Abstract
Background
Inflammatory cells play a major role in the pathology of heart failure by stimulating cardiac fibroblasts to regulate the extracellular matrix in an adverse way. In view of the fact that inflammatory cells have estrogen receptors, we hypothesized that estrogen provides cardioprotection by decreasing the ability of cardiac inflammatory cells to influence fibroblast function.
Methods
Male rats were assigned to either an untreated or estrogen-treated group. In the treated group, estrogen was delivered for 2 weeks via a subcutaneous implanted pellet containing 17β-estradiol. A mixed population of cardiac inflammatory cells, including T-lymphocytes (about 70%), macrophages (about 12%), and mast cells (about 12%), was isolated from each rat and cultured in a Boyden chamber with cardiac fibroblasts from untreated adult male rats for 24 hours. To examine if tumor necrosis factor-alpha (TNF-α) produced by inflammatory cells represents a mechanism contributing to the stimulatory effects of inflammatory cells on cardiac fibroblasts, inflammatory cells from the untreated group were incubated with cardiac fibroblasts in a Boyden chamber system for 24 hours in the presence of a TNF-α-neutralizing antibody. Cardiac fibroblasts were also incubated with 5 ng/mL of TNF-α for 24 hours. Fibro-blast proliferation, collagen synthesis, matrix metalloproteinase activity, β1 integrin protein levels, and the ability of fibroblasts to contract collagen gels were determined in all groups and statistically compared via one-way analysis of variance.
Results
Inflammatory cells from the untreated group resulted in: 1) an increased fibroblast proliferation, collagen production and matrix metalloproteinase activity; and 2) a loss of β1 integrin protein and a reduced ability to contract collagen gels. In contrast, inflammatory cells from the treated group resulted in: 1) an attenuated fibroblast proliferation; 2) a nonsignificant reduction in collagen production; 3) the prevention of matrix metalloproteinase activation and the loss of β1 integrin by fibroblasts and 4) a preservation of the fibroblasts’ ability to contract collagen gels. The TNF-α neutralizing antibody attenuated or prevented the untreated inflammatory cell-induced fibroblast proliferation, collagen production, matrix metalloproteinase activation and loss of β1 integrin protein as well as preserved fibroblast contractile ability. Incubation with TNF-α yielded changes in the cardiac fibroblast parameters that were directionally similar to the results obtained with untreated inflammatory cells.
Conclusion
These results and those of our previous in vivo studies suggest that a major mechanism by which estrogen provides cardioprotection is its ability to modulate synthesis of TNF-α by inflammatory cells, thereby preventing inflammatory cell induction of cardiac fibroblast events that contribute to adverse extracellular matrix remodeling.
Introduction
Sustained cardiac volume overload in male rats results in increased myocardial matrix metalloproteinase (MMP) activation and collagen degradation, which leads to ventricular dilatation. In contrast, female rats exposed to volume overload are cardioprotected, ie, collagen degradation does not occur and the ventricle does not dilate.Citation1–Citation3 However, following ovariectomy, volume overload does induce adverse cardiac remodeling almost identical to that in males.Citation1,Citation3 Estrogen supplementation in ovariectomized female rats restores cardioprotectionCitation2 and also confers cardioprotection in male rats.Citation4 Using a polymerase chain reaction array screening approach, we recently found the expression of several inflammatory genes to be regulated by estrogen in male hearts exposed to volume overload.Citation5 This raises the possibility that the cardioprotection provided by estrogen may, in part, be due to modulation of inflammatory cells by estrogen. There is now ample evidence to demonstrate that multiple types of inflammatory cells, including mast cells,Citation6–Citation16 macrophages,Citation17–Citation23 and T-cells,Citation19,Citation24–Citation26 are involved in myocardial remodeling in response to sustained elevations in myocardial stress, disease, or injury. While initially there may be differences in the temporal activation of these inflammatory cells, it is likely that a mixed population of these cells serves to amplify proinflammatory signaling pathways leading to adverse remodeling. Interestingly, mast cells,Citation27 macrophages,Citation28 and T-cellsCitation29 all have estrogen receptors, and estrogen has also been shown to regulate proinflammatory cytokines in noncardiac mast cells,Citation30 macrophages,Citation31 and T-cells.Citation32 Since cardiac fibroblasts are the cell type predominantly responsible for maintenance and repair of the extracellular matrix in the heart by actively producing collagen and MMPs, we hypothesized that estrogen may favorably modulate the function of cardiac inflammatory cells, thereby preventing their adverse effects on cardiac fibroblast phenotype, regulation, and function. The results indicate that estrogen prevents inflammatory cell induction of cardiac fibroblast events that contributes to the adverse extracellular matrix remodeling associated with degradation of the extracellular matrix and ventricular dilatation primarily via its ability to modulate the inflammatory cell release of tumor necrosis factor-alpha (TNF-α).
Materials and methods
Studies were performed using 8-week-old male (n = 16) Sprague Dawley (Hsd:SD) rats that were housed under standard environmental conditions and maintained on a commercially available estrogen-free diet and tap water ad libitum. All studies conformed to the principles of the National Institutes Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of South Carolina.
Experimental design
Male rats were chosen for this study for two reasons, ie, we have previously shown that treatment with estrogen attenuates cardiac remodeling in male ratsCitation4 and the fact that female rats are cardioprotected indicates that their cells have already acquired a protective phenotype. The rats were randomly allocated to an untreated group or an estrogen-treated group. Estrogen was delivered via a 21-day 17β-estradiol pellet (Innovative Research of America, Sarasota, FL, USA). The pellets were implanted subcutaneously between the shoulder blades for 2 weeks prior to isolation of cardiac inflammatory cells. We have shown that a daily dose of 0.02 mg/kg produces serum levels of approximately 35 pg/mL, which is comparable with the average estrogen level in female rats with ovaries.Citation2 All cardiac fibroblasts were isolated from male rats that were not exposed to exogenous estrogen.
Isolation of cardiac inflammatory cells
Cardiac inflammatory cells were isolated as described previouslyCitation33 This isolation method results in collection of a mixed population of inflammatory cells, including T-lymphocytes (about 70%), macrophages (about 12%), and mast cells (about 12%).Citation34 Briefly, a thoracotomy exposed the intact pericardial sac. The tip of a Teflon® catheter sleeve attached to a sterile syringe filled with Hank’s Balanced Salt Solution (7.4 pH, Gibco, Grand Island NY, USA) was then inserted into the pericardial sac, and the sac was filled with buffer. The buffer, which now contained the inflammatory cells, was aspirated into another sterile syringe. This procedure was repeated several times. The cells collected were then pelleted and the supernatant was removed. The pelleted cells were resuspended in HyClone buffer (Hank’s Balanced Salt Solution containing magnesium sulfate [1.1 mmol/L], calcium chloride [1.3 mmol/L], and phenol red; HyClone, Logan, UT, USA). The cells were then divided into 50,000 cell aliquots to be used for further experimentation. Inflammatory cells isolated from untreated male rats are henceforth referred to as “inflammatory cells” while those isolated from estrogen-treated male rats are referred to as “inflammatory cells + estrogen”.
Isolation of cardiac fibroblasts and Boyden chamber experiments
Primary cultures of cardiac fibroblasts were isolated from untreated adult male Hsd:SD rats (n = 4) as previously described.Citation10,Citation35 Accordingly, the fibroblasts were not exposed to the effects of exogenous estrogen. Briefly, the hearts were homogenized and digested with Liberase Blendzyme 3 (Roche Diagnostics, Roche, Indianapolis, IN, USA), with the fibroblasts purified by selective attachment to plastic culture ware. The cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Life Technologies, Carlsbad, CA, USA) containing 10% neonatal bovine serum and 5% fetal calf serum, and were used before passage number 3. Prior to experimentation, one million fibroblasts were allowed to adhere in DMEM with 10% neonatal bovine serum and 5% fetal calf serum for 24 hours before rinsing with Mosconas salt solution and serum starvation for 24 hours in DMEM/F-12. The medium was then replaced with DMEM containing 1.5% fetal bovine serum. To understand how inflammatory cells modulate fibroblast function, isolated cardiac inflammatory cells (50,000 cells), inflammatory cells + estrogen (50,000 cells), or inflammatory cells + rat recombinant anti-TNF-α neutralizing antibody (50,000 cells) were added to the upper portion of a Boyden chamber (1 μm inserts, BD Bioscience, Bedford, MA, USA), while the fibroblasts remained in the lower portion. A control group in the upper chamber contained no inflammatory cells. The neutralizing antibody to TNF-α was obtained from R&D Systems (Minneapolis, MN, USA) and used in a 1:1,000 dilution. The Boyden chamber arrangement allowed secretions from the inflammatory cells and the fibroblasts to pass through the filter while prohibiting direct contact between the fibroblasts and inflammatory cells. Accordingly, any change in fibroblast behavior would be directly related to secretions from the inflammatory cells. This inflammatory cell/fibroblast secretion exposure was continued for 24 hours at 37°C.
Collagen gel contraction
Cardiac fibroblasts were serum-starved by culture for 24 hours in DMEM/F-12. Fibroblasts were detached from the culture flasks by incubation in 0.25% trypsin and 0.1% ethylenediaminetetraacetic acid for several minutes. Cells were pelleted by centrifugation, resuspended in DMEM/F-12 at a concentration of 200,000 cells/mL, and mixed with an equal volume of collagen solution (Invitrogen, Carlsbad, CA, USA) as previously described.Citation36,Citation37 Briefly, 1 mL of the solution, at a final concentration of 1.28 mg/mL of collagen, and 100,000 fibroblasts/mL was added to each well of a 24-well plate precoated with 2% bovine serum albumin. The plate containing the collagen and cell mixture was incubated at 37°C for one hour to allow the collagen and cell mixture to polymerize. A 10 μL pipette tip was used to release the collagen gels from the sides of the plate following collagen polymerization. Inflammatory cells (n = 50,000), inflammatory cells + estrogen (n = 50,000), or inflammatory cells + anti-TNF-α-neutralizing antibody (50,000 cells) were added to the collagen gels. The controls had no inflammatory cells added. Following incubation for 6, 12, or 24 hours, images of the collagen gels were captured. The surface areas of the collagen gels were determined as a measure of the contractile ability of the fibroblasts.
Collagen assay
Collagen synthesis was determined by hydroxyproline (HPro) analysis of the collected medium, as described by Edwards and O’BrienCitation38 and our research group.Citation10,Citation35 Briefly, 6 N HCL was added to all media samples in a 1:1 ratio, and the samples were maintained at 108°C overnight, filtered, and subjected to vacuum centrifugation. The samples were reconstituted in citrate buffer (0.2 M) and reacted with chloramine-T for 20 minutes, followed by incubation with aldehyde for 20 minutes, before cooling on ice. Absorbance was read at 550 nm and compared with HPro standards ranging from 0 to 10 μg/mL.
Fibroblast proliferation
Additional fibroblasts were grown on coverslips until 50%–60% confluent for proliferation studies. As above, the fibroblasts and inflammatory cells, inflammatory cells + estrogen, or inflammatory cells + anti-TNF-α-neutralizing antibody were placed in the Boyden chamber. Control groups had no inflammatory cells added. Next, 5-bromo-2′-deoxyuridine (BrdU, Roche Applied Science, Indianapolis, IN, USA) labeling reagent was added to the medium under sterile conditions for the final 4 hours of the 24-hour treatment period. The culture medium was then aspirated and the fibroblasts were fixed with ethanol/glycine for one hour at −20°C. Anti-BrdU mouse immunoglobulin G (IgG) antibody was added and incubation was continued for another hour at 37°C. After washing, fluorescein isothiocyanate-conjugated mouse IgG secondary antibody was added to the fibroblasts for one hour at 37°C. Propidium iodide (Molecular Probes, Eugene, OR USA) served as the nuclear marker. The fibroblasts were mounted and examined for BrdU incorporation using a confocal laser scanning microscope (MRC-1024, Bio-Rad, Hercules, CA, USA) at 400× magnification. Proliferation is expressed as the percentage of BrdU-positive to total number of fibroblasts.
Matrix metalloproteinase activity
Total MMP and MMP-2 activities were determined from the cell culture media using assay kits from AnaSpec, Inc (Fremont, CA, USA).
Western blot analysis
Total protein was extracted from the cultured cardiac fibroblasts by homogenization of the cells with buffer containing a protease inhibitor cocktail (Pierce, Rockford IL, USA). Protein concentrations were determined by Bio-Rad protein assay. Next, 35 μg of total protein was loaded and separated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred onto a nitrocellulose membrane (Bio-Rad). Ponceau S staining (Sigma-Aldrich, St Louis, MO, USA) was used to confirm equal loading and accurate transfer of proteins from the SDS-PAGE gel to the nitrocellulose membrane. The membranes were blocked with 5% nonfat milk in Tris-buffered saline/0.01% Tween for 2 hours at room temperature. For detection of β1 integrin, the blots were probed with rabbit anti-β1 integrin (sc-8978, Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:200) for 2 hours at room temperature, and then goat antirabbit IgG (sc-2004, Santa Cruz Biotechnology, 1:3000) for 2 hours at room temperature. The blots were also probed for glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which served as a loading control (primary antibody, mouse anti-GAPDH, 1:3000 at room temperature for 2 hours, Santa Cruz Biotechnology; secondary antibody, goat anti-mouse IgG2a, 1:5000 at room temperature for 2 hours, Santa Cruz Biotechnology). Immunoreaction signals were visualized with enhanced chemiluminescence (Pierce) by exposure to hyperfilm (Phenix Research Products, Hayward CA, USA). Densitometry analysis was performed using a calibrated Bio-Rad GS-800 densitometer. The β1 integrin was quantified as a ratio to GAPDH.
Data and statistical analysis
All grouped data are expressed as the mean ± standard error of the mean. Grouped data comparisons were assessed by one-way analysis of variance. When a significant F ratio (P < 0.05) was obtained intergroup comparisons were made using the Bonferroni post hoc test.
Results
Effect of inflammatory cells and estrogen on cardiac fibroblast proliferation, collagen production, and matrix metalloproteinase activity
Secretions from the untreated group of cardiac inflammatory cells significantly increased the ability of the fibroblasts to proliferate (). Pretreatment of inflammatory cells with estrogen resulted in small but significant attenuation of this proliferation. Secretions from untreated cardiac inflammatory cells also significantly increased production of collagen by fibroblasts (). Inflammatory cells from rats receiving estrogen did not significantly alter the increased collagen obtained with untreated inflammatory cells. Untreated inflammatory cell secretions also significantly increased total MMP activity () and MMP-2 activity (). These increases in total MMP and MMP-2 (data not shown) activity did not occur when fibroblasts were exposed in the Boyden chamber to inflammatory cells pre-treated with estrogen.
Figure 1 Cardiac fibroblast proliferation (A), cardiac fibroblast hydroxyproline (HPro) release (B), and total matrix metalloproteinase (MMP) activity (C) in medium in response to coculture of cardiac fibroblasts with untreated cardiac inflammatory cells (IC) and estrogen-treated cardiac inflammatory cells (IC + E2). *P < 0.05 versus control; #P < 0.05 versus IC.
Abbreviation: BrdU, 5-bromo-2′-deoxyuridine.
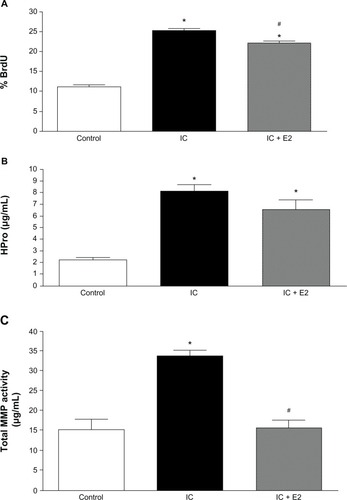
Figure 2 Cardiac fibroblast proliferation (A), cardiac fibroblast hydroxyproline (HPro) release (B), and matrix metalloproteinase-2 (MMP-2) activity (C) in medium in response to coculture of cardiac fibroblasts with untreated inflammatory cells (IC) and cardiac inflammatory cells in conjunction with a neutralizing antibody against TNF-α (IC + anti-TNF-α). *P < 0.05 versus control; #P < 0.05 versus IC.
Abbreviations: BrdU, 5-bromo-2′-deoxyuridine; TNF-α, tumor necrosis factor-alpha.
Effect of TNF-α neutralization on cardiac fibroblast proliferation, collagen production, and matrix metalloproteinase activity
Untreated inflammatory cell-induced proliferation of cardiac fibroblasts was slightly but significantly attenuated in the presence of a neutralizing antibody against TNF-α (). TNF-α neutralization also significantly attenuated the inflammatory cell-induced increase in collagen production by cardiac fibroblasts () and the increase in total MMP (data not shown) and MMP-2 () activity in the media.
Collagen gel contraction
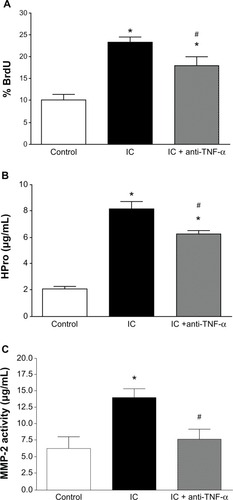
Untreated cardiac inflammatory cells significantly inhibited cardiac fibroblast-mediated contraction of collagen gels at 6, 12, and 24 hours (). This inhibition of gel contraction did not occur in the presence of estrogen-treated cardiac inflammatory cells () or in the presence of the TNF-α-neutralizing antibody when tested at 24 hours ().
Figure 3 Collagen gel contraction data for gels containing adult cardiac fibroblasts cocultured with cardiac inflammatory cells (IC) and estrogen-treated inflammatory cells (IC + E2) at 6, 12, and 24 hours (A) and collagen gel contraction data for gels containing adult cardiac fibroblasts cocultured with cardiac inflammatory cells (IC) and cardiac inflammatory cells in the presence of a neutralizing antibody against TNF-α (IC + anti-TNF-α) at 24 hours (B). *P < 0.05 versus control; #P < 0.05 versus IC.
Abbreviation: TNF-α, tumor necrosis factor-alpha.
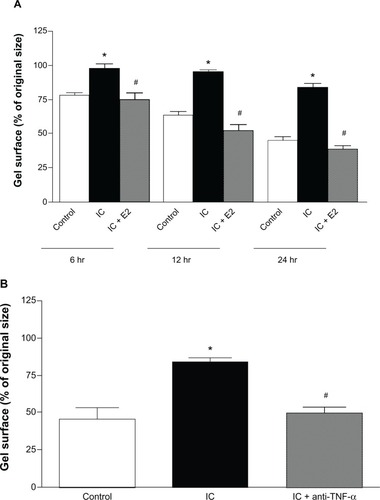
Western blot analysis of β1 integrin
In , the protein levels of β1 integrin are seen to be decreased in fibroblasts exposed to untreated inflammatory cell secretions when compared with the control. Boyden chamber exposure to estrogen-treated inflammatory cells prevented this decrease in β1 integrin levels. Inflammatory cell-induced loss of β1 integrin protein by cardiac fibroblasts was also prevented by the TNF-α-neutralizing antibody ().
Figure 4 (A) Western blot example and bar graph of β 1 integrin levels from isolated cardiac fibroblasts cocultured with cardiac inflammatory cells (IC) and estrogen-treated inflammatory cells (IC + E2). (B) Western blot example and bar graph of β1 integrin levels from isolated cardiac fibroblasts cocultured with cardiac inflammatory cells (IC) and cardiac inflammatory cells in the presence of a neutralizing antibody against TNF-α (IC + anti-TNF-α). *P < 0.05 versus control; *P < 0.05 versus IC.
Abbreviations: TNF-α, tumor necrosis factor-alpha; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Influence of TNF-α on fibroblasts
The findings with the neutralizing antibody ( to ) indicate that inflammatory cell-secreted TNF-α substantially contributes to many of the inflammatory cell-induced alterations in fibroblast function. To further investigate the role of TNF-α, cardiac fibroblasts were incubated with 5 ng/mL of TNF-α for 24 hours and the resulting percent changes in BrdU, HPro, total MMP, MMP-2, collagen gel contraction, and β1 integrin protein were determined. The TNF-α incubation results are compared to the percent changes following incubation with untreated inflammatory cells in . These comparisons together with the TNF-α neutralizing antibody results in to indicate that TNF-α accounted for most of the untreated inflammatory cell-induced percent changes in MMP-2, collagen gel contraction, and β1 integrin protein, while it contributed partially to the untreated inflammatory cell-induced changes in collagen synthesis and essentially nothing to fibroblast proliferation.
Table 1 Comparison of percent changes in cardiac fibroblast functional parameters resulting from incubation with either 5 ng/ml of TNF-α or untreated cardiac inflammatory cells for 24 hours
Discussion
There is now acceptance that inflammatory cells play a significant role in the adverse remodeling of the left ventricle that eventually leads to the development of heart failure. However, whether estrogen confers cardioprotection by modulating the influence of inflammatory cells is unknown. As a follow-up study to those reporting that estrogen provides cardioprotection in female and male rats in a model of chronic volume overload, we performed a polymerase chain reaction array analysis on hearts from untreated and estrogen-treated male rats exposed to chronic cardiac volume overload.Citation5 This array analysis identified several inflammation-related genes, including the genes for TNF-α, that were regulated by estrogen, raising the possibility that estrogen may modulate inflammatory cell function in the diseased heart. Interestingly, many inflammatory cells have estrogen receptors,Citation27–Citation29 and estrogen has been shown to regulate inflammatory cell cytokines and proteases.Citation30–Citation32 Since a critical part of the response of the heart to volume overload is an alteration to the extracellular matrix and cardiac fibroblasts are predominantly responsible for regulating the extracellular matrix, we hypothesized that estrogen modulates inflammatory cell function, leading to a change in the effect that these cells have on cardiac fibroblasts and ultimately on remodeling of the extracellular matrix.
A previous coculture study showed that peritoneal-derived mast cells were able to directly modulate neonatal cardiac fibroblast function by increasing collagen mRNA levels, inhibiting collagen gel contraction and increasing MMP protein levels and activity.Citation37 This is consistent with our in vivo findings in chronic volume overload where mast cells mediate MMP activation leading to collagen degradation.Citation6,Citation9 This mast cell-mediated effect occurs early in the volume overload-induced remodeling process and it is likely that, as the time course of remodeling progresses, multiple types of inflammatory cells are also involved. Therefore, this study addressed the following: the net effects that a mixed population of cardiac inflammatory cells have on adult cardiac fibroblasts; whether estrogen modulates the ability of cardiac inflammatory cells to affect fibroblast function; and the mediatory role of inflammatory cell-derived TNF-α on fibroblast function. Our findings show that Boyden chamber exposure of cardiac adult male fibroblasts with cardiac inflammatory cells increased fibroblast proliferation and collagen production. Interestingly, inflammatory cell exposure increased total and MMP-2 activity in the media. While collagen synthesis and MMP activation would seem to be paradoxical functions, regulation of the extracellular matrix is ultimately a balance between synthesis and degradation rates. For instance, increases in MMP activity and subsequent collagen degradation can eventually exceed collagen synthesis, which can cause a net loss in the percent of myocardial fibrillar collagen.Citation39 Therefore, the inflammatory cell-induced activation of MMPs and collagen synthesis by fibroblasts, as observed in this study, could still produce a net degradative effect in vivo. Inflammatory cell secretions also resulted in a 22%–25% decrease in the amount of fibroblast β1 integrin protein, which in all likelihood contributed to the reduction in ability of the fibroblasts to contract collagen gel. This suggests a trend toward a fibroblast degradative phenotype associated with ventricular dilatation.
Another novel aspect of this study is the finding that estrogen treatment of cardiac inflammatory cells altered the response of the cardiac fibroblasts to inflammatory cell secretions. Notably, estrogen treatment of the inflammatory cells only slightly, but significantly, attenuated the increased proliferation by adult cardiac fibroblasts. However, the most dramatic effect of estrogen was on MMP production and fibroblast β1 integrin protein levels. The increase in MMP activity seen with exposure to the non-estrogen-treated inflammatory cells was completely prevented by estrogen, as was the loss of β1 integrin by cardiac fibroblasts. These changes were manifested functionally by restoration of the ability of fibroblasts to contract collagen gels, which supports an in vivo preservation of the extracellular matrix. In fact, an increase in MMP activity is similar to what occurs in the male rat heart and in the hearts of ovariectomized female rats subjected to chronic cardiac volume overload, but does not occur in intact females.Citation3,Citation6,Citation40 In this regard, Chancey et alCitation41 showed that, as a result of cardiac mast cell chemical activation, ovariectomized rat hearts had a significant rapid increase in MMP-2 activity and a decrease in myocardial collagen concentration, which was not seen in the hearts of ovariectomized rats receiving supplementary estrogen.
Estrogen is likely either decreasing production of mediators by cardiac inflammatory cells or preventing their activation. Certainly estrogen can modulate inflammatory cell cytokine production and activation of inflammatory cells.Citation30–Citation32 Estrogen exerts rapid nongenomic responses via cell surface estrogen receptors linked to signal transduction pathways, such as mitogen-activated protein kinase,Citation32,Citation42 calcium cation influx,Citation43 or PI3K-AKT.Citation44 However, estrogen is known to modulate inflammatory cells at the transcriptional level since estrogen receptors are nuclear hormone transcription factors that subsequently bind estrogen response elements to activate or suppress specific gene targets.Citation45 We have recently shown in a model of chronic cardiac volume overload that estrogen downregulated gene transcription of TNF-α,Citation5 thus preventing the increased levels of TNF-α that are normally seen in the heart in response to volume overload.Citation3,Citation5 Since TNF-α causes MMP activation and collagen degradation in the volume-overloaded heart,Citation46,Citation47 actions consistent with what we observed in our inflammatory cell-fibroblast Boyden chamber study, we sought to determine if TNF-α mediated the effects of inflammatory cells on cardiac fibroblasts. Addition of a TNF-α-neutralizing antibody to the inflammatory cells attenuated collagen production and fibroblast proliferation; results very similar to those seen using inflammatory cells from the estrogen-treated group. However, the greatest effects of TNF-α neutralization were that it: prevented the increase in MMP activity; prevented the loss of β1 integrin on cardiac fibroblasts; and restored the ability of fibroblasts to contract collagen gels. The incubation of fibroblasts with TNF-α reinforces these observations and indicates that TNF-α accounted for most of the untreated inflammatory cell-induced percent changes in MMP-2, collagen gel contraction, and β1 integrin protein levels, while it only contributed partially to the untreated inflammatory cell-induced changes in collagen synthesis and essentially nothing to fibroblast proliferation. These responses are again consistent with what we observed with estrogen-treated inflammatory cells, suggesting that estrogen acts in part by downregulating TNF-α production by inflammatory cells. To this end, we have previously shown that around 90% of inflammatory cells obtained from the male rat heart contain TNF-α.Citation48
Taken together, the results of this in vitro study and our previous in vivo studies suggest that estrogen provides cardioprotection, at least in part, by modulating the cardiac inflammatory cell phenotype. This in turn affects the ability of cardiac fibroblasts to adversely modify the extracellular matrix. Specifically, the results of this study indicate that female cardioprotection is in part related to the ability of estrogen to target the synthesis and secretion of TNF-α by cardiac inflammatory cells.
Author contributions
JSJ contributed to the conception and design of the research, data analysis, interpretation of experimental results, edited and revised the manuscript, and approved the final version of manuscript, as did SPL (who also drafted the manuscript) and JLM (who also performed experiments). JL performed experiments and participated in the analysis of the data, interpretation of the results, construction of the figures and critical editing of the manuscript as well as approved the final version of manuscript.
Acknowledgments
This work was supported by a United Negro College Fund-Merck Graduate Science Research Dissertation Fellowship (to JLM) and National Heart, Lung, and Blood Institute grants R01-HL-62228 (to JSJ), R01-HL-073990 (to JSJ), and R00-HL-093215 (to SPL).
Disclosure
The authors declare that they have no competing interests in this work.
References
- BrowerGLGardnerJDJanickiJSGender mediated cardiac protection from adverse ventricular remodeling is abolished by ovariectomyMol Cell Biochem2003251899514575309
- GardnerJDBrowerGLVoloshenyukTGJanickiJSCardioprotection in female rats subjected to chronic volume overload: synergistic interaction of estrogen and phytoestrogensAm J Physiol Heart Circ Physiol2008294H198H20417965290
- LuHMelendezGCLevickSPJanickiJSPrevention of adverse cardiac remodeling to volume overload in female rats is the result of an estrogen-altered mast cell phenotypeAm J Physiol Heart Circ Physiol2012302H811H81722160000
- GardnerJDMurrayDBVoloshenyukTGBrowerGLBradleyJMJanickiJSEstrogen attenuates chronic volume overload induced structural and functional remodeling in male rat heartsAm J Physiol Heart Circ Physiol2010298H497H50419933421
- McLartyJLMelendezGCLevickSPEstrogenic modulation of inflammation-related genes in male rats following volume overloadPhysiol Genomics20124436237322274565
- BrowerGLChanceyALThanigarajSMatsubaraBBJanickiJSCause and effect relationship between myocardial mast cell number and matrix metalloproteinase activityAm J Physiol2002283H518H525
- BrowerGLJanickiJSPharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in ratsJ Cardiac Fail200511548556
- ChanceyALBrowerGLJanickiJSCardiac mast cell-mediated activation of gelatinase and alteration of ventricular diastolic functionAm J Physiol2002282H2152H2158
- LevickSPGardnerJDHollandMHauer-JensenMJanickiJSBrowerGLProtection from adverse myocardial remodeling secondary to chronic volume overload in mast cell deficient ratsJ Mol Cell Cardiol200845566118538342
- McLartyJLMelendezGCBrowerGLJanickiJSLevickSPTryptase/protease-activated receptor 2 interactions induce selective mitogen-activated protein kinase signaling and collagen synthesis by cardiac fibroblastsHypertension20115826427021730297
- HaraMOnoKHwangMWEvidence for a role of mast cells in the evolution to congestive heart failureJ Exp Med200219537538111828013
- MackinsCJKanoSSeyediNCardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusionJ Clin Invest20061161063107016585966
- ReidACSilverRBLeviRRenin: at the heart of the mast cellImmunol Rev200721712314017498056
- SilverRBReidACMackinsCJMast cells: a unique source of reninProc Natl Acad Sci U S A2004101136071361215342908
- FrangogiannisNGLindseyMLMichaelLHResident cardiac mast cells degranulate and release preformed TNF-α, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusionCirculation1998986997109715863
- SomasundaramPRenGNagarHMast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarctsJ Pathol200520510211115586361
- HinglaisNHuedesDNicolettiAColocalization of myocardial fibrosis and inflammatory cells in ratsLab Invest1994702862948139269
- KoyanagiMEgashiraKKitamotoSRole of monocyte chemoattractant protein-1 in cardiovascular remodeling induced by chronic blockade of nitric oxide synthesisCirculation20001022243224811056100
- KanzakiYTerasakiFOkabeMMyocardial inflammatory cell infiltrates in cases of dilated cardiomyopathy as a determinant of outcome following partial left ventriculectomyJpn Circ J20016579780211548879
- HayashidaniSTsutsuiHShiomiTAnti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarctionCirculation20031082134214014517168
- KuwaharaFKaiHTokudaKHypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation?Hypertension20044373974514967845
- KagitaniSUenoHHiradeSTakahashiTTakataMInoueHTranilast attenuates myocardial fibrosis in association with suppression of monocyte/macrophage infiltration in DOCA/salt hypertensive ratsJ Hypertens2004221007101515097242
- MaekawaYAnzaiTYoshikawaTEffect of granulocyte-macrophage colony-stimulating factor inducer on left ventricular remodeling after acute myocardial infarctionJ Am Coll Cardiol2004441510152015464336
- YuQWatsonRRMarchalonisJJLarsonDFA role for T lymphocytes in mediating cardiac diastolic functionAm J Physiol Heart Circ Physiol2005289H643H65116014617
- RatcliffeNRHutchinsJBarryBHickeyWFChronic myocarditis induced by T cells reactive to a single cardiac myosin peptide: persistent inflammation, cardiac dilatation, myocardial scarring and continuous myocyte apoptosisJ Autoimmun20001535936711040076
- YuQHorakKLarsonDFRole of T lymphocytes in hypertension-induced cardiac extracellular matrix remodelingHypertension2006489810416735642
- NicovaniSRudolphMIEstrogen receptors in mast cells from arterial wallsBiocell200226152412058378
- CapellinoSMontagnaPVillaggioBRole of estrogens in inflammatory response: expression of estrogen receptors in peritoneal fluid macrophages from endometriosisAnn N Y Acad Sci2006106926326716855153
- TornwallJCareyABFoxRIFoxHSEstrogen in autoimmunity: expression of estrogen receptors in thymic and autoimmune T cellsJ Gend Specif Med19992334011252833
- HarnishDCAlbertLMLeathurbyYBeneficial effects of estrogen treatment in the HLA-B27 transgenic rat model of inflammatory bowel diseaseAm J Physiol Gastrointest Liver Physiol2004286G118G12512958017
- CorcoranMPMeydaniMLichtensteinAHSchaeferEJDillardALamon-FavaSSex hormone modulation of proinflammatory cytokine and C-reactive protein expression in macrophages from older men and postmenopausal womenJ Endocrinol201020621722420484148
- SuzukiTYuHPHsiehYCChoudhryMABlandKIChaudryIHMitogen activated protein kinase (MAPK) mediates non-genomic pathway of estrogen on T cell cytokine production following trauma-hemorrhageCytokine200842323818343154
- McLartyJLMelendezGCSpencerWJLevickSPBrowerGLJanickiJSIsolation of functional cardiac immune cellsJ Vis Exp201158pii 3020
- LevickSPMurrayDBJanickiJSBrowerGLSympathetic nervous system modulation of inflammation and remodeling in the hypertensive heartHypertension20105527027620048196
- MelendezGCMcLartyJLLevickSPDuYJanickiJSBrowerGLInterleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in ratsHypertension20105622523120606113
- BurgessMLCarverWETerracioLWilsonSPWilsonMABorgTKIntegrin-mediated collagen gel contraction by cardiac fibroblasts. Effects of angiotensin IICirc Res1994742912988293568
- de AlmeidaAMustinDFormanMFBrowerGLJanickiJSCarverWEffects of mast cells on the behavior of isolated heart fibroblasts: modulation of collagen remodeling and gene expressionJ Cell Physiol2002191515911920681
- EdwardsCAO’BrienWDModified assay for determination of hydroxyproline in a tissue hydrolyzateClin Chim Acta19801041611677389130
- JanickiJSCollagen degradation in the heartEghbali-WebbMMolecular Biology of Collagen Matrix in the HeartAustin TXRG Landes Company1995
- VoloshenyukTGGardnerJDEstrogen improves TIMP-MMP balance and collagen distribution in volume-overloaded hearts of ovariectomized femalesAm J Physiol Regul Integr Comp Physiol2010299R683R69320504902
- ChanceyALGardnerJDMurrayDBBrowerGLJanickiJSModulation of cardiac mast cell-mediated extracellular matrix degradation by estrogenAm J Physiol Heart Circ Physiol2005289H316H32115722408
- MendelsohnMEKarasRHEstrogen and the blood vessel wallCurr Opin Cardiol199496196267987043
- ZhaoXMacBrideMMPetersonBRPfaffDWVasudevanNCalcium flux in neuroblastoma cells is a coupling mechanism between non-genomic and genomic modes of estrogensNeuroendocrinology20058117418216020926
- PattenRDPouratiIAronovitzMJ17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signalingCirc Res20049569269915345655
- KumarVGreenSStaubAChambonPLocalisation of the oestradiol-binding and putative DNA-binding domains of the human oestrogen receptorEMBO J19865223122363780678
- BozkurtBKribbsSBClubbFJJrPathophysiologically relevant concentrations of tumor necrosis factor-α promote progressive left ventricular dysfunction and remodeling in ratsCirculation199897138213919577950
- JobeLJMelendezGCLevickSPDuYBrowerGLJanickiJSTNF-α inhibition attenuates adverse myocardial remodeling in a rat model of volume overloadAm J Physiol Heart Circ Physiol2009297H1462H146819666842
- MurrayDBLevickSPBrowerGLJanickiJSInhibition of matrix metalloproteinase activity prevents increases in myocardial tumor necrosis factor-alphaJ Mol Cell Cardiol20104924525020403361