Abstract
Acute cerebrovascular disease can affect people at all stages of life, from neonates to the elderly, with devastating consequences. It is responsible for up to 10% of deaths worldwide, is a major cause of disability, and represents an area of real unmet clinical need. Acute cerebrovascular disease is multifactorial with many mechanisms contributing to a complex pathophysiology. One of the major processes worsening disease severity and outcome is inflammation. Pro-inflammatory cytokines of the interleukin (IL)-1 family are now known to drive damaging inflammatory processes in the brain. The aim of this review is to discuss the recent literature describing the role of IL-1 in acute cerebrovascular disease and to provide an update on our current understanding of the mechanisms of IL-1 production. We also discuss the recent literature where the effects of IL-1 have been targeted in animal models, thus reviewing potential future strategies that may limit the devastating effects of acute cerebrovascular disease.
Acute cerebrovascular disease
Acute cerebrovascular disease is a myriad of clinical conditions that can present in many different ways, from periventricular hemorrhage in preterm babies, through subarachnoid hemorrhage (SAH), ischemic and hemorrhagic stroke, and vascular dementia in middle and old age.
Evidence based treatments for acute stroke are limited to thrombolysis for up to 20% of all strokes, antiplatelet agents, and stroke unit care. There is some evidence for decompressive craniectomy in highly selected patients as a life saving measure, and ongoing trials are investigating the efficacy of endovascular interventions such as thrombectomy for acute ischemic stroke. For SAH, there is currently no treatment that addresses the ischemic injury occurring at ictus and the use of nimodipine still results in up to a third of patients developing delayed cerebral ischemia. In intracerebral hemorrhage (ICH), no current therapies other than those addressing hypertension at presentation improve the outcome,Citation1 and surgical trials including the recent Surgical Trial in Lobar Intracerebral Haemorrhage (STICH II) study have also failed to show any benefit.Citation2 It is therefore evident that treatment of acute cerebrovascular disease remains an area of unmet clinical need. While there have been numerous rodent studies targeting inflammation, particularly in stroke, only a few of these have successfully translated into clinical practice. Furthermore, there remains a paucity of research into the targeting of key molecular pathological mechanisms that underpin neurodegeneration occurring in all of these processes. The purpose of this review is to discuss the potential for targeting a pro-inflammatory cytokine called interleukin (IL)-1 and its regulatory processes as potential treatments for cerebrovascular disease.
Inflammation
Inflammation is an important host defense response to injury or infection. Key to an inflammatory response is the recruitment of polymorphonuclear cells to the site of injury or infection. This response is coordinated by cascades of cellular and molecular events that include the production of pro-inflammatory mediators such as cytokines, chemokines, and cell adhesion molecules. Many of the tools of the inflammatory response that target pathogens, such as proteases and reactive oxygen species, also kill healthy cells. This collateral damage may be acceptable when compared to the consequences of pathogen survival. However, when these responses occur in the absence of infection, during injury, or noncommunicable disease states, this collateral damage leads to a worsening of the condition and as such represents a therapeutic target.Citation3 Inflammation in the context of injury, and in the absence of infection, is considered as sterile inflammation. Acute brain cerebrovascular disease results primarily from a disruption to blood flow to a region of the brain and thus the inflammation stimulated by such injuries can be considered sterile. Cytokines of the IL-1 family, namely the IL-1α and IL-1β isoforms, are strongly implicated in sterile inflammatory responses that worsen acute cerebrovascular disease.Citation4
IL-1 production
In a healthy brain, IL-1 is undetectable, but it is expressed after an injury or during disease.Citation5 For example, in an animal model of stroke (induced by occlusion of the middle cerebral artery), a rapid increase in expression of both IL-1α,Citation6 and IL-1β are observed in microglia within hours of the insult and within areas of the brain that experience cell death.Citation7 Although IL-1α and IL-1β appear to be produced mainly by microglia in this model, there is evidence that astrocytes may also express IL-1.Citation7,Citation8 Microglia expressing IL-1α are also observed in regions adjacent to the bleed site in a rat model of SAH.Citation9 Preclinical pharmacological or genetic manipulation studies have demonstrated a consistently detrimental effect of IL-1 in experimental stroke.Citation10 In clinical studies, increased intrathecal concentrations of IL-1β are associated with increased injury,Citation11 and with increased incidence of vasospasm following SAH.Citation12 IL-1 induces release of the inflammatory mediator IL-6 from glia and neurons through type-1 receptor IL-1RI signaling.Citation13,Citation14 IL-6 concentrations are more readily detectable and tend to be more consistent over time. IL-6 is increased in cerebrovascular disease within the cerebrospinal fluid (CSF) and the brain extracellular fluid. In stroke, intrathecal IL-6 concentrations correlate with lesion size,Citation11,Citation15 while in SAH, increased intrathecal IL-6 concentrations are associated with demonstrable vasospasm.Citation12
IL-1 expression is stimulated by molecular motifs expressed by pathogens termed pathogen associated molecular patterns, or by molecules released from dead or injured tissue/cells, or that are modified during disease called damage associated molecular patterns (DAMPs). DAMPs are endogenous molecules normally retained within cells and thus not exposed to immune surveillance. Upon their release during cell death, DAMPs are sensed by pattern recognition receptors (PRRs) on the membranes of cells of the innate immune system, such as microglia, resulting in signaling cascades that induce the expression of pro-inflammatory cytokines.Citation16,Citation17 The specific DAMPs that drive the expression of IL-1 after an acute brain injury are unknown, although there are many potential candidates identified from other disease states including high mobility group protein B1, heat shock proteins, heparin sulfate, and many other endogenous molecules released by dying cells.Citation17 Acute phase proteins such as serum amyloid A may also induce expression of IL-1 in microglia after a brain injury after gaining access to the brain through a damaged blood-brain barrier.Citation18 Many of the DAMPs discovered stimulate PRRs of the toll-like receptor (TLR) family, resulting in the activation of signaling pathways such as nuclear factor- κB and p38 mitogen activated protein kinase that regulate the expression of multiple inflammatory genes in addition to IL1A and IL1B.Citation19 TLRs share a conserved toll- and IL-1 receptor related domain with IL-1RI, and thus TLRs and IL-1RI share many common signaling steps following stimulation.Citation19,Citation20
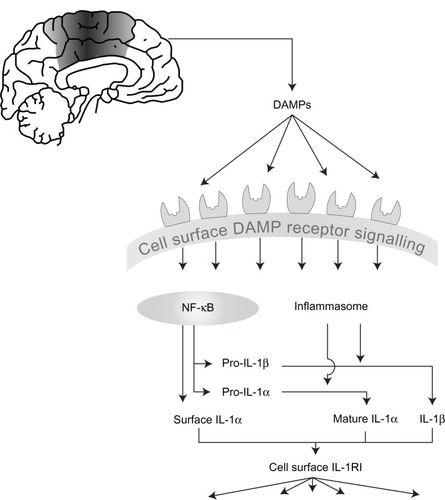
Many pro-inflammatory mediators have conventional secretory routes from the cell, but perhaps indicative of their position as master cytokines, IL-1α and IL-1 β are subject to an additional regulatory step. Stimulating cells to express IL-1 is often referred to as a priming step. A second stimulus is required to elicit their cellular release.Citation21 Both IL-1 forms are initially produced as precursors that reside in the cytoplasm of the cell following the initial priming step. Pro-IL-1β is inactive at IL-1RI, and requires proteolytic cleavage to yield a mature, secreted biologically active molecule. Pro-IL-1α has some biological activity, which is strongly enhanced following proteolytic cleavage ().Citation22,Citation23
Figure 1 The development of inflammation in acute cerebrovascular disease.
Notes: Cell death as a result of acute cerebrovascular disease results in the generation of numerous damage associated molecular patterns (DAMPs) that act on pattern recognition receptors on immune cells to initiate and shape the inflammatory response. DAMPs stimulate “priming,” resulting in increased expression of interleukin (IL)-1, and also stimulate inflammasome assembly, activation of caspase-1, and of other proteases capable of processing and activating IL-1. IL-1 (both IL-1 α and IL-1 β) is released and can then stimulate the type I IL-1 receptor on responsive cells to trigger inflammatory signaling pathways with pleiotropic effects.
Abbreviations: DAMPs, damage associated molecular patterns; NF-κB, nuclear factor-κB; IL, interleukin; IL-1R1, type I IL-1 receptor.
IL-1 processing and secretion
The protease most commonly reported to cleave pro-IL-1β is caspase-1. Caspase-1 is also produced as an inactive precursor however, and a “primed” cell must encounter a secondary stimulus that facilitates the activation of caspase-1. The secondary stimulus that drives IL-1β processing and release during sterile disease is an additional DAMP. During sterile inflammation the secondary stimulus is typically sensed by a cytosolic PRR called NACHT, LRR and PYD domains-containing protein (NLRP) 3.Citation24 Sterile activators of NLRP3 include (but not exclusively) molecules such as adenosine triphosphate (ATP),Citation25 sphingosine,Citation26 and monosodium urate.Citation27 There is limited evidence supporting a direct interaction between DAMP and NLRP3 and additional host cellular signals such as K+ efflux,Citation28 lysosomal destabilization and cathepsin activity,Citation29 and ROS production and/or mitochondrial dysfunction,Citation30 are suggested to be important. Once activated NLRP3 interacts via an interaction of pyrin domains with an adaptor protein called apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). ASC recruits pro-caspase-1 into the complex via an interaction of caspase activation and recruitment domain (CARD) that subsequently results in caspase-1 activation and processing of pro-IL-1β. This multimolecular IL-1β processing complex is called the inflammasome.Citation31 A role for the NLRP3 inflammasome in brain inflammation has been suggested using animal models of Alzheimer’s disease and multiple sclerosis.Citation32,Citation33 However, there is as yet no evidence to support a role of NLRP3 in ischemic brain injury. Another inflammasome forming PRR that is found in the brain is NLRP1,Citation34 which is expressed by neurons.Citation35 NLRP1 has a CARD and can thus interact with caspase-1 directly,Citation36 although the presence of ASC does appear to enhance its activity.Citation37 The NLRP1 inflammasome is suggested to be important in rodent models of acute CNS injury through studies using neutralizing antibodies in models of spinal cord injury and traumatic brain injury in the rat,Citation38,Citation39 and in a mouse model of thromboembolic stroke.Citation40
Once processed, IL-1β is secreted rapidly from the cell into the extracellular environment where it stimulates signaling pathways on IL-1RI expressing cells. The precise mechanisms of IL-1β secretion are poorly understood. IL-1β does not follow the conventional endoplasmic reticulum-golgi pathway of secretion and harnesses one or more of the nonconventional secretory routes. We have recently reviewed the literature for mechanisms of IL-1β secretion and postulated that the mechanism involved may depend on the strength of the inflammatory stimulus.Citation21 Based on this simple theory we classified IL-1β secretion mechanisms as (1) rescue and redirect where IL-1β targeted for degradation via lysosomal or autophagic routes could be rescued and secreted; (2) protected release where IL-1β is contained in secreted vesicles such as microvesicles shed from the plasma membrane, or exosomes released by the exocytosis of multivesicular bodies; and (3) terminal release where IL-1β is secreted through pores in the plasma membrane, with these pores ultimately resulting in an osmotic lysis of the cell. Whilst the vast majority of the literature reporting IL-1β release focuses on the inflammasome, it is important to note that additional caspase-1 independent mechanisms exist, and of particular relevance to sterile disease may be extracellular processing of pro-IL-1β by neutrophil derived proteases including proteinase 3.Citation41,Citation42 An extracellular cathepsin D dependent processing of pro-IL-1β has also been reported to occur from microglia stimulated with ATP under acidic conditions.Citation43
In contrast to IL-1β, there has been comparatively little research into the mechanisms of IL-1α processing and secretion. The secretion of IL-1α in response to DAMP stimulation is also reported to depend upon the inflammasome.Citation44 Pro-IL-1α is not a substrate for caspase-1 and it is not clear how inflammasome activation regulates its processing and release, although a non-catalytic role of caspase-1 and calpain protease dependent processing are implicated.Citation44,Citation45 An alternativeCitation23 to this model was proposed recently where pro-IL-1α binds tightly to an intracellular form of the type II IL-1 receptor (IL-1RII). Upon activation of the inflammasome, IL-1RII is cleaved by caspase-1 rendering pro-IL-1α available for calpain mediated cleavage.Citation23 Granzyme B is also reported to cleave pro-IL-1α leading to an increase in its biological activity.Citation22 In contrast to IL-1β, pro-IL-1α can traffic to the nucleus of the expressing cell and this mechanism may limit IL-1α release during cell death.Citation46
Consequences of IL-1 signaling
IL-1 released by microglia into the extracellular space can act directly on nearby neurons. IL-1β stimulates the activation of Src tyrosine kinases in neurons which results in phosphorylation of the NR2A and NR2B N-methyl-D-aspartate (NMDA) receptor subunits.Citation47 This subsequently results in increased calcium flux after NMDA receptor activation and increased excitotoxic cell death.Citation47 IL-1β can also exert neurotoxic effects via glial cell dependent production of reactive oxygen and proteases.Citation48,Citation49 In a microglia-neuron coculture model, aggregated amyloid-β (pathological feature of Alzheimer’s disease) induced neurotoxicity was lost when caspase-1 knockout microglia were used instead of wildtype.Citation50 Thus, “local” effects of IL-1 dependent signaling could contribute to brain injury.
We now know that in addition to centrally produced IL-1, peripheral sources also contribute to brain inflammation after experimental stroke.Citation51 Systemic IL-1β exacerbates neuronal injury after experimental stroke by increasing the production and infiltration of neutrophils into the ischemic tissue.Citation52 This neutrophil dependent toxicity is at least in part dependent upon a matrix metalloproteinase induced disruption of the blood–brain barrier.Citation53 In a culture system, platelet derived IL-1α can activate brain endothelial cells to produce chemokines and increase expression of adhesion molecules, thus facilitating the transmigration of neutrophils across an endothelial cell layer.Citation54 Transmigrated neutrophils were recently shown to be neurotoxic via a mechanism dependent upon proteases associated with neutrophil extracellular traps.Citation55 Thus, multiple sources and effects of IL-1 may contribute to the inflammation dependent damage that occurs during acute cerebrovascular disease.
Another consequence of IL-1 dependent inflammation in acute cerebrovascular disease is immune depression, which contributes to a susceptibility to infection.Citation56,Citation57 This is of particular clinical relevance, where infections following ischemic strokeCitation58,Citation59 and SAHCitation60 are observed with increased frequency, and are a major cause of morbidity in those surviving the initial insult. This phase of immune depression represents an unbalanced peripheral anti-inflammatory reaction following the initial insult,Citation56,Citation61 and results in altered lymphocyte functionCitation62 and depletion,Citation63 reduced natural killer cell function,Citation64 and a shift from Th1 to Th2 cytokine response.Citation65 The two main mechanisms that are thought to cause immune depression after an acute brain injury are (1) stimulation of the hypothalamic-pituitary-adrenal axis releasing adrenal cortisol, and (2) catecholamine mediated IL-10 release.Citation57,Citation66 IL-1β and IL-6 are direct inducers of both mechanisms.Citation56,Citation67 Our research group has also demonstrated the central role of IL-1 in poststroke immune depression by inducing its reversal with the IL-1 receptor antagonist (IL-1-Ra).Citation68 Further studies are currently underway in SAH and ICH. In ICH, although a reduction in white blood cells is not usually observed,Citation69 there is clinical evidence that plasma IL-10 concentrations are raised which may contribute to immune depression and increased risk of infection.Citation70
Targeting inflammation in acute cerebrovascular disease
A variety of pharmacological interventions against the IL-1 system have been studied in experimental models of acute cerebrovascular disease where they have been shown to limit neuronal injury.Citation71 We have recently reviewed these in more depth elsewhere,Citation71 but briefly, they range from biologicals such as IL-1Ra and neutralizing IL-1β antibodies, to small molecule inhibitors of inflammatory signaling pathways and caspase-1.Citation71 Although protective effects observed when targeting nuclear factor-κB or p38 mitogen activated protein kinase could be ascribed to a plethora of inflammatory signaling processes, at least comparable neuroprotective effects are observed with agents that specifically target the IL-1 pathway. Caspase-1 inhibitors are highly protective in rodent models of acute cerebrovascular disease.Citation72–Citation74 Antibodies that target IL-1β directly,Citation75 or components of the NLRP1 inflammasome are also protective in experimental models.Citation39,Citation40 For many years, IL-1Ra has been known to offer significant neuroprotection in experimental models of brain injury.Citation5 These studies have recently been extended to animals with comorbidities commonly seen in stroke patients, where similar neuroprotective effects of IL-1Ra are also reported.Citation76 Clinically, IL-1Ra has also been administered to patients with ischemic and hemorrhagic stroke where it was effective in reducing peripheral markers of inflammation.Citation77 In patients with SAH, IL-1Ra has been administered through both intravenousCitation78 and subcutaneous routes (Galea, unpublished data, 2012). Intravenous IL-1Ra has been demonstrated to penetrate CSF and achieve concentrations that are associated with neuroprotection in rodent studies.Citation79 Together these data suggest that targeting the IL-1 system could represent a real opportunity for treating acute cerebrovascular disease.
Conclusions, open questions, and future perspectives
In summary, there is compelling evidence to suggest that effective targeting of the IL-1 system could be therapeutic in the treatment of acute cerebrovascular disease. Despite the large number of potential DAMPs and signaling pathways involved in the regulation of IL-1, its actions depend entirely on IL-1RI activation, which is the only known signaling receptor for IL-1α and IL-1β (). Perhaps that is why IL-1Ra has proven to be so effective and is showing the greatest promise currently. Importantly IL-1Ra is also protective in comorbid models of stroke in rodents which better reflect the human condition.Citation76
In addition to nonrepresentative animal models, there are other reasons for the persistent failure of drugs that require an appreciation of the underlying biological pathways. Changes in the activity of single enzymes normally have only small effects on pathway fluxes, and multisite modulation is necessary to effect large changes in fluxes.Citation80 Targeting multiple nodes in a signaling network may therefore be a more effective therapeutic strategy.Citation81 The potential of this approach has already been demonstrated in a number of cellular models, including our own work on the expression of IL-1β.Citation82 The existence of such dynamic interactions and changes within the inflammatory cascade may thus be overcome by approaches that target multiple points within the inflammatory cascade.
At the fundamental level there exist a number of questions. Many arise from our yet incomplete understanding of inflammasome priming and activation and assembly, and the mechanism of secretion for IL-1. More specifically, the mechanisms and inflammasomes regulating IL-1 in acute brain injury remain unknown. Relative contributions of IL-1α and IL-1β to the expanding infarct or developing ischemia are also unknown. Understanding these processes will likely lead to the identification of new therapeutic targets that may further direct strategies to target the inflammation that occurs during acute cerebrovascular disease.
Acknowledgments
We are grateful to Professors Nancy Rothwell and Pippa Tyrrell, and to Dr Emmanuel Pinteaux (all from the University of Manchester), for providing detailed feedback on a draft version of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
- KeepRFHuaYXiGIntracerebral haemorrhage: mechanisms of injury and therapeutic targetsLancet Neurol20121172073122698888
- MendelowADGregsonBARowanENMurrayGDGholkarAMitchellPMSTICH II InvestigatorsEarly surgery versus initial conservative treatment in patients with spontaneous supratentorial lobar intracerebral haematomas (STICH II): a randomised trialLancet Epub5292013
- RockKLLatzEOntiverosFKonoHThe sterile inflammatory responseAnnu Rev Immunol20102832134220307211
- LucasSMRothwellNJGibsonRMThe role of inflammation in CNS injury and diseaseBr J Pharmacol2006147Suppl 1S232S24016402109
- AllanSMTyrrellPJRothwellNJInterleukin-1 and neuronal injuryNat Rev Immunol2005562964016034365
- LuheshiNMKovacsKJLopez-CastejonGBroughDDenesAInterleukin-1alpha expression precedes IL-1beta after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissuesJ Neuroinflammation2011818622206506
- AmanteaDBagettaGTassorelliCMercuriNBCorasanitiMTIdentification of distinct cellular pools of interleukin-1beta during the evolution of the neuroinflammatory response induced by transient middle cerebral artery occlusion in the brain of ratBrain Res2010131325926920025855
- DenesAFerencziSHalaszJKornyeiZKovacsKJRole of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouseJ Cereb Blood Flow Metab2008281707172118575457
- GreenhalghADBroughDRobinsonEMGirardSRothwellNJAllanSMInterleukin-1 receptor antagonist is beneficial after subarachnoid haemorrhage in rat by blocking haem-driven inflammatory pathologyDis Model Mech2012582383322679224
- LambertsenKLBiberKFinsenBInflammatory cytokines in experimental and human strokeJ Cereb Blood Flow Metab2012321677169822739623
- TarkowskiERosengrenLBlomstrandCEarly intrathecal production of interleukin-6 predicts the size of brain lesion in strokeStroke199526139313987631343
- HendrykSJarzabBJoskoJIncrease of the IL-1 beta and IL-6 levels in CSF in patients with vasospasm following aneurysmal SAHNeuro Endocrinol Lett20042514114715159698
- LeeSCLiuWDicksonDWBrosnanCFBermanJWCytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 betaJ Immunol1993150265926678454848
- RingheimGEBurgherKLHerouxJAInterleukin-6 mRNA expression by cortical neurons in culture: evidence for neuronal sources of interleukin-6 production in the brainJ Neuroimmunol1995631131238550808
- TarkowskiERosengrenLBlomstrandCIntrathecal release of pro- and anti-inflammatory cytokines during strokeClin Exp Immunol19971104924999409656
- ChenGYNunezGSterile inflammation: sensing and reacting to damageNat Rev Immunol20101082683721088683
- PiccininiAMMidwoodKSDAMPening inflammation by modulating TLR signallingMediators Inflamm2010 Article ID 672395
- SavageCDLopez-CastejonGDenesABroughDNLRP3-inflammasome activating DAMPs stimulate an inflammatory response in glia in the absence of priming which contributes to brain inflammation after injuryFront Immunol2012328823024646
- NewtonKDixitVMSignaling in innate immunity and inflammationCold Spring Harb Perspect Biol20124119
- WeberAWasiliewPKrachtMInterleukin-1 (IL-1) pathwaySci Signal2010193 (105):cm1.
- Lopez-CastejonGBroughDUnderstanding the mechanism of IL-1beta secretionCytokine Growth Factor Rev20112218919522019906
- AfoninaISTynanGALogueSEGranzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alphaMol Cell20114426527822017873
- ZhengYHumphryMMaguireJJBennettMRClarkeMCIntracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammationImmunity20133828529523395675
- CasselSLSutterwalaFSSterile inflammatory responses mediated by the NLRP3 inflammasomeEur J Immunol20104060761120201012
- MariathasanSWeissDSNewtonKCryopyrin activates the inflammasome in response to toxins and ATPNature200644022823216407890
- LuheshiNMGilesJALopez-CastejonGBroughDSphingosine regulates the NLRP3-inflammasome and IL-1beta release from macrophagesEur J Immunol20124271672522105559
- MartinonFPetrilliVMayorATardivelATschoppJGout-associated uric acid crystals activate the NALP3 inflammasomeNature200644023724116407889
- PetrilliVPapinSDostertCMayorAMartinonFTschoppJActivation of the NALP3 inflammasome is triggered by low intracellular potassium concentrationCell Death Differ2007141583158917599094
- HornungVLatzECritical functions of priming and lysosomal damage for NLRP3 activationEur J Immunol20104062062320201015
- ShimadaKCrotherTRKarlinJOxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosisImmunity20123640141422342844
- StrowigTHenao-MejiaJElinavEFlavellRInflammasomes in health and diseaseNature201248127828622258606
- HenekaMTKummerMPStutzANLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 miceNature201349367467823254930
- JhaSSrivastavaSYBrickeyWJThe inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18J Neurosci201030158111582021106820
- YinYYanYJiangXInflammasomes are differentially expressed in cardiovascular and other tissuesInt J Immunopathol Pharmacol20092231132219505385
- KummerJABroekhuizenREverettHInflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory responseJ Histochem Cytochem20075544345217164409
- SchroderKTschoppJThe inflammasomesCell201014082183220303873
- FaustinBLartigueLBrueyJMReconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activationMol Cell20072571372417349957
- de Rivero VaccariJPLotockiGMarcilloAEDietrichWDKeaneRWA molecular platform in neurons regulates inflammation after spinal cord injuryJ Neurosci2008283404341418367607
- de Rivero VaccariJPLotockiGAlonsoOFBramlettHMDietrichWDKeaneRWTherapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injuryJ Cereb Blood Flow Metab2009291251126119401709
- AbulafaDPde Rivero VaccariJPLozanoJDLotockiGKeaneRWDietrichWDInhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in miceJ Cereb Blood Flow Metab20092953454419066616
- FantuzziGKuGHardingMWResponse to local inflammation of IL-1 beta-converting enzyme-deficient miceJ Immunol1997158181818249029121
- JoostenLANeteaMGFantuzziGInflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1betaArthritis Rheum2009603651366219950280
- TakenouchiTIwamaruYSugamaSThe activation of P2X7 receptor induces cathepsin D-dependent production of a 20-kDa form of IL-1beta under acidic extracellular pH in LPS-primed microglial cellsJ Neurochem201111771272321395581
- GrossOYazdiASThomasCJInflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1Immunity20123638840022444631
- LuheshiNMRothwellNJBroughDDual functionality of interleukin-1 family cytokines: implications for anti-interleukin-1 therapyBr J Pharmacol20091571318132919681864
- LuheshiNMMcCollBWBroughDNuclear retention of IL-1 alpha by necrotic cells: a mechanism to dampen sterile inflammationEur J Immunol2009392973298019839011
- VivianiBBartesaghiSGardoniFInterleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinasesJ Neurosci2003238692870014507968
- ThorntonPPinteauxEGibsonRMAllanSMRothwellNJInterleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical releaseJ Neurochem20069825826616805812
- ThorntonPPinteauxEAllanSMRothwellNJMatrix metalloproteinase-9 and urokinase plasminogen activator mediate interleukin-1-induced neurotoxicityMol Cell Neurosci20083713514217939964
- HalleAHornungVPetzoldGCThe NALP3 inflammasome is involved in the innate immune response to amyloid-betaNat Immunol2008985786518604209
- DenesAWilkinsonFBiggerBChuMRothwellNJAllanSMCentral and haematopoietic interleukin-1 both contribute to ischaemic brain injury in miceDis Model Mech201361043104823519030
- McCollBWRothwellNJAllanSMSystemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1-and neutrophil-dependent mechanismsJ Neurosci2007274403441217442825
- McCollBWRothwellNJAllanSMSystemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in miceJ Neurosci2008289451946218799677
- ThorntonPMcCollBWGreenhalghADenesAAllanSMRothwellNJPlatelet interleukin-1{alpha} drives cerebrovascular inflammationBlood20101153632363920200351
- AllenCThorntonPDenesANeutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNAJ Immunol201218938139222661091
- MeiselCSchwabJMPrassKMeiselADirnaglUCentral nervous system injury-induced immune deficiency syndromeNat Rev Neurosci2005677578616163382
- WoiciechowskyCSchoningBLankschWRVolkHDDockeWDMechanisms of brain-mediated systemic anti-inflammatory syndrome causing immunodepressionJ Mol Med (Berl)19997776978010619437
- WeimarCRothMPZillessenGComplications following acute ischemic strokeEur Neurol20024813314012373029
- GrabskaKGromadzkaGCzlonkowskaAInfections and ischemic stroke outcomeNeurol Res Int2011201169134821766026
- StevensRDNyquistPAThe systemic implications of aneurysmal subarachnoid hemorrhageJ Neurol Sci200726114315617544451
- IadecolaCAnratherJThe immunology of stroke: from mechanisms to translationNat Med20111779680821738161
- WeissJMQuanNSundarSKWidespread activation and consequences of interleukin-1 in the brainAnn NY Acad Sci19947413383577825822
- OffnerHVandenbarkAAHurnPDEffect of experimental stroke on peripheral immunity: CNS ischemia induces profound immunosuppressionNeuroscience20091581098111118597949
- SundarSKBeckerKJCierpialMAIntracerebroventricular infusion of interleukin 1 rapidly decreases peripheral cellular immune responsesProc Natl Acad Sci U S A198986639864022548213
- CataniaALonatiCSordiAGattiSDetrimental consequences of brain injury on peripheral cellsBrain Behav Immun20092387788419394418
- ChamorroAAmaroSVargasMCatecholamines, infection, and death in acute ischemic strokeJ Neurol Sci2007252293517129587
- BethinKEVogtSKMugliaLJInterleukin-6 is an essential, corticotropin-releasing hormone-independent stimulator of the adrenal axis during immune system activationProc Natl Acad Sci U S A2000979317932210922080
- SmithCJEmsleyHCUdehCTInterleukin-1 receptor antagonist reverses stroke-associated peripheral immune suppressionCytokine20125838438922445501
- AgnihotriSCzapAStaffIFortunatoGMcCulloughLDPeripheral leukocyte counts and outcomes after intracerebral hemorrhageJ Neuroinflammation2011816022087759
- DziedzicTBartusSKlimkowiczAMotylMSlowikASzczudlikAIntracerebral hemorrhage triggers interleukin-6 and interleukin-10 release in bloodStroke2002332334233512215608
- BroughDTyrrellPJAllanSMRegulation of interleukin-1 in acute brain injuryTrends Pharmacol Sci20113261762221788085
- RossJBroughDGibsonRMLoddickSARothwellNJA selective, non-peptide caspase-1 inhibitor, VRT-018858, markedly reduces brain damage induced by transient ischemia in the ratNeuropharmacology20075363864217845807
- SuzukiHSozenTHasegawaYChenWZhangJHCaspase-1 inhibitor prevents neurogenic pulmonary edema after subarachnoid hemorrhage in miceStroke2009403872387519875734
- WuBMaQKhatibiNAc-YVAD-CMK decreases blood-brain barrier degradation by inhibiting caspase-1 activation of interleukin-1beta in intracerebral hemorrhage mouse modelTransl Stroke Res20101576420596246
- YamasakiYMatsuuraNShozuharaHOnoderaHItoyamaYKogureKInterleukin-1 as a pathogenetic mediator of ischemic brain damage in ratsStroke199526676680 discussion 6817709417
- PradilloJMDenesAGreenhalghADDelayed administration of interleukin-1 receptor antagonist reduces ischemic brain damage and inflammation in comorbid ratsJ Cereb Blood Flow Metab2012321810181922781338
- EmsleyHCSmithCJGeorgiouRFA randomised phase II study of interleukin-1 receptor antagonist in acute stroke patientsJ Neurol Neurosurg Psychiatry2005761366137216170078
- ClarkSRMcMahonCJGueorguievaIInterleukin-1 receptor antagonist penetrates human brain at experimentally therapeutic concentrationsJ Cereb Blood Flow Metab20082838739417684519
- GueorguievaIClarkSRMcMahonCJPharmacokinetic modelling of interleukin-1 receptor antagonist in plasma and cerebrospinal fluid of patients following subarachnoid haemorrhageBr J Clin Pharmacol20086531732517875190
- ThomasSFellDAThe role of multiple enzyme activation in metabolic flux controlAdv Enzyme Regul19983865859762347
- HopkinsALNetwork pharmacology: the next paradigm in drug discoveryNat Chem Biol2008468269018936753
- SmallBGMcCollBWAllmendingerREfficient discovery of anti-inflammatory small-molecule combinations using evolutionary computingNat Chem Biol2011790290822020553