMGB1 production and the associated inflammatory organ damage.Citation151,Citation152 In addition to canonical inflammasomes, the component of non-canonical inflammasome called CASP11 also has a great potential to modulate pyroptosis and the release of HMGB1 in the absence of CASP1.Citation155 This is because a genetic deletion of CASP11, rather than CASP1, in mice protected them from developing severe endotoxemia upon lethal lipopolysaccharide (LPS) challenge.Citation31 One of the contributing factors for the release of HMGB1 is the dead cells. However, it is not released by cells dying due to apoptosis; instead, it is rapidly released by cells dying due to pyroptosis that is induced by inflammasomes.Citation152,Citation156 The HMGB1 released due to pyroptotic cell death is more hyperacetylated at the nuclear localization sequences, as compared to the HMGB1 released due to necrotic cell death.Citation157 The hyperacetylated HMGB1 gets translocated easily and more frequently from the nucleus to the cytoplasm and from the cytoplasm to the secretory lysosomes by a default process; from there, it gets translocated into the extracellular environment upon appropriate stimulus, as compared to the hypoacetylated form.Citation158,Citation159 Further studies have indicated that HMGB1 also activates NLRP3 and CASP8 inflammasome via activating NF-κB.Citation160 Thus, both HMGB1 and inflammasome influence each other and targeting either of these during sepsis will provide a multi-target therapeutic approach that is essentially required. For example, glycyrrhizin-mediated inhibition of HMGB1 caused inhibition of the expression of adhesion molecules and oxidative stress on endothelial cells.Citation161 Thus, it will be interesting to observe the effect of glycyrrhizin on inflammasome activation during experimental sepsis and the associated immune response due to a strong interrelationship between HMGB1 release and inflammasome activation and vice versa.
ROS, oxidative stress, sepsis, and inflammasomes
Phagocytosis of pathogens by immune cells leads to the generation of ROS to kill the pathogen. For example, neutrophils and macrophages responsible for the phagocytic killing of the pathogen also undergo apoptotic cell death due to the higher production of ROSCitation162–Citation164 (). However, this increased generation of ROS also exerts bystander effects on cells responsible for its production along with the adjacent surrounding environment due to very high reactivity of these free radicals causing development of oxidative stress during sepsis.Citation165–Citation167 Circulating ROS responsible for the sepsis-associated oxidative stress induce a profound modification in the erythrocytes causing activation of the circulating leukocytes and maintains the pro-oxidative state even after the clearance of circulating bacteria.Citation168 Additionally, endothelial cells encountering proinflammatory signals (ie, cytokines and MPs released by neutrophils and monocytes/macrophages) and circulating pathogens during sepsis also produce various ROS that cause a profound change in their endothelial vascular function including vascular tone and their morphology, leading to severe coagulating defects and organ dysfunction (ie, cardiac dysfunction, liver and kidney failure, acute lung injury, etc),Citation126,Citation169–Citation171 as shown in . Thus, ROS plays an important role in the pathogenesis of sepsis and associated organ damage. However, a recent study has shown a lower production of ROS during early sepsis does not cause myocardial dysfunction.Citation172 This indicates that ROS generation starts during early sepsis and remains unchecked till late-stage sepsis called severe sepsis and septic shock, causing profound cardiac dysfunction and damage to the other vital organs leading to mortality. The late stage of sepsis is recognized as a stage of immunosuppression/immunoparalysis characterized by the susceptibility of the person to develop secondary infections. ROS generation also plays an important role in the induction of this stage by causing enhanced expression of NRF2-dependent ATF3.Citation173 Although this ROS-mediated induction of ATF3 protected the animals from developing endotoxic shock, it made them prone to develop severe bacterial and fungal infections by suppressing the release of IL-6.Citation173 Thus, the ROS levels impact both organ damage and susceptibility to secondary infections during sepsis.
The intracellular generation of ROS is also known to regulate the inflammasome activity; also, ROS are produced due to the activity of NLRP3 inflammasomes.Citation141,Citation174–Citation176 Thus, ROS generation in the macrophages provides a priming effect to activate NLRP3 inflammasomes required for the efficient clearance of the cytosolic pathogen that is blocked by treatment with ROS inhibitors.Citation141 Thus, it can be inferred that the higher levels of ROS generated during sepsis keep the NLRP3 inflammasomes activated (even after clearance of the infection) and the inflammatory process unchecked, causing the death of these immune cells. This exaggerated death of proinflammatory immune cells including M1 macrophages, CD4+, and CD8+ T-cells causes the development of a stage of immunosuppression.Citation22,Citation177 Additionally, ROS generation in endothelial cells serves as a local endogenous trigger for NLRP3 activationCitation178,Citation179 (). This NLRP3 inflammasome activation causes the release of cytokines IL-1β and IL-18 that act as chemoattractants to further recruit inflammatory immune cells including neutrophils, causing further inflammatory damage to the organs including kidneys.Citation178,Citation179 Furthermore, ROS-induced NLRP3 activity was found to induce acute liver injury.Citation180 Thus, regulating the generation of ROS during sepsis or inflammasome has a potential to target sepsis and associated organ damage effectively.
ATP, adenosine, sepsis, and inflammasomes
A higher plasma level of adenosine 5′-triphosphate (ATP) is reported in sepsis patients and in animal models of sepsisCitation181–Citation183 (). Furthermore, higher intracellular ATP is also observed in neutrophils isolated from sepsis patients.Citation184 Further study in this direction has indicated that higher levels of systemic ATP molecules cause an impairment in neutrophil chemotaxis and host defense.Citation185 The study further emphasizes that inhibition of endogenous ATP decreases the overactivation of neutrophils and the associated organ injury observed during sepsis. However, it exaggerates bacteremia and the spread of infection to distant organs and, thus, increases the mortality associated with sepsis.Citation185 But chelation or removal of systemic ATP enhanced the bacterial clearance from the circulation and increased survival by causing an improvement in chemotaxis of neutrophils at the site of infection.Citation185 The extracellular ATP is recognized by P2 purine receptors including ionotropic P2X receptors (P2X1–P2X7) and G-protein–coupled P2Y receptors (P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11–P2Y14) expressed by various innate immune cells and nonimmune cells.Citation186–Citation188 The recognition of extracellular ATP by P2X7 receptor is involved in several inflammatory diseases by causing the maturation and release of IL-1βCitation189–Citation191 ().
P2X7 receptor-mediated stimulation of NLRP3 inflammasome on the neutrophils was found to secrete IL-1β that may prove detrimental to the host.Citation191,Citation192 However, P2X7 receptor activation on the macrophages and DCs also causes activation of NLRP3 inflammasome and CASP1 activation, resulting in the release of IL-1β and IL-18Citation193–Citation196 (). This P2X7-mediated activation of NLRP3 due to extracellular ATP in these innate immune cells also increases their antimicrobial action against cytosolic pathogens.Citation191,Citation197 The systemic inhibition of P2X7 receptor with its antagonist called A740003 protected animals from sepsis-associated disruption in the intestinal barrierCitation198 (). Thus, the inhibition of ATP-mediated purinergic signaling mediated via P2X7 receptors has a potential inhibitory effect on overactivated inflammasomes during sepsis. Further studies in this direction are required to fill the unknown gaps in the field. The in vivo stimulation of NLRC4/NAIP inflammasome through flagellated pathogens in monocytes, macrophages, and neutrophils was reported to cause severe systemic inflammation as observed in sepsis.Citation199
Adenosine is an endogenous immunomodulatory metabolite produced under diverse inflammatory conditions including sepsis.Citation200–Citation202 Adenosine is constitutively present in the mammalian system in the extracellular environment in very low amount (≤1 µM).Citation200 However, its plasma level (4–10 µM in patients with septic shock) increases during different diseases causing metabolic stress such as acute tissue injury, sepsis, or septic shockCitation203,Citation204 (). Thus, high plasma adenosine levels can be used as a prognostic marker for sepsis and septic shock. However, adenosine acting via adenosine A2A receptor (A2AR) acts as a major regulator of inflammasome activity.Citation205 The higher plasma adenosine level during sepsis aggravates the maximal amplitude and duration of the inflammasome response.Citation205 This inflammasome regulation via adenosine is mediated by downstream signaling pathway initiated by A2AR activation which causes activation of cyclic adenosine monophosphate (cAMP)/PKA/CREB/hypoxia-inducible factor-1α signaling pathway.Citation205 This further causes the upregulation of pro-IL-1β, NLRP3 and increased activity of CASP1. This NLRP3 inflammasome pathway activation causes an increased production of IL-1β, IL-18, and pyroptosis of immune cells including macrophages to further aggravate the inflammatory process observed during sepsis, even in the absence of pathogen or associated PAMPs. This shows that these immune cells (ie, macrophages) do not exhibit tolerance, but remain in longer post-activation stage controlled by adenosine and cAMP.Citation205 Adenosine A2AR and A2BR targeting has been used as an immunomodulatory therapeutic approach to targeting sepsis.Citation206–Citation209 Thus, it will be interesting to observe the impact of this adenosine-based immunomodulatory therapeutic approach on inflammasome during sepsis.
Complement system, sepsis, and inflammasomes
Complement system (CS) is an evolutionarily conserved component of the innate immune system that plays a very important role in the host immune response and its regulation.Citation210–Citation213 Due to its important role in the recognition of potential pathogens, opsonization, directly killing the pathogens via forming membrane attack complex (MAC), and modulation of potential immune response, complement is recognized to play an important role in the pathogenesis of sepsis, septic shock, and associated organ damage and mortality.Citation214–Citation218 However, the CS components also influence NLRP3 inflammasomes.Citation219 For example, phagocytosis of the complement-opsonized pathogens by macrophages stimulates the activation of NLRP3 inflammasomes and CASP1, which cause the release of proinflammatory cytokines (IL-1β and IL-18) and promote leukocyte infiltration at the site of infection (source of sepsis), as shown in .Citation220 This inflammasome activation does not occur upon phagocytosis of unopsonized pathogens or other particles opsonized with serum deficient in the terminal components of the CS. Another study has indicated NLRP3 inflammasome and CASP1 activation due to the formation of sublethal MAC, but not by the complement components C3a and C5a during endotoxemia.Citation221 The deposition of sublethal MAC on the cell surface increases the intracellular Ca2+ which accumulates in the mitochondrial matrix and alters the mitochondrial transmembrane potential triggering NLRP3 inflammasome activation and CASP1 activity ().Citation222 This intracellular Ca2+ causes increased generation of mitochondrial ROS (mtROS) and the death of mitochondria causes the release of mitochondrial DNA (mtDNA) into the cytosol.Citation223,Citation224 This oxidized mtDNA activates NLRP3 inflammasome during the apoptosis of macrophages and other leukocytes which is a well-recognized phenomenon observed during sepsis or other acute infection.Citation225–Citation227
Figure 4 Role of complement in the activation of inflammasomes in innate immune cells during sepsis.
Notes: The complement (C3b)-opsonized bacteria phagocytosed via MAC cause the binding of MAC to the macrophage membrane, which leads to the initiation of bystander damage via the upregulation and activation of NLRP3 inflammasome. The activation of NLRP3 causes activation of CASP1, which leads to the maturation and release of proinflammatory cytokines, IL-1β and IL-18. On the other hand, the sublethal form of the MAC called sublethal MAC also induces the activation of NLRP3 inflammasome and the release of IL-1β and IL-18 via increasing the intracellular Ca2+ which enters into the mitochondrial matrix. The increase of Ca2+ in the mitochondrial matrix leads to increased mtROS generation and alters the mitochondrial membrane potential, causing mitochondrial damage and the release of mitochondrial DNA into the cytosol. All these factors activate NLRP3 inflammasome. Additionally, binding of C3a to its cognate receptor C3aR also activates NLRP3 inflammasome via activating the ERK1/ERK2 pathway which causes the efflux of ATP that activates NLRP3 inflammasome via binding to P2X7. The ATP that gets converted into adenosine via the enzymatic action of CD39 and CD73 also activates NLRP3 inflammasome by binding to A2AR.
Abbreviations: A2AR, A2A receptor; ATP, adenosine triphosphate; MAC, membrane attack complex; mtDNA, mitochondrial DNA; mtROS, mitochondrial reactive oxygen species.
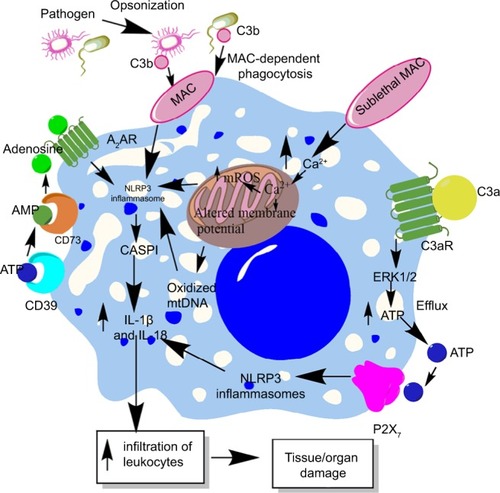
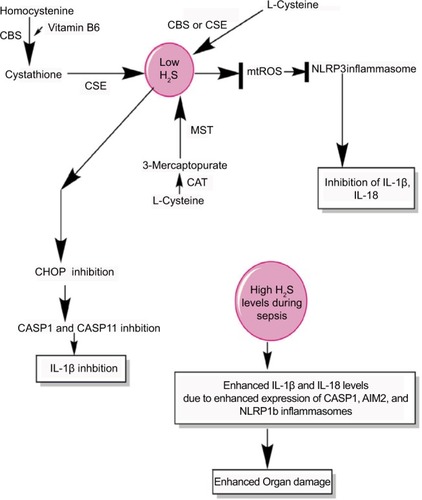
Figure 5 H2S production during sepsis and its impact on inflammasomes.
Notes: H2S is produced by the enzymatic action of CSE, CBS, CAT, and MST on the amino acids l-cysteine and homocysteine (for details, see wangCitation271) At a lower level, H2S inhibits NLRP3 inflammasome activation and the release of IL-1β and IL-18 via inhibiting the generation of mtROS and CHOP activity required for the activation of CASP1 and CASP11. However, at its higher levels observed during severe sepsis or septic shock, H2S exerts proinflammatory action via enhancing the levels of IL-1β and IL-18 through activating the CASP1, AIM2, and NLRP1b inflammasomes.
Abbreviation: mtROS, mitochondrial reactive oxygen species.
Intracellular CS components, that is, C3aR and C5aR1, are shown to regulate the Th1 (CD4+ T-cells) immune response via generating intracellular ROS and effectively upregulating NLRP3 inflammasome, causing the release of IL-1β and IFN-γ in response to the pathogen.Citation228,Citation229 Studies have indicated early death and an alteration in the function of CD4+ and CD8+ T-cells during sepsis.Citation16,Citation22,Citation230 Thus, it will be interesting to study the intracellular C5aR1 and C3aR and NLRP3 inflammasome in the T-cell population during early sepsis. The complement component C5a also plays an important role in the activation of NLRP3 inflammasomes and IL-1β release.Citation231 The cecal ligation and puncture (CLP) model of sepsis in C5aR1 or C5aR2 (C5L2) −/− mice showed decreased activation of NLRP3 inflammasome and decreased production of IL-1β.Citation231 Further study indicates an increased production of ROS (both cytosolic and mitochondrial) in cardiomyocytes upon challenging with recombinant C5a, an inducer of NLRP3 inflammasome.Citation231 The NLRP3−/− mice showed decreased defect in echo/Doppler parameters in the heart after CLP; thus, complement-mediated activation of NLRP3 inflammasome plays an important role in cardiac dysfunction observed during sepsis.Citation231
Furthermore, C3a component of CS also enhances the production of IL-1β from the macrophages and DCs treated with LPS via regulating the efflux of ATP and subsequent activation of NLRP3 ().Citation232 The binding of C3a to C3aR on macrophages induces ERK-1/2 phosphorylation which further promotes the efflux of ATP from the macrophages ().Citation232,Citation233 Thereafter, the binding of extracellular ATP to a P2X7 receptor or the conversion of ATP into adenosine via CD39 and CD73 and its binding to adenosine A2 receptors both cause activation of NLRP3 inflammasome and CASP1 during sepsis.Citation191,Citation200,Citation205 The C1q component of CS acts as a negative regulator of inflammasome activation during the uptake of apoptotic cells and directs macrophage polarization via decreasing the cleavage of proCASP1 to CASP1 required for the release of IL-1β.Citation234 Thus, the CS components influence inflammasome during sepsis in a wide variety of immune cells, and therapies targeting CS in a cell-specific manner have the potential to target sepsis via inflammasome modulation.
Autophagy, sepsis, and inflammasomes
Autophagy is considered one of the crucial innate immune mechanisms against infectious agents invading the host and plays an important role in inflammation.Citation235,Citation236 The major intracellular signaling mechanisms controlling the induction of autophagy comprise AMPK, JNK/p38 MAPK pathways generating ROS and regulating NF-κB transcription factor regulated by TLRs (ie, TLR4, TLR9, etc).Citation237–Citation239 The induction of autophagy during sepsis provides protection to the host against multiorgan dysfunction syndrome by preventing apoptotic cell death of immune cells, maintaining the homeostatic cytokine balance between the production of pro- and anti-inflammatory cytokines, and preserving mitochondrial functions,Citation240–Citation243 whereas any decline in the process of autophagy during sepsis is associated with an increased tissue and organ injury (ie, liver, brain, kidneys, etc).Citation244,Citation245 The polymorphisms of the autophagy-related loci in the IRGM gene are associated with high mortality in sepsis.Citation246 However, the induction of autophagy starts at the beginning of sepsis, but its flux is lost after a short duration of hyperactivity.Citation241,Citation247 Various strategies are used to restore the immune homeostasis via modulating the autophagy during sepsis.Citation248,Citation249 Thus, a regulated process of autophagy is essential to prevent exaggerated organ damage during sepsis.
It was found that loss of Atg16L1 protein required for the initiation of autophagy causes an increased production of IL-1β and IL-18 by the macrophages upon LPS challenge or during endotoxemia, due to overactivation of NLRP3 inflammasome and CASP1 in the presence of higher ROS and oxidized mtDNA.Citation225,Citation250–Citation252 Thus, increased autophagy prevents an upregulated inflammasome activity responsible for proinflammatory damage observed during sepsis. For example, the inflammasome components, including NLRP3, NLRP1, and pro-caspase 1, can also be degraded via the process of autophagy through TRIM20 in the macrophages stimulated with IFN-γ.Citation253 Autophagy also controls the release of IL-1β via targeting the degradation of pro-IL-1β as it sequesters into autophagosomes, and further increasing the process of autophagy via rapamycin (an mTOR inhibitor) caused its degradation and inhibited the release of mature IL-1β during endotoxemia.Citation254,Citation255 The inhibition of autophagy increased the release of IL-1β in an NLRP3 inflammasome-dependent manner due to increased generation of ROS.Citation254,Citation255 Also, activation of NLRP3 and NLRP4 inflammasomes inhibits autophagy in a beclin1- and PINK1-dependent manner during group A streptococcal infection responsible for sepsis in humans and hyperoxia that causes an increased ROS generation.Citation256–Citation258
An enhanced activation of NLRP3 inflammasome during Pseudomonas aeruginosa infection in human macrophages prevents their intracellular killing without affecting the generation of antimicrobial peptides, ROS, and NO.Citation259 This pathogen escape via activation of NLRP3 involves an increase in LC-3-II protein and the formation of autophagosomes leading to enhanced autophagy.Citation259 The deficiency of Atg7 during P. aeruginosa-induced abdominal infection causes defective bacterial clearance, increased mortality, bacteremia, and development of sepsis.Citation260 The deficiency of Atg7 causes an aggravated release of IL-1β and pyroptosis of macrophages due to the upregulation of NLRC4 inflammasome and its activity.Citation260 NLRP6 inflammasome deficiency was also found to cause defective autophagy among intestinal epithelial cells and goblet cells, making them more susceptible to persistent Citrobacter rodentium infection and their dysbiosis to peripheral circulation causing sepsis.Citation261 SESN2 or Sestrin 2, a stress-inducible protein, suppresses sepsis by inhibiting the NLRP3 inflammasome activation via inducing mitophagy through mitochondrial priming to clear damaged mitochondria in activated macrophages.Citation262 SESN2 promotes the perinuclear clustering of mitochondria via mediating the aggregation of SQSTM1 and its binding to Lys63-linked ubiquitins on the mitochondrial surface.Citation262 Thereafter, SESN2 activates the specific autophagic machinery to degrade the primed mitochondria through an increased expression of ULK1 protein, a mitophagy inducer. The increased SESN2 expression during sepsis/endotoxemia is further enhanced by NO production through iNOS2 in the macrophages. Thus, NO production during sepsis exerts an inhibitory action of NLRP3 inflammasomes in both human and mice myeloid immune cells via induction of SESN2.Citation262,Citation263 SESN2−/− mice subjected to sepsis exhibit defective mitophagy which causes increased activation of inflammasomes and enhanced mortality.Citation262 Additionally, RIPK2 was also found to exert a negative impact on NLRP3 inflammasome activation during acute infections via inducing autophagy of mitochondria (mitophagy) in a kinase-dependent manner by phosphorylating ULK1 protein and reducing CASP1 activation.Citation264 Thus, autophagy and inflammasome activation are interrelated processes playing an important role in the pathogenesis of sepsis and its outcome.Citation265,Citation266 For example, autophagy inhibits inflammasome activation and inflammasome activation inhibits autophagy.Citation267–Citation269 Therefore, therapeutics enhancing autophagy may indirectly decrease overactivation of the inflammatory function of inflammasomes or inhibiting inflammasomes during sepsis may enhance autophagy to maintain the immune homeostasis during sepsis. This area needs an urgent research priority to target sepsis.
H2S, sepsis, and inflammasomes
H2S is a gaseous transmitter/gasotransmitter that is produced endogenously in many cell types, including the macrophages and endothelial cells, from cysteine due to the activity of enzymes called CBS, CSE, CAT, and MST.Citation270,Citation271 H2S plays an important role in the modulation of cardiovascular function including hypertension and ischemia–reperfusion injury.Citation272,Citation273 For example, H2S protects from ischemia–reperfusion injury via preserving the structure and function of myocardial mitochondria and decreasing the neutrophil infiltration, IL-1β secretion, and cellular necrosis.Citation272 However, the cardiac dysfunction is a major problem in sepsis patients who had undergone septic shock.Citation274–Citation278 H2S is shown to protect the mitochondrial function via inhibiting the generation of mtROS that activates the NLPR3 inflammasomeCitation279 (). The decrease in mtROS by H2S involves S-sulfhydration of c-Jun that enhances its transcriptional activity of SIRT3 and p62. The H2S-mediated anti-inflammatory action in both murine and human macrophages involves the inhibition of NLPR3 inflammasome via the inhibition mtROS production and ASC oligomerization in vitro and in a mouse model of peritonitisCitation280 (). H2S also inhibits the TLR4/NF-κB activation-mediated inflammatory pathway, along with reducing the expression of NLRP3, ASC, pro-caspase-1, CASP-1, IL-1β, IL-18, and CASP-3 that is regulated by TLR4 signaling.Citation281 Thus, H2S exerts its protective action via inhibiting NLRP3 inflammasome-mediated proinflammatory action.
In animal model of sepsis, the inhibition of H2S or its activation is proven beneficial to the hosts in terms of their survival and reduced tissue/organ injury, including myocardial injury, via improving neutrophil infiltration, K+ATP channel activation, reducing TNF-α, and increasing IL-10 levels.Citation282–Citation284 The CLP-induced sepsis in mice causes a significant increase in the plasma level of H2S along with its synthesis in the liver 8 hours post-CLP.Citation282 Thus, it can be inferred that the beneficial and harmful effects of H2S depend on its concentration and duration or stage of sepsis. The beneficial effect of H2S during sepsis depends on the inhibition of CHOP, which is a transcription factor and mediates the unfolded protein response that plays a major role in the induction of apoptosis.Citation285–Citation287 CHOP is also involved in the generation of IL-1β during endotoxemia or sepsis via regulating the induction of CASP11 that is involved in the maturation of pro-IL-1β via CASP1.Citation288,Citation289 Thus, CHOP inhibition via H2S is also another mechanism of inflammasome inhibition during sepsis to prevent exaggerated inflammation and organ damage. Hence, H2S inhibits NLRP3 activation via inhibiting mtROS generation and CHOP. However, at later stages of sepsis including septic shock, its circulating levels are increased due to the hepatic dysfunction causing an impaired oxygen (O2) availability to the tissues.Citation290–Citation292 The acute exposure of higher amount of H2S causes an increased expression of IL-18, MyD88, and TLR9 in addition to a decreased expression of CCL12 and CXCR4.Citation293 Furthermore, this acute expression of H2S increases the expression of genes associated with inflammasome, including IL-1β, CASP-1, and AIM2 (). However, NLRP3 expression was decreased, but NLRP1b was upregulated upon H2S treatment.Citation293 H2S has a great potential to act as a prognostic marker of septic shock and its severity level becomes higher in septic patients within 12 hours of onset of sepsis.Citation294,Citation295 Even H2S can be detected in the exhaled pulmonary gases of patients with sepsis.Citation296 Thus, H2S negatively regulates NLRP3 at all concentrations, but increases the proinflammatory action of NLRP1b inflammasome at its higher level, aggravating the sepsis-associated organ damage. Thus, H2S acts as a novel gasotransmitter regulating the function of inflammasomes during sepsis at all levels. Future studies will prove beneficial in this direction as H2S donors have therapeutic potential for both acute and chronic inflammatory diseases.Citation297
Prostanoids, eicosanoids, inflammasomes, and sepsis
Prostanoids are lipid mediators synthesized from a 20-carbon unsaturated fatty acid called arachidonic acid by the action of enzymes called cyclooxygenases (COX-1 and COX-2) or PG G/H synthase.Citation298,Citation299 COX-1 plays an important role in the generation of prostanoids during homeostasis, while COX-2 is induced by proinflammatory cytokines, shear stress, and tumor promoters and serves as a major source for prostanoid biosynthesis during inflammation.Citation299 Sepsis is shown to alter vasoconstriction via modulating prostanoids’ levels ().Citation300 The generation of prostanoids is also regulated by IL-1β generated due to the activation of inflammasomes, and mice lacking COX-2 are defective in producing IL-18 cytokine.Citation69,Citation301 Bacterial flagellin or anthrax lethal toxin-mediated systemic activation of NAIP5/NLRC4 inflammasomes causes dysregulated production of eicosanoids called eicosanoid storm (a profound or pathological release of signaling lipids, including PGs and leukotrienes, causing a rapid initiation of inflammation and vascular fluid loss), which proves highly lethal to the host due to the rapid loss of vascular fluid into the intestine and peritoneal cavity ().Citation302,Citation303 The eicosanoid generation due to the inflammasome activation occurs due to the activation of cytosolic PLA2 in the peritoneal macrophages.Citation303 The NLRP3 inflammasome activation is also shown to activate leukotriene B4 that further activates PGE2 via IL-1β/IL-1R signaling.Citation304 However, an exogenous administration of leukotriene B4 inhibits NLRP3 inflammasome and IL-1β release.Citation304 Furthermore, 15-deoxy-ΔCitation12,Citation14-PGJ2 and related cyclopentenone PGs inhibit CASP1 activation via inhibiting NLRP1 and NLRP3 inflammasome activation through preventing the autoproteolytic cleavage of CASP-1 and, thus, the maturation of IL-1β through the induction of a cellular state inhibitory to CASP-1 proteolytic function.Citation305 Thus, cyclopentenone PGs acts as anti-inflammatory molecules via inhibiting CASP1, NLRP1, and NLRP3 inflammasomes. The PGE2 via PGE2 receptor E-prostanoid 4 enhances the activity of PKA that directly inhibits NLRP3 inflammasome activation via direct phosphorylation of cytoplasmic NLRP3 at Ser295 and inhibits its ATPase activity ().Citation306 Additionally, PGE2 via E-prostanoid 4 also increases the intracellular levels of cAMP which further decreases NLPR3 inflammasome activation.Citation307 Thus, eicosanoids both inhibit and enhance inflammasome activation and this mechanism needs to be identified clearly.
Figure 6 Schematic representation of the relationship between prostanoids, eicosanoids, sepsis, and inflammasomes.
Notes: Overactivation of inflammasomes is seen in sepsis, which also exhibits overproduction of prostanoids and eicosanoids. These prostanoids and eicosanoids exert their impact on vascular tone and vascular contractility and induce vascular leakage. Additionally, at the immune cell level, a prostanoid called PGE2 causes induction of PKA via acting through EP4 receptors expressed on the macrophages, which directly inhibits NLRP3 inflammasome by binding to Ser295. Also, PGE2 activates RIPK2 via EP3, which activates MAP kinases and NF-κB causing activation of NLRC4 inflammasomes. The activation of NLRC4 inflammasome increases the systemic levels of IL-18 and IL-1β cytokines, while PGD2, via acting through DP1, induces the production of PYDC3 protein that directly inhibits NLRP3 inflammasome overactivation. See text for detail.
Abbreviations: 5,14-HEDGE, N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine; 20-HETE, 20-hydroxyeicosatetraenoic acid; DP1, D-prostanoid receptor 1; PG, prostaglandin.
![Figure 6 Schematic representation of the relationship between prostanoids, eicosanoids, sepsis, and inflammasomes.Notes: Overactivation of inflammasomes is seen in sepsis, which also exhibits overproduction of prostanoids and eicosanoids. These prostanoids and eicosanoids exert their impact on vascular tone and vascular contractility and induce vascular leakage. Additionally, at the immune cell level, a prostanoid called PGE2 causes induction of PKA via acting through EP4 receptors expressed on the macrophages, which directly inhibits NLRP3 inflammasome by binding to Ser295. Also, PGE2 activates RIPK2 via EP3, which activates MAP kinases and NF-κB causing activation of NLRC4 inflammasomes. The activation of NLRC4 inflammasome increases the systemic levels of IL-18 and IL-1β cytokines, while PGD2, via acting through DP1, induces the production of PYDC3 protein that directly inhibits NLRP3 inflammasome overactivation. See text for detail.Abbreviations: 5,14-HEDGE, N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine; 20-HETE, 20-hydroxyeicosatetraenoic acid; DP1, D-prostanoid receptor 1; PG, prostaglandin.](/cms/asset/ee925bbc-f3a6-4579-91ad-ae7d5c1ec8d4/djir_a_178084_f0006_c.jpg)
The PG E2-E type PG receptor 3 (PGE2-EP3) signaling is shown to activate NLRC4 inflammasome and the secretion of IL-1β and IL-18 via RIPK2 that activates NF-κB signaling and MAP kinases upon infection.Citation301,Citation308 In addition, the COX-2-dependent generation of prostanoids in endothelial cells (ECs) plays an important role in the vascular functions of ECs, which gets dysregulated during sepsis and results in septic shock and organ damage.Citation309 Thus, it will be interesting to delineate the relationship between inflammasome activation and prostanoid generation during sepsis to develop future therapeutic to prevent vascular dysfunction or vascular leakage seen during sepsis. Inhibition of prostanoids by 20-hydroxyeicosatetraenoic acid (an ω-hydroxylation product of arachidonic acid that is produced by CYP/CYP450 [CYP] enzymes, mainly by the CYP4A and CYP4F isoforms, in the kidney, heart, liver, brain, lung, and vasculature) and N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine during sepsis is shown to inhibit vasodilation, hypotension, tachycardia, and inflammation in a rat model of septic shock.Citation310,Citation311 This can be attributed to the inhibition of NLRP3 and NLRC4 inflammasome also and needs to be investigated. Another study has indicated the negative effect of PG D2 on inflammasome activation during certain infections via D-prostanoid receptor 1 that stimulates the induction of a putative inflammasome inhibitor called PYDC3 in CD11b+ myeloid cells including monocytes/macrophages and microglial cells ().Citation312 PYDC3 acts as PYD only protein lacking other ligand-binding domains and inhibits the process of inflammasome nucleation via direct binding.Citation312 In addition to PYDC3, POP1 and POP3 (pyrin domain-only proteins) also act as negative regulators of NLRP3 and ALR inflammasomes and prevent their overactivation.Citation313,Citation314 POP1 inhibits ASC-dependent inflammasome assembly through prevention of inflammasome nucleation, which interferes with CASP-1 activation, IL-1β and IL-18 release, pyroptosis, and the release of ASC particles.Citation313 POP1 expression is regulated by other inflammatory signals mediated by TLR and IL-1R signaling that also affect prostanoid or PG synthesis. Thus, it will be interesting to observe the impact of prostanoids on POP1 and POP3 expression during sepsis. It can be suggested that inflammasomes and prostanoids impact each other in both directions and the regulation of one affects the other. The relationship between inflammasomes and prostanoids needs to be understood for forming better therapeutics to manage sepsis.
Immunosuppression during sepsis and inflammasomes
Sepsis-associated immunosuppression/immunoparalysis proves detrimental to the host and makes it prone to develop secondary infections even after full recovery.Citation8,Citation23,Citation315 This is because it lasts for longer duration due to severe damage acquired by the host immune system, including paralyzed DCs and macrophages.Citation316,Citation317 Activation of inflammasomes is a major regulator of the release of proinflammatory cytokines IL-1β and IL-18 and the induction of death of macrophages and DCs via pyroptosis or necroptosis. However, the death of these immune cells contributes to the development of immunosuppression/immunoparalysis observed during the late stage of sepsis.Citation8 Additionally, conventional T-cells (CD4+, CD8+, and CD4+CD8+ T-cells) are also lost during early sepsis and further aggravate the immunoparalysis.Citation318 Also, an upregulation and profound activation of Tregs at the later stages of sepsis is the major contributor to the development of immunosuppression.Citation319,Citation320 A recent study has identified the existence of NLRP3 inflammasome and its activation for the normal Th1 immune response mediated by CD4+ T-cells.Citation229 Another study has indicated the role of NLRP3 inflammasome activation in the migration of CD4+ T-cells to the distant organs due to the upregulation of chemotactic proteins including osteopontin, CCR2, and CXCR6.Citation124 Thus, it would be interesting to investigate the role of the inflammasome in T-cell function, migration, and their death during sepsis.
Another mechanism that can contribute to the sepsis-associated immunosuppression involves the Foxp3− type one regulatory (Tr1) cells which suppress NLRP3 activation via an IL-10–dependent mechanism.Citation321 The activation of Tr1 cells causes the release of IL-10 that, in turn, inhibits the release of maturation of pro-IL-1β, release of mature IL-1β, and inflammasome-mediated activation of CASP1 in macrophages stimulated with LPS and ATP.Citation321 It should be noted that IL-10 is a major immunosuppressive cytokine that gets elevated during sepsis.Citation322–Citation325 Also, a higher level of IL-10 is shown to regulate the transition from early reversible sepsis to the late phase of irreversible septic shock in laboratory animals.Citation326 Thus, inflammasomes play an essential role during both the stages of immune response (ie, exaggerated activation of immune response and immunosuppression) observed during sepsis.
Therapeutic targeting of inflammasomes to manage sepsis
Inflammasomes play an important role in inflammation via regulating the initiation of the inflammatory cascade through regulating the release of IL-1β and IL-18 cytokines, immune cell (ie, neutrophils, monocytes etc) chemotaxis and infiltration, and direct recognition of cytosolic pathogens and their PAMPs. Thus, molecules targeting inflammasomes will prove beneficial to control the exaggerated systemic inflammation and inflammatory organ damage during sepsis. In addition to increasing the release of IL-1β and IL-18 cytokines via activating CASP1, the NLRP3/ASC inflammasome complex inhibits the maturation and release of IL-33 cytokine.Citation327,Citation328 Thus, IL-33 levels are decreased in the circulation during sepsis. IL-33 works by binding to IL1RL1 or ST2 that belongs to IL-1 superfamily.Citation329,Citation330 The binding of IL-33 to membrane-bound ST2 initiates MyD88/NF-κB signaling which increases the mast cell function and Th2- and Treg-mediated immune response.Citation330,Citation331 However, this protective action of IL-33 is lost due to the inhibition of its maturation and release by NLRP3 inflammasome-mediated activation of CASP1 that inactivates IL-33. This is because activation of mast cells causes prompt release of TNF-α due to their property of storing presynthesized cytokine during acute bacterial peritonitis.Citation332 In addition to a decrease in circulating IL-33 during sepsis, the serum levels of soluble ST2 also increases, which acts as decoy receptor for IL-33 that further lowers IL-33 levels by sequestering it.Citation330,Citation333–Citation335 Exogenous administration of soluble ST2 in septic mice leads to decreased IL-6 and TNF-α levels,Citation336 which further indicates that the IL-33–mediated activation of mast cells is inhibited giving an initial proinflammatory signal via releasing presynthesized TNF-α and IL-6. IL-33 cytokine therapy in a mouse model of sepsis exhibits protective action.Citation337 IL-33–treated animals exhibited increased neutrophil infiltration that is required to kill the pathogens efficiently when their number can be easily controlled. This can be explained as the IL-33–mediated activation of peritoneal mast cells that initiate the very first signal for neutrophil infiltration via releasing the presynthesized TNF-α and IL-6.Citation332,Citation338 Furthermore, IL-33 treatment decreased systemic inflammation, leaving the local proinflammatory response intact, which is required to kill the pathogen at its niche acting as a source for bacteremia and, thus, the sepsis. The increased neutrophil infiltration into the peritoneum can also be expected due to the upregulation of CXCR2 on neutrophils as a result of inhibition of GRK2 expression.Citation337 GRK2 is a serine–threonine protein kinase that induces internalization of chemokine receptors. However, it does not activate an immunologic shift from a Th1 immune response to a Th2 immune response.Citation337 The activation of peritoneal mast cells during bacterial peritonitis inhibits the phagocytic capacity of peritoneal macrophages via releasing prestored IL-4.Citation339 Thus, it will be interesting to observe the effect of IL-33 treatment on the levels of IL-4.
Various phytochemicals and chemicals (MCC950) have been identified and developed that target inflammasomes during diverse sterile inflammatory conditions mentioned.Citation340–Citation342 For example, MCC950 is shown to control the endothelial overexpression of adhesion molecules during the inflammatory process observed during sepsis.Citation136 Furthermore, MCC950 regulates inflammation via targeting both NLRP3-dependent canonical CASP1 and non-canonical activation of CASP11 required for IL-1β release and pyroptosis during endotoxemia in mice.Citation343 Thus, this molecule has a great potential to be used during sepsis for controlling severe systemic inflammation and inflammatory organ damage. In addition, a ketone body called β-hydroxybutyrate is also shown to inhibit NLRP3 inflammasome via inhibiting K+ efflux and ASC oligomerization and prevent the synthesis and release of proinflammatory cytokines IL-1β and IL-18.Citation344 Ketone bodies are small lipid-derived molecules that serve as a circulating energy source for tissues during shortage of energy, that is, due to fasting or prolonged exercise.Citation345,Citation346 An altered ketone body metabolism and changes in their levels in the systemic circulation due to its impaired synthesis by the liver during sepsis are observed.Citation347–Citation349 These ketone bodies exert anti-inflammatory action under diverse inflammatory conditions via different mechanisms including targeting and modulating HDACs and activating GPCRs and PPAR-α.Citation346
PPARs are nuclear receptors that have three isoforms (ie, PPAR-α, PPAR-β, and PPAR-γ) and play an active role in various cellular processes (ie, lipid metabolism, glucose homeostasis, implantation, embryonic development, bone formation, adipogenesis, insulin sensitivity, inflammation, etc).Citation350 The activation of PPAR-α and PPAR-γ via drugs such as fenofibrate and pioglitazone (PIO) has been found to inhibit NLRP3 inflammasome and CASP1 activation under diverse inflammatory conditions.Citation351,Citation352 Previously, it was found that the activation of PPAR-γ with rosiglitazone and PIO during sepsis prevented cardiac dysfunction and mortality, along with increasing bacterial clearance.Citation353–Citation356 A reduced expression of PPAR-α is associated with decreased survival and increased bacterial load in a polymicrobial sepsis model.Citation357 This can be attributed to the decreased activation of NLRP3 inflammasome and CASP1 required for IL-1β and clearance of intracellular bacteria.Citation357 Thus, β-hydroxybutyrate analogs, MCC950, and PPAR agonists including fenofibrate and PIO have a novel potential to be used during sepsis to prevent inflammatory organ damage and mortality.
Thalidomide has been found to exhibit protective action in a mouse model of sepsis and pneumonia due to its immunomodulatory action alone or in combination with antibiotics.Citation358–Citation361 A recent study has also shown that thalidomide exerts its immunomodulatory action by inhibiting NLRP3 inflammasome and CASP1 required to release IL-1β and IL-18.Citation362 Thus, thalidomide is a broad-spectrum immunomodulatory agent that also inhibits inflammation via targeting NLRP3 inflammasome and CASP1 activation. Another molecule called cytokine release inhibitory drug 3 (CP-456773) blocks ASC oligomerization, a crucial step required for the activation of canonical inflammasome signaling pathway, and is also found to exert immunomodulatory action.Citation363,Citation364 Recently, a new molecule called OLT1177 (β-sulfonyl nitrile compound) was found to be orally active in inhibiting NLRP3 inflammasome oligomerization and activation through inhibiting the interaction between NLRP3 and ASC and the interaction between NLRP3 and CASP1.Citation365 Treatment of a mouse model of endotoxemia/sepsis with the compound caused a significant decrease in IL-1β and IL-18 levels in tissue homogenates, oxidative stress, and an enhanced oxidative metabolism in the muscle tissues.Citation365 Interestingly, this compound is considered safe in humans.
Shikonin is a highly lipophilic naphthoquinone derivative derived from the roots of the plant called Lithospermum erythrorhizon belonging to the Boraginaceae family.Citation366 Shikonin and its derivatives are the major active components in the Chinese traditional medicine called Zicao which is used due to its antibacterial, antiviral, anti-inflammatory, and antipyretic action. A study by the authors has shown that shikonin inhibits the NLRP3 inflammasome-dependent maturation of proIL-1β as well as AIM2 inflammasome.Citation366 Thus, it inhibits the process of ASC oligomerization along with directly inhibiting the activation of CASP1. Another plant extract derived from the plant Artemisia princeps of the Asteraceae family has also shown NLRP3 and AIM2 inhibitory action via reducing ASC phosphorylation, its oligomerization, and speck formation required for the activation of canonical inflammasome signaling.Citation367,Citation368 Treatment of animals with A. princeps extract also reduced the levels of the proinflammatory cytokines including IL-1β in a mouse model of peritonitis.Citation368 Additionally, another naturally occurring compound known as sulforaphane isolated from the vegetables of Brassicaceae or Cruciferae was also found to inhibit NLRC4 and NLRP3 inflammasomes without affecting AIM2 in a mouse model of peritonitis.Citation369 Sulforaphane directly inhibits the expression of NLRP3 gene and proIL-1β during the priming step and further inhibits the secretion of IL-1β and mtROS generation.Citation369 Thus, the continuous development in molecular biology and immunology tools along with the advances in pharmacology and natural chemistry have led to the development and identification of novel inflammasome inhibitors with negligible toxic effects. Hence, molecules directly targeting inflammasomes can be ascribed as future therapeutic agents to target sepsis-associated inflammation and inflammatory organ damage, alone or in combination with other potential drugs and antibiotics. Inflammasome targeting comprises an area of sepsis research with great therapeutic potential.
Future perspective
Sepsis is a disease of dysregulation of immune response against an infection that can be easily contained in humans with normal immune response, but not in newborns or older group of people or persons having some chronic diseases (ie, type 2 diabetes mellitus [T2DM], obesity, etc) where it impacts the effectiveness of the innate immune response.Citation370–Citation374 Both diabetes and obesity are also associated with impaired functions of inflammasomes. For example, an overactivated NLRP3 inflammasome in the immune cells is associated with both obesity and insulin resistance and T2DM.Citation375–Citation380 Thus, one can speculate that pre-existing dysregulated inflammasome function may predispose these persons to develop sepsis from an infection that can be easily taken care of in otherwise healthy persons. One reason can be decreased autophagy during obesity and T2DM that has been observed in both conditions.Citation381–Citation385 Even the activation of FXR or BAR is shown to negatively regulate NLRP3 inflammasome and CASP1 activation via physical interaction during cholestasis-associated sepsis.Citation386 The targeting of FXR (Fexaramine) also has a great potential to target obesity that have potential to impact NLRP3.Citation387 Thus, these FXR agonists can be used to target NLRP3 inflammasome due to the negative effect of FXR activation on NLRP3 inflammasome and CASP1. For example, GW4064 is a molecular agonist for FXR and inhibits NLRP3 inflammasome in FXR-independent manner.Citation388 GW4064 inhibits the oligomerization and ubiquitination of ASC, a critical step for NLRP3 inflammasome activation, and partially protects from the symptoms of NLRP3-dependent inflammatory diseases including sepsis in animal models. Hence, targeting specific inflammasomes, FXR, and autophagy during sepsis can be a future patient-based specific therapeutic approach. In addition to autophagy, other specific molecules or mediators released during early or late stages (ie, C5a, C3a, adenosine, ROS, HMGB1, H2S, prostanoids, eicosanoids, etc) of sepsis have great potential to impact inflammasome function and vice versa during the pathogenesis of sepsis.
Inflammasomes were first discovered by Dr Jürg Tschopp in the year 2002 to identify the LPS (responsible for endotoxemia and endotoxic shock during gram-negative bacterial sepsis)-mediated activation of CASP1 as an inflammatory complex playing a role in the release of IL-1β. Since their discovery, inflammasomes have been studied in detail in various chronic sterile inflammatory conditions including different tumors, neurodegenerative diseases, atherosclerosis, and autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, juvenile idiopathic arthritis, experimental autoimmune encephalomyelitis, and multiple sclerosis in humans. But continuous research in the field targeting host–pathogen interaction has also established them as potent intracellular PRRs for a wide variety of pathogens that can lead to the development of sepsis if remained unchecked without an effective clearance. Thus, a perfect time has come where we are equipped with advanced molecular biology, omics, and immunology tools for closing the Pandora’s box of inflammasomes to understand more precisely their role in the immunopathogenesis of sepsis and its targeting. For example, NLRP3 inflammasome-deficient mice show resistance toward polymicrobial sepsis due to increased production of proresolving lipid mediator lipoxin B4 that exerts anti-inflammatory and anti-pyroptotic effects.Citation389 Future studies in the direction are immediately required as targeting inflammasomes has a novel potential to serve as a panacea for sepsis.
Disclosure
The author reports no conflicts of interest in this work.
References
- GeroulanosSDoukaETHistorical perspective of the word “sepsis”Intensive Care Med200632207717131165
- FunkDJParrilloJEKumarASepsis and septic shock: a historyCrit Care Clin2009258310119268796
- RittirschDFlierlMAWardPAHarmful molecular mechanisms in sepsisNat Rev Immunol200881077678718802444
- BudelmannGHugo Schottmuller, 1867–1936. The problem of sepsisInternist196910921014903434
- BoneRCDefinitions for sepsis and organ failureCrit Care Med1992207247261597021
- BoneRCBalkRACerraFBDefinitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care MedicineChest1992101164416551303622
- SingerMDeutschmanCSSeymourCWThe third international consensus definitions for sepsis and septic shock (sepsis-3)JAMA2016315880181026903338
- HotchkissRSMonneretGPayenDImmunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approachLancet Infect Dis2013a1326026823427891
- HotchkissRSMonneretGPayenDSepsis-induced immunosuppression: from cellular dysfunctions to immunotherapyNat Rev Immunol2013131286287424232462
- HotchkissRSMoldawerLLOpalSMReinhartKTurnbullIRVincentJLSepsis and septic shockNat Rev Dis Primers201621604528117397
- GülFArslantaşMKCinelİKumarAChanging definitions of sepsisTurk J Anaesthesiol Reanim20174512913828752002
- MartinGSSepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomesExpert Rev Anti Infect Ther201210670170622734959
- AngusDCvan der PollTSevere sepsis and septic shockN Engl J Med201336984085123984731
- CollingKPBantonKLBeilmanGJVasopressors in sepsisSurg Infect (Larchmt)20181920220729336676
- Stearns-KurosawaDJOsuchowskiMFValentineCKurosawaSRemickDGThe pathogenesis of sepsisAnnu Rev Pathol20116194820887193
- Cabrera-PerezJCondottaSABadovinacVPGriffithTSImpact of sepsis on CD4 T cell immunityJ Leukoc Biol20149676777724791959
- WiersingaWJLeopoldSJCranendonkDRvan der PollTHost innate immune responses to sepsisVirulence201451364423774844
- CaoCMaTChaiYFShouSTThe role of regulatory T cells in immune dysfunction during sepsisWorld J Emerg Med2015615925802559
- DelanoMJWardPAThe immune system’s role in sepsis progression, resolution, and long-term outcomeImmunol Rev2016274133035327782333
- DelanoMJWardPASepsis-induced immune dysfunction: can immune therapies reduce mortality?J Clin Invest20161261233126727230
- van der PollTvan de VeerdonkFLSciclunaBPNeteaMGThe immunopathology of sepsis and potential therapeutic targetsNat Rev Immunol201717740742028436424
- KumarVT cells and their immunometabolism: a novel way to understanding sepsis immunopathogenesis and future therapeuticsEur J Cell Biol2018a
- SundarKMSiresMSepsis induced immunosuppression: implications for secondary infections and complicationsIndian J Crit Care Med20131716216924082613
- DanahyDBStrotherRKBadovinacVPGriffithTSClinical and experimental sepsis impairs CD8 T-cell-mediated immunityCrit Rev Immunol201636577427480902
- LamkanfiMDixitVMInflammasomes: guardians of cytosolic sanctityImmunol Rev20092279510519120479
- FranchiLMunoz-PlanilloRReimerTEigenbrodTNunezGInflammasomes as microbial sensorsEur J Immunol20104061161520201013
- LamkanfiMvande WalleLKannegantiTDDeregulated inflammasome signaling in diseaseImmunol Rev201124316317321884175
- FranchiLMunoz-PlanilloRNunezGSensing and reacting to microbes through the inflammasomesNat Immunol20121332533222430785
- ZmoraNLevyMPevsner-FishcerMElinavEInflammasomes and intestinal inflammationMucosal Immunol20171086588328401932
- AlhallafRAghaZMillerCMThe NLRP3 inflammasome suppresses protective immunity to gastrointestinal helminth infectionCell Rep2018231085109829694887
- KayagakiNWarmingSLamkanfiMNon-canonical inflammasome activation targets caspase-11Nature201147911712122002608
- BrozPMonackDMNoncanonical inflammasomes: caspase-11 activation and effector mechanismsPLoS Pathog20139e100314423468620
- LatzEXiaoTSStutzAActivation and regulation of the inflammasomesNat Rev Immunol201313639741123702978
- von MoltkeJAyresJSKofoedEMChavarría-SmithJVanceRERecognition of bacteria by inflammasomesAnnu Rev Immunol2013317310623215645
- MonieTPThe canonical inflammasome: a macromolecular complex driving inflammationSubcell Biochem201783437328271472
- PellegriniCAntonioliLLopez-CastejonGBlandizziCFornaiMCanonical and non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammationFront Immunol2017883628179906
- PróchnickiTLatzEInflammasomes on the crossroads of innate immune recognition and metabolic controlCell Metab2017261719328683296
- KerurNFukudaSBanerjeeDcGAS drives noncanonical-inflammasome activation in age-related macular degenerationNat Med201824506129176737
- MartinonFBurnsKTschoppJThe inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-betaMol Cell20021041742612191486
- SinghalGJaehneEJCorriganFTobenCBauneBTInflammasomes in neuroinflammation and changes in brain function: a focused reviewFront Neurosci201483
- FreemanLCTingJPThe pathogenic role of the inflammasome in neurodegenerative diseasesJ Neurochem2016136Suppl 1293826119245
- GuoBFuSZhangJLiuBLiZTargeting inflammasome/IL-1 pathways for cancer immunotherapySci Rep201663610727786298
- NikolettLDavidBÁdámDInflammasomes link vascular disease with neuroinflammation and brain disordersJ Cereb Blood Flow Metab2016361668168527486046
- SaresellaMLa RosaFPianconeFThe NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s diseaseMol Neurodegener20161112326939933
- KantonoMGuoBInflammasomes and cancer: the dynamic role of the inflammasome in tumor developmentFront Immunol20178113228955343
- LinCZhangJInflammasomes in inflammation-induced cancerFront Immunol20178Pt 127128360909
- MamikMKPowerCInflammasomes in neurological diseases: emerging pathogenic and therapeutic conceptsBrain201714092273228529050380
- ElinavEThaissCAFlavellRAAnalysis of microbiota alterations in inflammasome-deficient miceMethods Mol Biol2013104018519423852605
- SesterDPSagulenkoVThygesenSJDeficient NLRP3 and AIM2 inflammasome function in autoimmune NZB miceJ Immunol201519531233124126116505
- ThygesenSJSesterDPCridlandSOWiltonSDStaceyKJCorrecting the NLRP3 inflammasome deficiency in macrophages from autoimmune NZB mice with exon skipping antisense oligonucleotidesImmunol Cell Biol201694552026833024
- KimYMBrinkmannMMPaquetMEPloeghHLUNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomesNature200845223423818305481
- MogensenTHPathogen recognition and inflammatory signaling in innate immune defensesClin Microbiol Rev200922224027319366914
- TakeuchiOAkiraSPattern recognition receptors and inflammationCell2010140680582020303872
- WangYYangCMaoKChenSMengGSunBCellular localization of NLRP3 inflammasomeProtein Cell20134642543123609011
- LamkanfiMDixitVMMechanisms and functions of inflammasomesCell20141571013102224855941
- ManSMKannegantiT-DRegulation of inflammasome activationImmunol Rev2015265162125879280
- de ZoeteMRPalmNWZhuSFlavellRAInflammasomesCold Spring Harb Perspect Biol20146a01628725324215
- RathinamVAKFitzgeraldKAinflammasome complexes: emerging mechanisms and effector functionsCell2016165479280027153493
- Lopez-CastejonGBroughDUnderstanding the mechanism of IL-1β secretionCytokine Growth Factor Rev201122418919522019906
- KangSFernandes-AlnemriTRogersCCaspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3Nat Commun20156751526104484
- ZhangXFanCZhangHMLKL and FADD are critical for suppressing progressive lymphoproliferative disease and activating the NLRP3 inflammasomeCell Rep201616123247325927498868
- ConosSAChenKWde NardoDActive MLKL triggers the NLRP3 inflammasome in a cell-intrinsic mannerProc Natl Acad Sci U S A2017114E961e96928096356
- GutierrezKDDavisMADanielsBPMLKL activation triggers NLRP3-mediated processing and release of IL-1beta independently of gasdermin-DJ Immunol20171982156216428130493
- WeinlichROberstABeereHMGreenDRNecroptosis in development, inflammation and diseaseNat Rev Mol Cell Biol201718212713627999438
- SharmaAAJenRKanBImpaired NLRP3 inflammasome activity during fetal development regulates IL-1β production in human monocytesEur J Immunol201545123824925311115
- StouchANMccoyAMGreerRMIL-1β and inflammasome activity link inflammation to abnormal fetal airway developmentJ Immunol201619683411342026951798
- Arango DuqueGDescoteauxAMacrophage cytokines: involvement in immunity and infectious diseasesFront Immunol2014549125339958
- DinarelloCACannonJGMierJWMultiple biological activities of human recombinant interleukin 1J Clin Invest198677173417393519678
- RadekeHHMartinMTopleyNKaeverVReschKDifferential biological activities of human interleukin-1 alpha and interleukin-1 betaEur Cytokine Netw1991251591873492
- ReznikovLLKimS-HWestcottJYIL-18 binding protein increases spontaneous and IL-1-induced prostaglandin production via inhibition of IFN-gammaProc Natl Acad Sci U S A20009752174217910681439
- LeeJKKimSHLewisECAzamTReznikovLLDinarelloCADifferences in signaling pathways by IL-1beta and IL-18Proc Natl Acad Sci U S A20041018815882015161979
- KeyelPAHow is inflammation initiated? Individual influences of IL-1, IL-18 and HMGB1Cytokine20146913614524746243
- GattiSBeckJFantuzziGBartfaiTDinarelloCAEffect of interleukin-18 on mouse core body temperatureAm J Physiol Regul Integr Comp Physiol2002282R70270911832389
- Komai-KomaMGracieJAWeiX-QChemoattraction of human T cells by IL-18J Immunol20031701084109012517977
- GutcherIBecherBAPC-derived cytokines and T cell polarization in autoimmune inflammationJ Clin Invest20071171119112717476341
- NeteaMGJoostenLABLewisEDeficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistanceNat Med200612665065616732281
- ZorrillaEPSanchez-AlavezMSugamaSInterleukin-18 controls energy homeostasis by suppressing appetite and feed efficiencyProc Natl Acad Sci U S A2007104110971110217578927
- GaglianiNPalmNWde ZoeteMRFlavellRAInflammasomes and intestinal homeostasis: regulating and connecting infection, inflammation and the microbiotaInt Immunol20142649549924948595
- RathinamVAKChanFK-MInflammasome, inflammation, and tissue homeostasisTrends Mol Med201824330431829433944
- ElinavEStrowigTKauALNLRP6 inflammasome regulates colonic microbial ecology and risk for colitisCell201114574575721565393
- LevyMThaissCAZeeviDMicrobiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signalingCell20151631428144326638072
- LoboLABenjamimCFOliveiraACThe interplay between micro-biota and inflammation: lessons from peritonitis and sepsisClin Transl Immunology201657e9027525063
- Cabrera-PerezJBadovinacVPGriffithTSEnteric immunity, the gut microbiome, and sepsis: rethinking the germ theory of diseaseExp Biol Med (Maywood)201724212713927633573
- Lei-LestonACMurphyAGMaloyKJEpithelial cell inflammasomes in intestinal immunity and inflammationFront Immunol20178
- MedzhitovRPattern recognition theory and the launch of modern innate immunityJ Immunol201319194473447424141853
- JanewayCAJrApproaching the asymptote? Evolution and revolution in immunologyCold Spring Harb Symp Quant Biol198954Pt1113
- HartSPSmithJRDransfieldIPhagocytosis of opsonized apoptotic cells: roles for ‘old-fashioned’ receptors for antibody and complementClin Exp Immunol200413518118514738443
- TaylorPRMartinez-PomaresLStaceyMLinHHBrownGDGordonSMacrophage receptors and immune recognitionAnnu Rev Immunol20052390194415771589
- ChenGShawMHKimYGNunezGNOD-like receptors: role in innate immunity and inflammatory diseaseAnnu Rev Pathol2009436539818928408
- MartinonFMayorATschoppJThe inflammasomes: guardians of the bodyAnnu Rev Immunol200927122926519302040
- NakhaeiPGeninPCivasAHiscottJRIG-I-like receptors: sensing and responding to RNA virus infectionSemin Immunol200921421522219539500
- JacobsSRDamaniaBNLRs, inflammasomes, and viral infectionJ Leukoc Biol20129246947722581934
- SchmidtARothenfusserSHopfnerK-PSensing of viral nucleic acids by RIG-I: from translocation to translationEur J Cell Biol2012911788521496944
- BauernfeindFHornungVOf inflammasomes and pathogens – sensing of microbes by the inflammasomeEMBO Mol Med2013581482623666718
- TakedaKAkiraSToll-like receptorsCurr Protoc Immunol201510912141110
- SatohTAkiraSToll-like receptor signaling and its inducible proteinsMicrobiol Spectr20164
- TarteySTakeuchiOPathogen recognition and Toll-like receptor targeted therapeutics in innate immune cellsInt Rev Immunol2017362577328060562
- ZhangHLuoJAlcornJFAIM2 inflammasome is critical for influenza-induced lung injury and mortalityJ Immunol2017198114383439328424239
- KumarVSharmaANeutrophils: Cinderella of innate immune systemInt Immunopharmacol201010111325133420828640
- KumarVSharmaAMast cells: emerging sentinel innate immune cells with diverse role in immunityMol Immunol2010a48142520678798
- KumarVAhmadARole of MAIT cells in the immunopathogenesis of inflammatory diseases: new players in old gameInt Rev Immunol2018379011029106304
- BoydJHDivangahiMYahiaouiLGvozdicDQureshiSPetrofBJToll-like receptors differentially regulate CC and CXC chemokines in skeletal muscle via NF-κB and calcineurinInfect Immun2006746829683816982839
- SalomãoRMartinsPSBrunialtiMKCTLR signaling pathway in patients with sepsisShock200830Suppl 1737718704004
- TsujimotoHOnoSEfronPAScumpiaPOMoldawerLLMochizukiHRole of Toll-like receptors in the development of sepsisShock200829331532118277854
- SavvaARogerTTargeting Toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseasesFront Immunol2013184387
- TurnerMDNedjaiBHurstTPenningtonDJCytokines and chemokines: at the crossroads of cell signalling and inflammatory diseaseBiochim Biophys Acta20141843112563258224892271
- FranchiLEigenbrodTMuñoz-PlanilloRNuñezGThe inflammasome: a caspase-1 activation platform regulating immune responses and disease pathogenesisNat Immunol20091024119221555
- TaxmanDJHuangMTTingJPInflammasome inhibition as a pathogenic stealth mechanismCell Host Microbe20108171120638636
- MenuPVinceJEThe NLRP3 inflammasome in health and disease: the good, the bad and the uglyClin Exp Immunol2011166111521762124
- Chavarria-SmithJVanceREThe NLRP1 inflammasomesImmunol Rev2015265223425879281
- ZhaoYShaoFThe NAIP–NLRC4 inflammasome in innate immune detection of bacterial flagellin and type III secretion apparatusImmunol Rev20152658510225879286
- KruegerMHeinzmannANauckMAdhesion molecules in pediatric intensive care patients with organ dysfunction syndromeIntensive Care Med20073335936317124613
- ZonneveldRMartinelliRShapiroNIKuijpersTWPlötzFBCarmanCVSoluble adhesion molecules as markers for sepsis and the potential pathophysiological discrepancy in neonates, children and adultsCrit Care20141820420424602331
- AmalakuhanBHabibSAMangatMEndothelial adhesion molecules and multiple organ failure in patients with severe sepsisCytokine20168826727327701021
- WhalenMJDoughtyLACarlosTMWisniewskiSRKochanekPMCarcilloJAIntercellular adhesion molecule-1 and vascular cell adhesion molecule-1 are increased in the plasma of children with sepsis-induced multiple organ failureCrit Care Med20002872600260710921602
- Sosa-BustamanteGPAmador-LiconaNBarbosa-SabaneroGIntercellular adhesion molecules and mortality for sepsis in infants younger than 1 year of lifeRev Invest Clin20116360160623650672
- SuCMChengHHTsaiTCElevated serum vascular cell adhesion molecule-1 is associated with septic encephalopathy in adult community-onset severe sepsis patientsBiomed Res Int2014201410
- SagerHBDuttaPDahlmanJERNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarctionSci Transl Med20168342342ra380
- HildebrandFPapeH-CHarwoodPRole of adhesion molecule ICAM in the pathogenesis of polymicrobial sepsisExp Toxicol Pathol20055628129015816357
- ChiZMelendezAJRole of cell adhesion molecules and immune-cell migration in the initiation, onset and development of atherosclerosisCell Adh Migr2007117117519262139
- ChaeYKChoiWMBaeWHOverexpression of adhesion molecules and barrier molecules is associated with differential infiltration of immune cells in non-small cell lung cancerSci Rep20188102329348685
- BauerPRMicrovascular responses to sepsis: clinical significancePathophysiology2002814114812039645
- ReinhartKBayerOBrunkhorstFMeisnerMMarkers of endothelial damage in organ dysfunction and sepsisCrit Care Med200230SupplementS302S31212004252
- InoueMWilliamsKLGunnMDShinoharaMLNLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitisProc Natl Acad Sci U S A2012109104801048522699511
- WangJGWilliamsJCDavisBKMonocytic microparticles activate endothelial cells in an IL-1β-dependent mannerBlood201111882366237421700772
- Boisrame-HelmsJKremerHSchini-KerthVMezianiFEndothelial dysfunction in sepsisCurr Vasc Pharmacol20131115016023506494
- InceCMayeuxPRNguyenTThe endothelium in sepsisShock20164525927026871664
- ChenYLiXBoiniKMEndothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritisBiochim Biophys Acta2015185339640825450976
- LuoS-FChinC-YHoL-JTsengW-YKuoC-FLaiJHMonosodium urate crystals induced ICAM-1 expression and cell–cell adhesion in renal mesangial cells: implications for the pathogenesis of gouty nephropathyJ Microbiol Immunol Infect2018S16841182(18)30003
- HewettJASchultzeAEVanciseSRothRANeutrophil depletion protects against liver injury from bacterial endotoxinLab Invest1992663473611538588
- TsujiCMinhazMUShioyaSFukahoriMTanigakiTNakazawaHThe importance of polymorphonuclear leukocytes in lipopolysaccharide-induced superoxide anion production and lung injury: ex vivo observation in rat lungsLung199817611139436173
- RaeburnCDCalkinsCMZimmermanMAICAM-1 and VCAM-1 mediate endotoxemic myocardial dysfunction independent of neutrophil accumulationAm J Physiol Regul Integr Comp Physiol20022832R477R48612121861
- YangLFroioRMSciutoTEDvorakAMAlonRLuscinskasFWICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flowBlood2005106258459215811956
- XuRHuangHZhangZWangFSThe role of neutrophils in the development of liver diseasesCell Mol Immunol201411322423124633014
- WoodfinABeyrauMVoisinMBICAM-1-expressing neutrophils exhibit enhanced effector functions in murine models of endotoxemiaBlood2016127789890726647392
- van der HeijdenTKritikouEVenemaWNLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E–deficient miceArterioscler Thromb Vasc Biol20173781457146128596375
- BakeleMJoosMBurdiSLocalization and functionality of the inflammasome in neutrophilsJ Biol Chem20142895320532924398679
- Pérez-FigueroaETorresJSánchez-ZaucoNContreras-RamosAAlvarez-ArellanoLMaldonado-BernalCActivation of NLRP3 inflammasome in human neutrophils by Helicobacter pylori infectionInnate Immun201622210311226610398
- Alfonso-LoechesSUreña-PeraltaJRMorillo-BarguesMJOliver-de La CruzJGuerriCRole of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cellsFront Cell Neurosci201418216
- HarijithAEbenezerDLNatarajanVReactive oxygen species at the crossroads of inflammasome and inflammationFront Physiol2014535225324778
- AbaisJMXiaMZhangYBoiniKMLiP-LRedox regulation of NLRP3 inflammasomes: ROS as trigger or effector?Antioxid Redox Signal2015221111112925330206
- AnderssonUTraceyKJHMGB1 in sepsisScand J Infect Dis20033557758414620138
- HuangWTangYLiLHMGB1, a potent proinflammatory cytokine in sepsisCytokine20105111912620347329
- KaczmarekAVandenabeelePKryskoDVNecroptosis: the release of damage-associated molecular patterns and its physiological relevanceImmunity20133820922323438821
- ZhaoHJafferTEguchiSWangZLinkermannAMaDRole of necroptosis in the pathogenesis of solid organ injuryCell Death Dis20156e197526583318
- FiuzaCBustinMTalwarSInflammation-promoting activity of HMGB1 on human microvascular endothelial cellsBlood20031012652266012456506
- AsavarutPZhaoHGuJMaDThe role of HMGB1 in inflammation-mediated organ injuryActa Anaesthesiol Taiwan201351283323711603
- LuiGWongCKIpMHMGB1/RAGE signaling and pro-inflammatory cytokine responses in non-HIV adults with active pulmonary tuberculosisPLoS One2016117e015913227434276
- AbdulmahdiWPatelDRabadiMMHMGB1 redox during sepsisRedox Biol20171360060728806702
- StevensNEChapmanMJFraserCKKuchelTRHayballJDDienerKRTherapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomesSci Rep201771585028724977
- WillinghamSBAllenICBergstralhDTNLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathwaysJ Immunol200918332008201519587006
- LamkanfiMSarkarAvande WalleLInflammasome-dependent release of the alarmin HMGB1 in endotoxemiaJ Immunol20101854385439220802146
- vande WalleLKannegantiTDLamkanfiMHMGB1 release by inflammasomesVirulence20112216216521422809
- LeblancPMDoggettTAChoiJAn immunogenic peptide in the A-box of HMGB1 protein reverses apoptosis-induced tolerance through RAGE receptorJ Biol Chem20142897777778624474694
- LuBWangHAnderssonUTraceyKJRegulation of HMGB1 release by inflammasomesProtein Cell20134316316723483477
- ScaffidiPMisteliTBianchiMERelease of chromatin protein HMGB1 by necrotic cells triggers inflammationNature2002418689419119512110890
- LuBNakamuraTInouyeKNovel role of PKR in inflammasome activation and HMGB1 releaseNature2012488741367067422801494
- BonaldiTTalamoFScaffidiPMonocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretionEMBO J2003225551556014532127
- EvankovichJChoSWZhangRHigh mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activityJ Biol Chem2010285398883989720937823
- ChiWChenHLiFZhuYYinWZhuoYHMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-kappaB pathway in acute glaucomaJ Neuroinflammation20151213726224068
- Abu El-AsrarAMAlamKGarcia-RamirezMAssociation of HMGB1 with oxidative stress markers and regulators in PDRMol Vis20172385387129259392
- WatsonRWRedmondHPWangJHCondronCBouchier-HayesDNeutrophils undergo apoptosis following ingestion of Escherichia coliJ Immunol199615610398639928621940
- DeleoFRModulation of phagocyte apoptosis by bacterial pathogensApoptosis2004939941315192322
- WilkieRPVissersMCDragunowMHamptonMBA functional NADPH oxidase prevents caspase involvement in the clearance of phagocytic neutrophilsInfect Immun20077573256326317438039
- VictorVMRochaMde La FuenteMImmune cells: free radicals and antioxidants in sepsisInt Immunopharmacol20044332734715037211
- VíctorVMEspulguesJVHernández-MijaresARochaMOxidative stress and mitochondrial dysfunction in sepsis: a potential therapy with mitochondria-targeted antioxidantsInfect Disord Drug Targets20099437638919689380
- BimeCZhouTWangTSlepianMJGarciaJGNHeckerLReactive oxygen species–associated molecular signature predicts survival in patients with sepsisPulm Circ2016619620127252846
- OliveiraYpadePontes-de-CarvalhoLCCoutoRDNoronha-DutraAAOxidative stress in sepsis. Possible production of free radicals through an erythrocyte-mediated positive feedback mechanismBraz J Infect Dis2017211192627916603
- CepinskasGWilsonJXInflammatory response in microvascular endothelium in sepsis: role of oxidantsJ Clin Biochem Nutr20084217518418545638
- HuetODupicLHarroisADuranteauJOxidative stress and endothelial dysfunction during sepsisFront Biosci (Landmark Ed)1620111986199521196278
- PotzBASellkeFWAbidMREndothelial ROS and impaired myocardial oxygen consumption in sepsis-induced cardiac dysfunctionJ Intensive Crit Care2016020120
- LégerTCharrierAMoreauCEarly sepsis does not stimulate reactive oxygen species production and does not reduce cardiac function despite an increased inflammation statusPhysiol Rep20175e1323128684640
- HoetzeneckerWEchtenacherBGuenovaEROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppressionNat Med20111812822179317
- CruzCMRinnaAFormanHJVenturaALPersechiniPMOjciusDMATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophagesJ Biol Chem20072822871287917132626
- MartinonFSignaling by ROS drives inflammasome activationEur J Immunol201040361661920201014
- HeYHaraHNúñezGMechanism and regulation of NLRP3 inflammasome activationTrends Biochem Sci2016411012102127669650
- KumarVTargeting macrophage immunometabolism: dawn in the darkness of sepsisInt Immunopharmacol20185817318529625385
- AbaisJMZhangCXiaMNADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemiaAntioxid Redox Signal2013181537154823088210
- BoiniKMXiaMAbaisJMActivation of inflammasomes in podocyte injury of mice on the high fat diet: effects of ASC gene deletion and silencingBiochim Biophys Acta2014184383684524508291
- HanSCaiWYangXROS-mediated NLRP3 inflammasome activity is essential for burn-induced acute lung injuryMediators Inflamm2015201516
- SumiYWoehrleTChenYPlasma ATP is required for neutrophil activation in a mouse sepsis modelShock201442214214724675414
- LedderoseCLiXBaoYSumiYShapiroNJungerWSystemic ATP levels suppress the function of CD4+ T cells in sepsis by impairing autocrine purinergic signalingFASEB J201529972976
- LedderoseCBaoYKondoYPurinergic signaling and immune responses in sepsisClin Ther2016381054106527156007
- SueyoshiKSumiYInoueYFluorescence imaging of ATP in neutrophils from patients with sepsis using organelle-localizable fluorescent chemosensorsAnn Intensive Care2016616427422255
- LiXKondoYBaoYSystemic adenosine triphosphate impairs neutrophil chemotaxis and host defense in sepsisCrit Care Med2017451e97e10427548819
- DubyakGRSignal transduction by P2-purinergic receptors for extracellular ATPAm J Respir Cell Mol Biol199142953001707633
- BuzziNBilbaoPSBolandRde BolandARExtracellular ATP activates MAP kinase cascades through a P2Y purinergic receptor in the human intestinal Caco-2 cell lineBiochim Biophys Acta200917901651165919836435
- SäveSPerssonKExtracellular ATP and P2Y receptor activation induce a proinflammatory host response in the human urinary tractInfect Immun20107883609361520515921
- RiteauNGassePFauconnierLExtracellular ATP is a danger signal activating P2X 7 receptor in lung inflammation and fibrosisAm J Respir Crit Care Med2010182677478320522787
- GargADKryskoDVVandenabeelePAgostinisPExtracellular ATP and P2X7 receptor exert context-specific immunogenic effects after immunogenic cancer cell deathCell Death Dis20167e209726890136
- di VirgilioFdal BenDSartiACGiulianiALFalzoniSThe P2X7 receptor in infection and inflammationImmunity201747153128723547
- KarmakarMKatsnelsonMADubyakGRPearlmanENeutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATPNat Commun201671055526877061
- di VirgilioFLiaisons dangereuses: P2X(7) and the inflammasomeTrends Pharmacol Sci20072846547217692395
- Coutinho-SilvaROjciusDMRole of extracellular nucleotides in the immune response against intracellular bacteria and protozoan parasitesMicrobes Infect2012141271127722634346
- GombaultABaronLCouillinIATP release and purinergic signaling in NLRP3 inflammasome activationFront Immunol2012341423316199
- IdzkoMFerrariDEltzschigHKNucleotide signalling during inflammationNature201450931031724828189
- CsokaBNemethZHToroGExtracellular ATP protects against sepsis through macrophage P2X7 purinergic receptors by enhancing intracellular bacterial killingFASEB J2015293626363726060214
- WuXRenJChenGSystemic blockade of P2X7 receptor protects against sepsis-induced intestinal barrier disruptionSci Rep201771436428663567
- NicholsRDvon MoltkeJVanceRENAIP/NLRC4 inflammasome activation in MRP8+ cells is sufficient to cause systemic inflammatory diseaseNat Commun201781220929263322
- KumarVSharmaAAdenosine: an endogenous modulator of innate immune system with therapeutic potentialEur J Pharmacol200961671519464286
- KumarVAdenosine as an endogenous immunoregulator in cancer pathogenesis: where to go?Purinergic Signal2013914516523271562
- BaoRHouJLiYAdenosine promotes Foxp3 expression in Treg cells in sepsis model by activating JNK/AP-1 pathwayAm J Transl Res201682284229227347335
- MartinCLeoneMViviandXAyemM-LGuieuRHigh adenosine plasma concentration as a prognostic index for outcome in patients with septic shockCrit Care Med20002893198320211008982
- SperlaghBDodaMBaranyiMHaskoGIschemic-like condition releases norepinephrine and purines from different sources in super-fused rat spleen stripsJ Neuroimmunol2000111455411063820
- OuyangXGhaniAMalikAAdenosine is required for sustained inflammasome activation via the A2A receptor and the HIF-1α pathwayNat Commun201341290924352507
- KumarVChhibberSIntravenous 2-chloroadenosine protects BALB/c mice from Klebsiella pneumoniae B5055-induced sepsis by modulating the pro-inflammatory immune responseJ Chemother20092163964520071287
- CsokaBNemethZHRosenbergerPA2B adenosine receptors protect against sepsis-induced mortality by dampening excessive inflammationJ Immunol201018554255020505145
- KumarVHarjaiKChhibberS2-Chloroadenosine (2-CADO) treatment modulates the pro-inflammatory immune response to prevent acute lung inflammation in BALB/c mice suffering from Klebsiella pneumoniae B5055-induced pneumoniaInt J Antimicrob Agents201035659960220189776
- SivakKVVasinAVEgorovVVAdenosine A2A receptor as a drug target for treatment of sepsisMol Biol2016502200212
- NonakaMKimuraAGenomic view of the evolution of the complement systemImmunogenetics200658970171316896831
- NonakaMEvolution of the complement systemSubcell Biochem201480314324798006
- MerleNSChurchSEFremeaux-BacchiVRoumeninaLTComplement system part I – molecular mechanisms of activation and regulationFront Immunol20156426226082779
- MerleNSNoeRHalbwachs-MecarelliLFremeaux-BacchiVRoumeninaLTComplement system part II: role in immunityFront Immunol2015618025726074922
- WardPAThe dark side of C5a in sepsisNat Rev Immunol20044213315040586
- GressnerOAKochASansonETrautweinCTackeFHigh C5a levels are associated with increased mortality in sepsis patients-no enhancing effect by actin-free Gc-globulinClin Biochem20084197498018538666
- MarkiewskiMMDeangelisRALambrisJDComplexity of complement activation in sepsisJ Cell Mol Med2008126a2245225418798865
- CharchafliehJWeiJLabazeGThe role of complement system in septic shockClin Dev Immunol201220128
- YanCGaoHNew insights for C5a and C5a receptors in sepsisFront Immunol2012336823233853
- RicklinDReisESLambrisJDComplement in disease: a defence system turning offensiveNat Rev Nephrol201612738340127211870
- SureshRChandrasekaranPSutterwalaFSMosserDMComplement-mediated ‘bystander’ damage initiates host NLRP3 inflammasome activationJ Cell Sci201612991928193927006116
- LaudisiFSpreaficoREvrardMCutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β releaseJ Immunol20131911006101023817414
- TriantafilouMHughesTRMorganBPTriantafilouKComplementing the inflammasomeImmunology2016147215216426572245
- MurakamiTOckingerJYuJCritical role for calcium mobilization in activation of the NLRP3 inflammasomeProc Natl Acad Sci U S A201210928112821128722733741
- HorngTCalcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasomeTrends Immunol20143525326124646829
- ShimadaKCrotherTRKarlinJOxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosisImmunity201236340141422342844
- LuanY-YYaoY-MXiaoX-ZShengZ-YInsights into the apoptotic death of immune cells in sepsisJ Interferon Cytokine Res2015351172225007137
- FangCWeiXWeiYMitochondrial DNA in the regulation of innate immune responsesProtein Cell20167111626498951
- LiszewskiMKKolevMLeFriecGIntracellular complement activation sustains T cell homeostasis and mediates effector differentiationImmunity20133961143115724315997
- ArboreGWestEESpolskiRT helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cellsScience20163526292aad1210aad121027313051
- IwasakaHNoguchiTTh1/Th2 balance in systemic inflammatory response syndrome (SIRS)Nihon Rinsho2004622237224315597790
- KalbitzMFattahiFGrailerJJComplement-induced activation of the cardiac NLRP3 inflammasome in sepsisFASEB J2016303997400627543123
- AsgariELe FriecGYamamotoHC3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activationBlood20131223473348123878142
- DinarelloCAThe C3a receptor, caspase-1, and release of IL-1betaBlood20131223394339524235127
- BenoitMEClarkeEVMorgadoPFraserDATennerAJComplement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cellsJ Immunol20121885682569322523386
- NakagawaIAmanoAMizushimaNAutophagy defends cells against invading group A StreptococcusScience200430656981037104015528445
- Matsuzawa-IshimotoYHwangSCadwellKAutophagy and inflammationAnnu Rev Immunol20183617310129144836
- Djavaheri-MergnyMAmelottiMMathieuJBesanconFBauvyCCodognoPRegulation of autophagy by NFkappaB transcription factor and reactive oxygen speciesAutophagy2007339039217471012
- XuYJagannathCLiuXDSharafkhanehAKolodziejskaKEEissaNTToll-like receptor 4 is a sensor for autophagy associated with innate immunityImmunity200727113514417658277
- WuHMWangJZhangBFangLXuKLiuRYCpG-ODN promotes phagocytosis and autophagy through JNK/P38 signal pathway in Staphylococcus aureus-stimulated macrophageLife Sci2016161515927476088
- PiquereauJGodinRDeschênesSProtective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunctionAutophagy20139111837185124121678
- TakahashiWWatanabeEFujimuraLKinetics and protective role of autophagy in a mouse cecal ligation and puncture-induced sepsisCrit Care2013174R16023883625
- YenYTYangHRLoHCEnhancing autophagy with activated protein C and rapamycin protects against sepsis-induced acute lung injurySurgery2013153568969823434181
- HoJYuJWongSHAutophagy in sepsis: degradation into exhaustion?Autophagy2016121073108227172163
- HsiaoHWTsaiKLWangLFThe decline of autophagy contributes to proximal tubular dysfunction during sepsisShock20123728929622089196
- SuYQuYZhaoFLiHMuDLiXRegulation of autophagy by the nuclear factor κB signaling pathway in the hippocampus of rats with sepsisJ Neuroinflammation20151211626067996
- KimuraTWatanabeESakamotoTAutophagy-related IRGM polymorphism is associated with mortality of patients with severe sepsisPLoS One20149e9152224626347
- LinC-WLoSPerngD-SComplete activation of autophagic process attenuates liver injury and improves survival in septic miceShock201441324124924365881
- ZhangLAiYTsungAClinical application: restoration of immune homeostasis by autophagy as a potential therapeutic target in sepsisExp Ther Med20161141159116727073416
- RenCZhangHWuT-TYaoYMAutophagy: a potential therapeutic target for reversing sepsis-induced immunosuppressionFront Immunol20178
- SaitohTFujitaNJangMHLoss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β productionNature2008456721926426818849965
- ZhouRTardivelAThorensBChoiITschoppJThioredoxin-interacting protein links oxidative stress to inflammasome activationNat Immunol20101113614020023662
- ZhouRYazdiASMenuPTschoppJA role for mitochondria in NLRP3 inflammasome activationNature201146922122521124315
- KimuraTJainAChoiSWTRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunityJ Cell Biol201521097398926347139
- HarrisJHartmanMRocheCAutophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradationJ Biol Chem20112869587959721228274
- HarrisJLangTThomasJPWSukkarMBNabarNRKehrlJHAutophagy and inflammasomesMol Immunol201786101528249679
- SuzukiTFranchiLTomaCDifferential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophagesPLoS Pathog200738e11117696608
- JounaiNKobiyamaKShiinaMOgataKIshiiKJTakeshitaFNLRP4 negatively regulates autophagic processes through an association with beclin1J Immunol20111861646165521209283
- ZhangYSaulerMShinnASEndothelial PINK1 mediates the protective effects of NLRP3 deficiency during lethal oxidant injuryJ Immunol2014192115296530424778451
- DengQWangYZhangYPseudomonas aeruginosa triggers macrophage autophagy to escape intracellular killing by activation of the NLRP3 inflammasomeInfect Immun201684566626467446
- PuQGanCLiRAtg7 deficiency intensifies inflammasome activation and pyroptosis in Pseudomonas sepsisJ Immunol201719883205321328258192
- WlodarskaMThaissCANowarskiRNLRP6 inflammasome orchestrates the colonic host–microbial interface by regulating goblet cell mucus secretionCell201415651045105924581500
- KimMJBaeSHRyuJCSESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophagesAutophagy20161281272129127337507
- MaoKChenSChenMNitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shockCell Res201323220121223318584
- LupferCThomasPGAnandPKReceptor interacting protein kinase 2–mediated mitophagy regulates inflammasome activation during virus infectionNat Immunol201314548048823525089
- SaitohTAkiraSRegulation of inflammasomes by autophagyJ Allergy Clin Immunol20161381283627373323
- QianSFanJBilliarTRScottMJInflammasome and autophagy regulation: a two-way streetMol Med20172318819528741645
- ShiC-SShenderovKHuangN-NActivation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destructionNat Immunol201213325526322286270
- YukJMJoEKCrosstalk between autophagy and inflammasomesMol Cells201336539339924213677
- TakahamaMAkiraSSaitohTAutophagy limits activation of the inflammasomesImmunol Rev20182811627329248000
- ShibuyaNMikamiYKimuraYNagaharaNKimuraHVascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfideJ Biochem2009146562362619605461
- WangRPhysiological implications of hydrogen sulfide: a whiff exploration that blossomedPhysiol Rev201292279189622535897
- ElrodJWCalvertJWMorrisonJHydrogen sulfide attenuates myocardial ischemia–reperfusion injury by preservation of mitochondrial functionProc Natl Acad Sci U S A2007104155601556517878306
- YangGWuLJiangBH2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyaseScience2008322590158759018948540
- FernandesCJJrde AssuncaoMSMyocardial dysfunction in sepsis: a large, unsolved puzzleCrit Care Res Pract2012201289643022482045
- AntonucciEFiaccadoriEDonadelloKTacconeFSFranchiFScollettaSMyocardial depression in sepsis: from pathogenesis to clinical manifestations and treatmentJ Crit Care20142950051124794044
- KakihanaYItoTNakaharaMYamaguchiKYasudaTSepsis-induced myocardial dysfunction: pathophysiology and managementJ Intensive Care201642227011791
- LvXWangHPathophysiology of sepsis-induced myocardial dysfunctionMil Med Res2016313027708836
- VallabhajosyulaSPruthiSShahSWileyBMMankadSVJentzerJCBasic and advanced echocardiographic evaluation of myocardial dysfunction in sepsis and septic shockAnaesth Intensive Care2018461132429361252
- LinZAltafNLiCHydrogen sulfide attenuates oxidative stress-induced NLRP3 inflammasome activation via S-sulfhydrating c-Jun at Cys269 in macrophagesBiochim Biophys Acta201818649 Pt B28902900
- CastelblancoMLugrinJEhirchiouDHydrogen sulfide inhibits the NLRP3 inflammasome and reduces cytokine production both in vitro and in a mouse model of inflammationJ Biol Chem201829372546255729279328
- HuangZZhuangXXieCExogenous hydrogen sulfide attenuates high glucose-induced cardiotoxicity by inhibiting NLRP3 inflammasome activation by suppressing TLR4/NF-kappaB pathway in H9c2 CellsCell Physiol Biochem2016401578159027997926
- ZhangHZhiLMoorePKBhatiaMRole of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouseAm J Physiol Lung Cell Mol Physiol20062906L1193120116428267
- SpillerFOrricoMINascimentoDCHydrogen sulfide improves neutrophil migration and survival in sepsis via K+ATP channel activationAm J Respir Crit Care Med2010182336036820339148
- LiXChengQLiJHeYTianPXuCSignificance of hydrogen sulfide in sepsis-induced myocardial injury in ratsExp Ther Med20171432153216128962136
- OyadomariSMoriMRoles of CHOP/GADD153 in endoplasmic reticulum stressCell Death Differ200411438138914685163
- TabasIRonDIntegrating the mechanisms of apoptosis induced by endoplasmic reticulum stressNat Cell Biol201113318419021364565
- FerlitoMWangQFultonWBHydrogen sulfide increases survival during sepsis: protective effect of CHOP inhibitionJ Immunol20141921806181424403532
- EndoMOyadomariSSugaMMoriMGotohTThe ER stress pathway involving CHOP is activated in the lungs of LPS-treated miceJ Biochem200513850150716272146
- EndoMMoriMAkiraSGotohTC/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammationJ Immunol20061766245625316670335
- HuiYduJTangCBinGJiangHChanges in arterial hydrogen sulfide (H(2)S) content during septic shock and endotoxin shock in ratsJ Infect20034715516012860150
- NorrisENarasimhanSCulbersonCClemensMGThe liver is a critical site for hydrogen sulfide clearance in sepsisFASEB J20112511171118
- ColettaCSzaboCPotential role of hydrogen sulfide in the pathogenesis of vascular dysfunction in septic shockCurr Vasc Pharmacol20131120822123506499
- KimDAnantharamPVWhitleyEMAcute exposure to high level of hydrogen sulfide induces neurodegeneration via activation of inflammasomes in brainFASEB J2016a3011911195
- GaddamRRChambersSMurdochDShawGBhatiaMCirculating levels of hydrogen sulfide and substance P in patients with sepsisJ Infect20177529330028760413
- KoširMPodbregarMAdvances in the diagnosis of sepsis: hydrogen sulfide as a prognostic marker of septic shock severityEJIFCC20172813414128757821
- BeeNWhiteRPetrosAJHydrogen sulfide in exhaled gases from ventilated septic neonates and children: a preliminary reportPediatr Crit Care Med201718e327e33228622279
- BeltowskiJHydrogen sulfide in pharmacology and medicine – an updatePharmacol Rep20156764765825933982
- TilleySLCoffmanTMKollerBHMixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanesJ Clin Invest20011081152311435451
- SmythEMGrosserTWangMYuYFitzgeraldGAProstanoids in health and diseaseJ Lipid Res200950SupplementS423S42819095631
- KawabeTHarrisPDZakariaELGarrisonRNSepsis alters vessel contraction by adrenoceptor-induced nitric oxide and prostanoidJ Surg Res200311035235912788665
- WangXShawDKHammondHLThe prostaglandin E2-EP3 receptor axis regulates Anaplasma phagocytophilum-mediated NLRC4 inflammasome activationPLoS Pathog2016128e100580327482714
- RicciottiEFitzgeraldGAProstaglandins and inflammationArterioscler Thromb Vasc Biol2011315986100021508345
- von MoltkeJTrinidadNJMoayeriMRapid induction of inflammatory lipid mediators by the inflammasome in vivoNature2012490741810711122902502
- ZoccalKFSorgiCAHoriJIOpposing roles of LTB4 and PGE2 in regulating the inflammasome-dependent scorpion venom-induced mortalityNat Commun201671076026907476
- MaierNKLepplaSHMoayeriMThe cyclopentenone prostaglandin 15d-PGJ2 inhibits the NLRP1 and NLRP3 inflammasomesJ Immunol201519462776278525681332
- MortimerLMoreauFMacdonaldJAChadeeKNLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutationsNat Immunol201617101176118627548431
- SokolowskaMChenL-YLiuYProstaglandin E2 inhibits NLRP3 inflammasome activation through EP4 receptor and intracellular cyclic AMP in human macrophagesJ Immunol2015194115472548725917098
- TingJPDuncanJALeiYHow the noninflammasome NLRs function in the innate immune systemScience2010327596328629020075243
- LiZZhangYLiuBLuoWLiHZhouYRole of E-type prostaglandin receptor EP3 in the vasoconstrictor activity evoked by prostacyclin in thromboxane-prostanoid receptor deficient miceSci Rep2017714216728165064
- TunctanBKorkmazBSariANA novel treatment strategy for sepsis and septic shock based on the interactions between prostanoids, nitric oxide, and 20-hydroxyeicosatetraenoic acidAntiinflamm Antiallergy Agents Med Chem201211212115023013331
- TunctanBKorkmazBSariANContribution of iNOS/sGC/PKG pathway, COX-2, CYP4A1, and gp91(phox) to the protective effect of 5,14-HEDGE, a 20-HETE mimetic, against vasodilation, hypotension, tachycardia, and inflammation in a rat model of septic shockNitric Oxide201333184123684565
- VijayRFehrARJanowskiAMVirus-induced inflammasome activation is suppressed by prostaglandin D2/DP1 signalingProc Natl Acad Sci U S A201711427E5444e545328630327
- KhareSRatsimandresyRAde AlmeidaLThe PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA virusesNat Immunol20141534335324531343
- de AlmeidaLKhareSMisharinAVThe PYRIN domain-only protein POP1 inhibits inflammasome assembly and ameliorates inflammatory diseaseImmunity20154326427626275995
- AngusDCOpalSImmunosuppression and secondary infection in sepsis: part, not all, of the storyJAMA20163151457145926975243
- FattahiFWardPAUnderstanding immunosuppression after sepsisImmunity2017473528723551
- RoquillyAMcwilliamHEGJacquelineCLocal modulation of antigen-presenting cell development after resolution of pneumonia induces long-term susceptibility to secondary infectionsImmunity2017471e135147
- JensenIJSjaastadFVGriffithTSBadovinacVPSepsis-induced T cell immunoparalysis: the ins and outs of impaired T cell immunityJ Immunol20182001543155329463691
- CaoCMaTChaiY-FShouS-TThe role of regulatory T cells in immune dysfunction during sepsisWorld J Emerg Med2015a65925802559
- TaturaRZeschnigkMHansenWRelevance of Foxp3+ regulatory T cells for early and late phases of murine sepsisImmunology2015146114415626059660
- YaoYVent-SchmidtJMcgeoughMDTr1 cells, but not Foxp3+ regulatory T cells, suppress NLRP3 inflammasome activation via an IL-10-dependent mechanismJ Immunol2015195248849726056255
- FriedmanGJankowskiSMarchantAGoldmanMKahnRJVincentJLBlood interleukin 10 levels parallel the severity of septic shockJ Crit Care1997121831879459114
- GogosCADrosouEBassarisHPSkoutelisAPro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic optionsJ Infect Dis200018117618010608764
- PileriDAccardo PalomboAD’AmelioLConcentrations of cytokines Il-6 and Il-10 in plasma of burn patients: their relationship to sepsis and outcomeAnn Burns Fire Disasters20082118218521991134
- SurbatovicMPopovicNVojvodicDCytokine profile in severe Gram-positive and Gram-negative abdominal sepsisSci Rep201551135526079127
- LatifiSQO’RiordanMALevineADInterleukin-10 controls the onset of irreversible septic shockInfect Immun2002704441444612117955
- CayrolCGirardJ-PThe IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1Proc Natl Acad Sci U S A20091069021902619439663
- MadouriFGuillouNFauconnierLCaspase-1 activation by NLRP3 inflammasome dampens IL-33-dependent house dust mite-induced allergic lung inflammationJ Mol Cell Biol20157435136525714839
- TominagaSIOhtaSTagoKSoluble form of the ST2 gene product exhibits growth promoting activity in NIH-3T3 cellsBiochem Biophys Rep2016581528955802
- GriesenauerBPaczesnySThe ST2/IL-33 axis in immune cells during inflammatory diseasesFront Immunol20178
- KakkarRLeeRTThe IL-33/ST2 pathway: therapeutic target and novel biomarkerNat Rev Drug Discov2008782784018827826
- EchtenacherBMannelDNHultnerLCritical protective role of mast cells in a model of acute septic peritonitisNature199638175778609992
- HoogerwerfJJTanckMWTvan ZoelenMadWitteboleXLaterreP-Fvan der PollTSoluble ST2 plasma concentrations predict mortality in severe sepsisIntensive Care Med20103663063720151106
- ParenicaJMalaskaJJarkovskyJSoluble ST2 levels in patients with cardiogenic and septic shock are not predictors of mortalityExp Clin Cardiol20121720520923592937
- HurMKimHKimHJSoluble ST2 has a prognostic role in patients with suspected sepsisAnn Lab Med20153557057726354344
- SweetMJLeungBPKangDA novel pathway regulating lipopolysaccharide-induced shock by ST2/T1 via inhibition of Toll-like receptor 4 expressionJ Immunol2001166116633663911359817
- Alves-FilhoJCSônegoFSoutoFInterleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infectionNat Med2010
- SutherlandREOlsenJSMckinstryAVillaltaSAWoltersPJMast cell IL-6 improves survival from Klebsiella pneumonia and sepsis by enhancing neutrophil killingJ Immunol200818185598560518832718
- DahdahAGautierGAttoutTMast cells aggravate sepsis by inhibiting peritoneal macrophage phagocytosisJ Clin Invest20141244577458925180604
- NeteaMGJoostenLABInflammasome inhibition: putting out the fireCell Metab201521451351425863243
- ShaoB-ZXuZ-QHanB-ZSuD-FLiuCNLRP3 inflammasome and its inhibitors: a reviewFront Pharmacol20156378
- JahanSKumarDChaturvediSTherapeutic targeting of NLRP3 inflammasomes by natural products and pharmaceuticals: a novel mechanistic approach for inflammatory diseasesCurr Med Chem2017241645167028245768
- CollRCRobertsonAabChaeJJA small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseasesNat Med20152124825686105
- YoumYHNguyenKYGrantRWThe ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory diseaseNat Med201521326325686106
- NewmanJCVerdinEKetone bodies as signaling metabolitesTrends Endocrinol Metab2014251425224140022
- GrabackaMPierzchalskaMDeanMReissKRegulation of ketone body metabolism and the role of PPARαInt J Mol Sci2016172093
- OhtoshiMJikkoAAsanoMUchidaKOzawaKTobeTKetogenesis during sepsis in relation to hepatic energy metabolismRes Exp Med19841844209219
- Lanza-JacobySRosatoEBracciaGTabaresAAltered ketone body metabolism during gram-negative sepsis in the ratMetabolism199039115111572233276
- LevyBSadouneLOGelotAMBollaertPENabetPLarcanAEvolution of lactate/pyruvate and arterial ketone body ratios in the early course of catecholamine-treated septic shockCrit Care Med20002811411910667509
- GuanYBreyerMDPeroxisome proliferator-activated receptors (PPARs): novel therapeutic targets in renal diseaseKidney Int200160143011422732
- DengYHanXYaoZPPARalpha agonist stimulated angiogenesis by improving endothelial precursor cell function via a NLRP3 inflammasome pathwayCell Physiol Biochem2017422255226628817808
- WangYYuBWangLPioglitazone ameliorates glomerular NLRP3 inflammasome activation in apolipoprotein E knockout mice with diabetes mellitusPLoS One2017127e018124828708885
- AronoffDMSerezaniCHCarstensJKStimulatory effects of peroxisome proliferator-activated receptor-γ on Fcγ receptor-mediated phagocytosis by alveolar macrophagesPPAR Res200720078
- DrosatosKKhanRSTrentCMPPARγ activation prevents sepsis-related cardiac dysfunction and mortality in miceCirc Heart Fail2013655056223572494
- KaplanJNowellMChimaRZingarelliBPioglitazone reduces inflammation through inhibition of NF-κB in polymicrobial sepsisInnate Immun20142051952824029145
- ReddyATLakshmiSPReddyRCPPAR γ in bacterial infections: a friend or foe?PPAR Res2016379635407
- StandageSWCaldwellCCZingarelliBWongHRReduced peroxisome proliferator-activated receptor α expression is associated with decreased survival and increased tissue bacterial load in sepsisShock201237216416922089192
- KumarVChhibberSAnti-inflammatory effect of thalidomide alone or in combination with augmentin in Klebsiella pneumoniae B5055 induced acute lung infection in BALB/c miceEur J Pharmacol200859214615018662682
- KumarVHarjaiKChhibberSA combination of thalidomide and augmentin protects BALB/c mice suffering from Klebsiella pneumoniae B5055-induced sepsisJ Chemother20092115916419423468
- KumarVHarjaiKChhibberSThalidomide treatment modulates macrophage pro-inflammatory function and cytokine levels in Klebsiella pneumoniae B5055 induced pneumonia in BALB/c miceInt Immunopharmacol201010777778320399910
- KumarVChhibberSThalidomide: an old drug with new actionJ Chemother20112332633422233815
- KellerMSollbergerGBeerHDThalidomide inhibits activation of caspase-1J Immunol20091835593559919843943
- López-CastejónGPelegrínPCurrent status of inflammasome blockers as anti-inflammatory drugsExpert Opin Investig Drugs20122179951007
- Ludwig-PortugallIBartokEDhanaEAn NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in miceKidney Int201690352553927262364
- MarchettiCSwartzwelterBGamboniFOLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammationProc Natl Acad Sci U S A20181157E1530E153929378952
- ZormanJSušjanPHafner-BratkovičIShikonin suppresses NLRP3 and AIM2 inflammasomes by direct inhibition of caspase-1PLoS One201611e015982627467658
- HaraHTsuchiyaKKawamuraIPhosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activityNat Immunol2013141247125524185614
- KwakS-BKoppulaSInE-JArtemisia extract suppresses NLRP3 and AIM2 inflammasome activation by inhibition of ASC phosphorylationMediators Inflamm20182018605406929686531
- LeeJAhnHHongEJAnBSJeungEBLeeGSSulforaphane attenuates activation of NLRP3 and NLRC4 inflammasomes but not AIM2 inflammasomeCell Immunol20163065330760
- YendeSvan der PollTDiabetes and sepsis outcomes –it is not all bad newsCrit Care200913111719291261
- KohGCKWPeacockSJvan der PollTWiersingaWJThe impact of diabetes on the pathogenesis of sepsisEur J Clin Microbiol Infect Dis20123137938821805196
- KolyvaASZolotaVMpatsoulisDThe role of obesity in the immune response during sepsisNutr Diabetes20144e13725244356
- Papadimitriou-OlivgerisMArethaDZotouAThe role of obesity in sepsis outcome among critically ill patients: a retrospective cohort analysisBiomed Res Int20162016599
- NgPYEikermannMThe obesity conundrum in sepsisBMC Anesthesiol201717114729070011
- DixitVDNlrp3 inflammasome activation in type 2 diabetes: is it clinically relevant?Diabetes201362222423258906
- LeeH-MKimJ-JKimHJShongMKuBJJoE-KUpregulated NLRP3 inflammasome activation in patients with type 2 diabetesDiabetes20136219420423086037
- VolpeCMAnjosPMNogueira-MachadoJAInflammasome as a new therapeutic target for diabetic complicationsRecent Pat Endocr Metab Immune Drug Discov2016101566226899852
- RheinheimerJde SouzaBMCardosoNSBauerACCrispimDCurrent role of the NLRP3 inflammasome on obesity and insulin resistance: a systematic reviewMetabolism2017741928764843
- SepehriZKianiZAfshariMKohanFDalvandAGhavamiSInflammasomes and type 2 diabetes: an updated systematic reviewImmunol Lett20171929710329079203
- ZhangXDaiJLiLChenHChaiYNLRP3 inflammasome expression and signaling in human diabetic wounds and in high glucose induced macrophagesJ Diabetes Res 20172017b7
- FujitaniYKawamoriRWatadaHThe role of autophagy in pancreatic beta-cell and diabetesAutophagy2009528028219158492
- NuñezCERodriguesVSGomesFSDefective regulation of adipose tissue autophagy in obesityInt J Obes2013371114731480
- BarlowADThomasDCAutophagy in diabetes: beta-cell dysfunction, insulin resistance, and complicationsDNA Cell Biol20153425226025665094
- SarparantaJGarcía-MaciaMSinghRAutophagy and mitochondria in obesity and type 2 diabetesCurr Diabetes Rev201713435236926900135
- YangJSLuCCKuoSCAutophagy and its link to type II diabetes mellitusBiomedicine201772828612706
- HaoHCaoLJiangCFarnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsisCell Metab201725856–867e855
- de Magalhaes FilhoCDDownesMEvansRMFarnesoid X receptor an emerging target to combat obesityDig Dis20173518519028249279
- XieSGuoCChiZA rapid administration of GW4064 inhibits the NLRP3 inflammasome activation independent of farnesoid X receptor agonismFEBS Lett2017591182836284728787755
- LeeSNakahiraKDalliJNLRP3 inflammasome deficiency protects against microbial sepsis via increased lipoxin B4 synthesisAm J Respir Crit Care Med201719671372628245134