
Abstract
Hemolytic uremic syndrome (HUS) is a thrombotic microangiopathy (TMA) defined by the triad of hemolytic anemia, thrombocytopenia, and acute kidney injury. Microthrombi develop in the glomerular capillaries secondary to endothelial damage and exert shear stress on red blood cells, consume platelets, and contribute to renal dysfunction and failure. Per current understanding of pathophysiology, HUS is classified into infectious, secondary, and atypical disease. The most common etiology is infectious sequelae of Shiga toxin-producing Escherichia coli (STEC); other causative organisms include shigella and salmonella. Secondary HUS arises from cancer, chemotherapy, solid organ and hematopoietic stem cell transplant, pregnancy, or autoimmune disorders. Primary atypical hemolytic-uremic syndrome (aHUS) is associated with genetic mutations in complement and complement regulatory proteins. Under physiologic conditions, complement regulators keep the alternative complement system continuously active at low levels. In times of inflammation, mutations in complement-related proteins lead to uncontrolled complement activity. The hyperactive inflammatory state leads to glomerular endothelial damage, activation of the coagulation cascade, and TMA findings. Atypical hemolytic-uremic syndrome is a rare disorder with a prevalence of 2.21 to 9.4 per million people aged 20 years or younger; children between the ages of 0 and 4 are most affected. Multidisciplinary health care is necessary for timely management of its extra-renal manifestations. These include vascular disease of the heart, brain, and skin, pulmonary hypertension and hemorrhage, and pregnancy complications. Adequate screening is required to monitor for sequelae. First-line treatment is the monoclonal antibody eculizumab, but several organ systems may require specialized interventions and coordination of care with sub-specialists.
Introduction
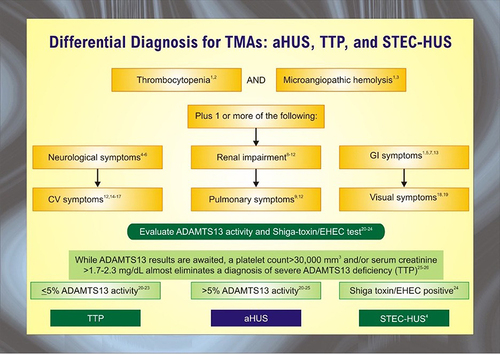
Thrombotic microangiopathies (TMA) are a constellation of disorders characterized by injury to the endothelium of micro-vessels, resulting sequentially in platelet activation, micro-thrombosis, Coombs-negative hemolytic anemia, and thrombocytopenia.Citation1,Citation2 The differential for TMA disorders is demonstrated in . The development of microthrombi can lead to small vessel occlusion, downstream ischemia, and end-organ damage. TMA encompasses thrombotic thrombocytopenic purpura (TTP), hemolytic-uremic syndrome (HUS), and atypical hemolytic-uremic syndrome (aHUS). The overlap between these conditions makes a definitive diagnosis challenging and demands extensive work-up.Citation3
Figure 1 The differential diagnosis for thrombotic microangiopathies (TMAs) is broad and includes infection-associated hemolytic uremic syndrome (HUS), atypical hemolytic-uremic syndrome (aHUS), and thrombotic thrombocytopenic purpura (TTP). The most common infectious source of HUS is Shiga-toxin producing Escherichia coli (STEC). Activity of the ADAMTS13 enzyme is a key diagnostic variable.
The goals of this review are to provide an update on the genetic mechanisms behind complement dysfunction in aHUS, detail organ-specific clinical manifestations, and outline current management strategies. Multi-organ disease involvement belies the need for a multidisciplinary approach to management.
Pathophysiology
The complement system has three pathways: classical, alternative, and lectin-binding. These pathways have different initial steps but converge on forming C3 convertase to propagate complement activity and destroy pathogens. The classical pathway is initiated by binding of the first complement cascade protein (C1q) to pathogen surfaces or antibody-antigen immune complexes. The C1 complex activates C4 and furthers the cascade. In the lectin-binding pathway, mannose-binding lectin in the serum binds to mannose polysaccharides on pathogen cell surfaces, forming a C1-like complex.Citation4
The alternative complement pathway is a component of the innate immune system and involves several serum and cell surface proteins. It does not require a specific antigen for activation. Instead, a permissive host cell surface or serum microorganisms spontaneously hydrolyze the C3 thioester bond. Hydrolysis of complement proteins into active components via all pathways culminates in formation of the membrane-attack complex (MAC).Citation5 MAC perforates the cell membrane and lyses target cells. The complement system also opsonizes targets by the covalent binding of C3b for chemotaxis of innate immune cells and phagocytosis.Citation6 Without tight control of the alternative complement pathway, extensive damage is inflicted with endothelial damage and activation of the coagulation cascade including platelets. In aHUS, the innate immune system loses its ability to regulate the pathway, resulting in uncontrolled complement activation. This is most commonly caused by mutations in genes encoding alternative complement pathway initiators (eg, C3, Factor B), regulatory proteins (eg, Factor H, Factor I, Membrane Cofactor Protein), and autoantibodies against Factor H.Citation7,Citation8
Genetic Etiology of aHUS
Causal variants in alternative complement pathway proteins are detected in up to 60% of aHUS cases (); mutations may also be found in coagulation proteins and enzymes that indirectly contribute to the coagulation cascade. The absence of identifiable mutations does not preclude a diagnosis of aHUS.
Figure 2 Underlying genetics play a key role in pathophysiology and extra-renal manifestations of atypical hemolytic-uremic syndrome. The CFH gene is the most common site of mutations.
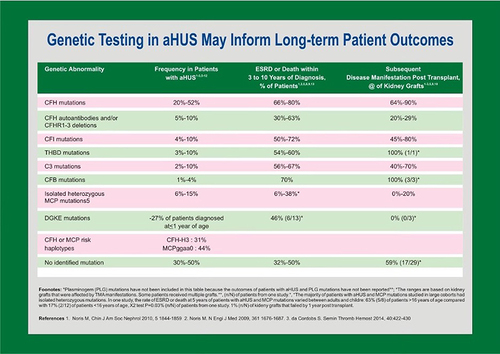
Nearly half of aHUS patients with an identified genetic cause carry a pathogenic variant in the CFH gene, residing in the regulators of complement activation (RCA) cluster on chromosome 1. The gene encodes Factor H (FH), a serum protein that binds to glycan structures on host cells and blocks complement activation.Citation9 FH is the main regulatory protein of the alternative complement pathway.Citation10 It comprises 20 units of a common structural motif known as the complement control protein (CCP) unit. CCPs at the N-terminal domain are sufficient for regulating C3 in the fluid phase. At the C-terminal domain, CCP units 19 and 20 are essential for the binding of FH to cell surface glycosaminoglycans in order to regulate alternative pathway activation. FH supports cleavage of C3b deposited on cell surfaces to iC3b and prevents autolysis of C3 in the serum. CFH pathogenic variants are spread throughout the gene, but most are found in the C-terminus short consensus repeats 19 and 20. Mutant FH molecules do not bind to the cell surface and, in turn, cannot regulate alternative complement activation. This leads to complement-induced damage and microthrombi in the microvasculature, especially the glomerular capillaries.Citation11
The proximity of CFH and genes for FH-related proteins provides an opportune environment for genetic recombination. Factor H-related proteins comprise five plasma proteins that bind to complement component C3b. They may serve a role in complement regulation, but the precise role of each protein is unknown. Genetic variations of the individual genes are associated with aHUS, lupus, age-related macular degeneration, IgA nephropathy, and C3 glomerulopathies.Citation12 Homozygous deletion or duplication of CFHR1 in particular is found in 85–90% of patients with anti-factor H-associated aHUS. These anti-FH autoantibodies target the C-terminus of FH and mirror the disruption of complement pathway regulation seen in CFH genetic defects.Citation13
Further pathogenic variants in aHUS are found in genes coding for complement regulatory proteins Factor I (FI) and membrane cofactor protein (MCP). FI is a plasma serine protease responsible for degrading C3b to iC3b and other degradation products. Without sufficient levels of active C3b, the complement pathway does not generate C3 convertase. FI protease activity requires concurrent binding of C3b to FH or MCP. With C3b bound to FH on the cell surface or plasma, the proper scaffolding is in place for FI to cleave C3b.Citation11 MCP is a membrane protein expressed on nucleated cells that acts as a complement regulator by serving as a cofactor for FI to cleave C3b and C4b, thereby modulating both the alternative and classical complement pathways. The majority of MCP pathogenic variants decrease the expression of MCP on cell surfaces and can be detected by flow cytometry. MCP mutations are associated with frequent relapses, but the clinical course is overall mild with less than 20% of patients progressing to kidney failure.Citation4
Most CFH mutations are loss-of-function; gain-of-function variants affect activating factors such as C3 and complement factor B (FB), a key mediator of C3 convertase function.Citation14 Mutations affecting FB and C3 are found in less than 10% of aHUS patients. C3 mutations reduce the binding affinity of C3b to FH, decreasing the degradation of C3b to iC3b. CFB mutations increase the affinity of FB to C3b, resulting in a more stable C3 convertase.Citation15 Both C3 and CFB mutations result in a state of complement hyperactivation. Mutant C3 and FB are associated with aggressive aHUS disease progression. Amongst all patients with aHUS, those with CFH-aHUS have the highest risk of developing kidney failure, usually within one year of diagnosis. More than 80% of patients with pathogen variants of C3 and FB also develop kidney failure, and 80% of post-transplant patients relapse despite kidney grafts.Citation16
Renal Manifestations
The clinical presentation of aHUS depends on the extent of microvascular injury and ensuing ischemic damage to organs. Diagnosis is dependent on lab values demonstrating hemolytic anemia (hemoglobin <10 g/dL), thrombocytopenia (platelet count <150,000 mm3), and acute kidney injury (AKI). Despite the importance of complement proteins in aHUS pathogenesis, they are not reliable diagnostic markers. Patients typically present with nonspecific symptoms including fatigue, pallor, and somnolence. In addition to AKI, hematuria and proteinuria may be identified in urine studies. Oliguria/anuria and uremia can develop as patients progress to kidney failure; the risk of progression to CKD stages 3–5 is high.Citation17 Fifty percent of aHUS cases progress to end-stage renal disease (ESRD) requiring chronic dialysis, and the overall mortality rate is 25%.Citation18 These rates vary based on the causative mutation; for example, ESRD rates are 60–80% for mutations affecting C3, FH, FB, and FI but only 30–40% in cases of MCP mutations. Disease prognosis depends on ESRD progression, delay from symptom onset to initiation of therapy (eg, eculizumab), and involvement of extra-renal systems.Citation19
Kidney injury in aHUS has various clinical courses and histological findings. In the case of a 37-year-old female, initial CT findings raised suspicion for a complicated urinary tract infection. The patient failed to improve with several days of standard management until renal biopsy revealed marked glomerular capillary congestion with intraluminal thrombus formation. In aHUS, a cascade of cellular damage and inflammation stimulates endothelial cell proliferation and microthrombi formation in the capillaries. Low C3 levels on complement studies confirmed the diagnosis, and treatment was initiated with a platelet infusion and eculizumab, leading to clinical improvement.Citation20 Biopsy findings in other aHUS cases include fibrinoid necrosis, severe vacuolar degeneration of the tubular epithelium, and high-density brown granules in the lumen consistent with hemoglobin casts. The hemoglobin casts occluding tubules likely precipitated acute tubular necrosis.Citation21,Citation22
Extra-Renal Manifestations
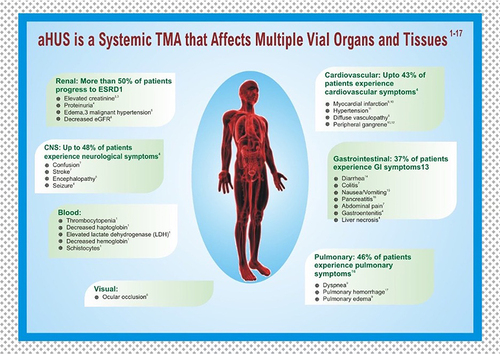
Extra-renal systems involved in aHUS include the cardiovascular system, central nervous system, skin, visceral organs, and reproductive organs. The full spectrum of manifestations is listed in . Cardiovascular and neurologic involvement is particularly noteworthy due to the morbidity and mortality associated with heart failure, pulmonary hypertension, altered mental status, seizures, coma, and blindness. Timely intervention with eculizumab and system-specific interventions is necessary for favorable outcomes. In a literature review of 259 aHUS patients across 176 articles, 45% developed hypertension and 14% demonstrated cardiac sequelae over a 5-year period.Citation23 In a multicenter French study of 156 adult aHUS patients treated with plasma exchange, approximately half exhibited signs of neurologic impairment, including seizures in 11% and coma in 8%. Gastrointestinal (GI) involvement was also common (~30% showing signs of abdominal pain and diarrhea each) albeit not life-threatening.Citation24 Skin complications are rare, but ulceration and peripheral gangrene secondary to macrovascular stenosis are described in case reports.
Figure 3 Atypical hemolytic uremic syndrome may have multi-system manifestations affecting the renal, cardiovascular, gastrointestinal, pulmonary, ophthalmic, and central nervous systems.
Cardiac Manifestations
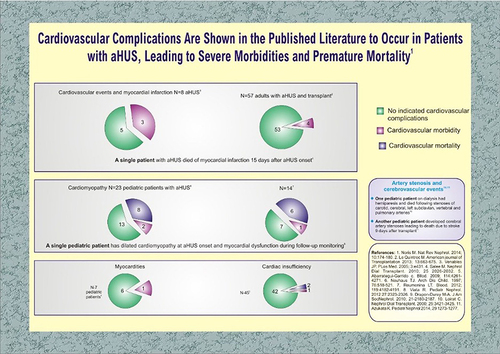
Complement-mediated microangiopathy in aHUS can injure the coronary microvasculature, resulting in cardiac complications in 3–14% of patients.Citation17,Citation23 As shown in , there is considerable variety in presentations based on the mutations involved. Life-threatening sequelae include dilated cardiomyopathy, congestive heart failure, and myocardial infarction. Onset of symptoms may be sudden and terminal. For example, an adult female with a CFH mutation developed pulmonary edema, pericardial effusion with tamponade, and cardiac arrest two weeks into plasma therapy, culminating in patient death. Autopsy revealed a myocardial infarction and multiple microscopic infarctions ranging from 24 hours to 15 days old.Citation25 In another case with CFH mutation, a 1-year-old girl developed dilated cardiomyopathy, heart failure with reduced ejection fraction, and pulmonary edema. Initial treatment included plasma exchange and hemodialysis but were tapered due to concern of fluid overload. Cardiac dysfunction did not resolve until eculizumab was administered.Citation26
Figure 4 Cardiovascular complications of atypical hemolytic-uremic syndrome include myocardial infarction, dilated cardiomyopathy, cardiac tamponade, myocarditis, and heart failure.
Neurologic Manifestations
The central nervous system (CNS) is commonly involved in aHUS with symptoms reported in half of cases.Citation24 Vaso-occlusive lesions induced by inappropriate complement activation can deposit in the cerebral, carotid, and vertebral arteries, leading to ischemic damage to the brain.Citation27 Neurological symptoms in aHUS include encephalopathy, coma, stupor, seizures, hemiparesis, and cortical blindness.Citation17 Symmetrical hyperintense lesions in the basal ganglia and brain stem on diffusion-weighted imaging are pathognomonic. However, magnetic resonance angiography (MRA) is often necessary to differentiate aHUS from posterior reversible encephalopathy syndrome (PRES), a rare condition of non-occlusive, vasogenic edema in the brain. PRES may have a similar presentation (eg, seizures, blindness, AKI) to aHUS, but management is different. Proper diagnosis is necessary to thwart severe morbidity and mortality.Citation28
MRI findings may vary in aHUS depending on the mechanism of CNS involvement. In the case of a four-year-old male with CFH mutation, partial complex seizures developed days after bilateral nephrectomy for uncontrolled hypertension. Initial brain MRIs and repeats after a month remained normal. The patient suffered another bout of seizures with violent agitation after eight months. Due to persistent hypertension, concern was raised for hypertensive encephalopathy. A third MRI revealed bilateral symmetrical lesions in the cerebral peduncles, caudate nuclei, putamen, thalami, and hippocampi, suggesting aHUS relapse with neurologic complications. Immediate treatment with daily plasma exchanges led to complete recovery of mental status.Citation29
Dermatologic Manifestations
Dermatologic findings in aHUS are uncommon and poorly recognized as disease sequelae. Nonetheless, skin lesions of unknown origin in a patient with kidney failure should raise the possibility of TMA injury. Macrovascular stenosis may contribute to severe lesions in aHUS such as gangrene.Citation30 Early recognition of complement-mediated injury and initiation of eculizumab can avert progression of these lesions; timely identification is critical given the positive outcomes with treatment. Eculizumab, plasma exchange, and dialysis have all shown to be effective. In the case of a 4-year-old girl with anti-FH autoantibodies, gangrene of the fingertips developed two days after initial presentation of aHUS and was stabilized with peritoneal dialysis. Another case of a Middle Eastern infant with aHUS complicated by ESRD developed gangrene in the fingertips despite plasma exchange. Eculizumab therapy rapidly restored perfusion to the non-necrotic digits, showing effectiveness of the agent against macrovascular injury.Citation31
Pulmonary Manifestations
Pulmonary complications of aHUS may be seen in severe cases with multi-organ dysfunction. Manifestations include pulmonary hypertension and hemorrhage. Pulmonary edema may also occur secondary to cardiac dysfunction and volume overload. In one case, a 9-year-old boy presented with MAHA, thrombocytopenia, hypertension, and nephrotic syndrome. The hypertension remained uncontrolled despite multimodal treatment with anti-hypertensive agents and HD. Despite the aggressive management, the patient suffered three hypertensive crises complicated by congestive heart failure and pulmonary edema. Bilateral nephrectomy was performed as a last resort to manage the hypertension to no avail; the crises culminated in cardiac arrest.Citation32
Pulmonary hemorrhage and embolism are uncommon but similarly associated with poor patient outcomes in aHUS. In a study of 46 pediatric patients with identified complement mutations, only one had a pulmonary complication (hemorrhage); however, that patient was one of the four with demise within months of disease onset.Citation33 A report on long-term sequelae in patients with anti-FH antibodies also reported pulmonary hypertension in an infant.Citation34 Acute respiratory failure necessitating mechanical ventilation is a severe complication, and a review identified an incidence rate as high as 21% in pediatric aHUS.Citation35 Therefore, careful fluid balance and respiratory monitoring are essential to reduce the risk of pulmonary compromise in aHUS patients.
Gastrointestinal Manifestations
Gastrointestinal (GI) complications in aHUS are frequently associated with homozygous genetic deletions of FHR1 or FH3, leading to the formation of FH autoantibodies. FH autoantibodies may even serve as a biomarker for risk and severity of GI involvement.Citation35 Diarrhea is a classic prodromal finding and is observed in half of patients.Citation36 In a study of 32 aHUS patients positive for anti-FH IgG, 53% had diarrhea, 84% developed vomiting, and two progressed to Mallory-Weiss tears.Citation34 Incidence of GI prodromal symptoms (abdominal pain, diarrhea, vomiting) was similarly high in another study of 16 aHUS patients with elevated anti-FH antibody titers.Citation37 A Turkish study of 146 pediatric patients also noted bleeding, transaminitis, cholelithiasis, and pancreatitis.Citation38
Chronic GI symptoms as seen in inflammatory bowel disease (IBD) have been noted in reports of aHUS patients on occasion. In one case, a 16-year-old male with treatment-refractory ulcerative colitis presented with MAHA and AKI secondary to glomerulonephritis. The patient experienced prodromal symptoms of decreased appetite and bloody diarrhea. A working diagnosis of aHUS was made based on decreased C3 levels and renal biopsy with positive immunostaining for MAC. Eculizumab was administered for seven weeks and GI symptoms resolved completely.Citation39 Additional cases of concurrent aHUS and IBD report improvement in GI manifestations with eculizumab.Citation40 While these cases are uncommon, treatment-refractory IBD in patients with MAHA and AKI should prompt evaluation for TMA and potential treatment with eculizumab.
Gynecologic Manifestations
Pregnancy-associated thrombotic microangiopathy (P-TMA) is a rare disorder with an estimated incidence of 1 in 25,000 pregnancies. It is associated with significant perinatal and maternal morbidity and mortality. Pregnancy can act as a precipitating factor in TTP; however, the pathogenesis and presentation of pregnancy-associated aHUS (P-aHUS) remain unclear due to limited data. Recent findings suggest pregnancy may trigger complement dysregulation in individuals with predisposing genetic abnormalities in the alternative complement pathway. Data show that fetal losses and preeclampsia occur more often in patients with genetic complement abnormalities than in the general population; understanding complement dysregulation in P-aHUS is essential to mitigate complications.Citation41
The diagnosis of P-aHUS is complicated by its similar presentation and frequent co-occurrence with preeclampsia and hemolysis with elevated liver enzymes and low platelet (HELLP) syndrome. Given the rarity of aHUS, it is often mistaken for these more common diagnoses.Citation41 While all of these conditions present with MAHA and kidney injury, P-aHUS is unlikely to affect the liver enzyme levels. Patients with aHUS also have a steadily rising creatinine rather than the abrupt change in serum creatinine seen in preeclampsia. Of note, a retrospective study of 105 pregnant patients with AKI found no significant difference in liver enzymes between patients with preeclampsia or TMA at days 0–3 of admission. However, this study did identify a dynamic pattern of platelet levels that were significantly decreased in primary TMA-related AKI versus the other groups in the study.Citation42
Given that preeclampsia, HELLP, TTP, and P-aHUS have similar clinical features, an essential issue for evaluating a pregnant/postpartum patient with severe MAHA and thrombocytopenia is to appreciate the nuances between these diagnoses. Recognizing the severity of neurologic abnormalities, kidney injury, and thrombocytopenia can help the clinician narrow the differential. Preeclampsia and HELLP patients are severely ill, and the neurologic abnormalities are minor compared to TTP and aHUS. The former two can present with headache and vision changes, but TTP and aHUS have more severe neurologic manifestations including weakness and aphasia. Cognitive impairment is most common in TTP.Citation43
The degree of kidney injury also differs between syndromes. Preeclampsia and HELLP may have elevated creatinine levels, but the need for dialysis upon initial presentation suggests aHUS. Discriminating between these clinical features directs appropriate treatment. The management of preeclampsia and HELLP syndrome is to control the hypertension and immediate delivery the infant, but there is no evidence to suggest that delivery is helpful in P-aHUS patients. Instead, P-aHUS is treated with anti-complement therapy.Citation43 The long-term outcomes of anti-complement therapy in pregnant patients with aHUS have been outlined in a study of 31 pregnancies in the Global aHUS Registry. Live births occurred at a higher rate in non-eculizumab exposed vs eculizumab-exposed patients (78% versus 55%). However, the eculizumab group fared better in terms of aHUS relapse at 5% versus 22% for the non-eculizumab treated group.Citation44
To characterize the genetic abnormalities found in P-aHUS and define its pregnancy-associated outcomes, Fakhouri et al retrospectively analyzed 100 female aHUS patients; P-aHUS occurred in 21 total. In 18 of the 21, mutations were detected in genes encoding components of the alternative complement pathway: ten patients had a CFH mutation, two had an IF mutation, two had a C3 mutation, one had an MCP mutation, and three had more than one mutation (CFH and C3, CFH and MCP, IF/IF). The three remaining patients had no identified gene defects. Fifteen of the twenty-one P-aHUS patients developed disease in the peripartum period, which may be explained by trophoblastic loss of complement regulatory proteins (eg, DAF, MCP, CD59) and FH sequestration after delivery. DAF, MCP, and FH typically down-regulate C3 convertase, providing adequate control of complement activation at the feto-maternal interface in pregnancy. However, an increase in inflammation after delivery due to hemorrhage, fetal cells in circulation, and infection leads to activation of the alternative pathway and aHUS.Citation44
A cohort of 22 patients with P-aHUS in the Spanish aHUS database was examined. Nine were positive for inherited complement abnormalities in CFH. Sixteen of the twenty-two presented with aHUS for the first time during their first pregnancy, and nine required HD at diagnosis. Four of the six patients with prior pregnancies had successful live births. Most patients developed P-aHUS around week one of the postpartum period after caesarean section deliveries. C-sections may prove more stressful on the body, leading to potent induction of the complement system. Patients were either treated with eculizumab or plasmapheresis. Of the 13 treated with eculizumab, all achieved normalization of hematologic parameters and preservation of renal function. In the plasmapheresis group, only 2 of the 17 patients had a renal response. Improved outcomes with eculizumab suggest that anti-complement therapy ought to be first-line in P-aHUS.Citation45
Ophthalmologic Manifestations
Ocular manifestations are sporadic but potentially severe complications of aHUS. Although CNS involvement is a common issue in up to 48% of patients, ophthalmic involvement is seldom reported.Citation24 Acute visual symptoms include decreased acuity, scotomata, blurriness, ocular pain, and diplopia. These manifestations present with sudden onset and can lead to permanent visual defects. Interestingly, mutations in CFH are associated with age-related macular degeneration, suggesting ocular complications in aHUS may be secondary to FH dysfunction. Changes in tissue-specific ocular sulfated glycosaminoglycans altering the binding affinity of FH to host tissues may contribute to ophthalmic complications.Citation46
A variety of findings have been observed on ophthalmologic exam in aHUS patients with pertinent symptoms. In a pediatric case study, findings included vitreous bleeding, elevated ocular pressure, choroidal hemorrhage, and retinal ischemia. The patient was a 7-month-old boy with FH deficiency presenting with hypertension, acute renal failure, and hemolytic anemia without thrombocytopenia. He was first stabilized with anti-hypertensive therapy, then with plasma infusions. Following another relapse, bilateral nephrectomy was performed and the patient was started on HD. After three years, he unexpectedly developed pain and blurry vision. TMA was suspected due to FH deficiency, and the patient was treated with plasma exchange and FFP. After five weeks, the vitreous hemorrhage, ocular pain, and visual impairment resolved.Citation47 Regular monitoring of ocular involvement in aHUS patients with CFH mutations may help minimize complications and intractable damage.
Pediatric case reports have described bilateral intra-retinal flame hemorrhage, optic disc edema, and tortuosity of retinal vessels on funduscopic exam. In one such, an 11-year-old girl with aHUS presented with bilateral central retinal vein occlusions with macular sub-hyaloid hemorrhage and reduced visual acuity. Her ocular exam was notable for flame hemorrhage, venous tortuosity, and optic disc edema with central retinal vein occlusions and venous stasis retinopathy. No other extra-renal complications were noted, and full recovery of 20/20 vision only occurred after treatment with plasma fusion, steroids, and supportive care.Citation48
Additional reports detail the impact of eculizumab on outcomes in aHUS patients with ocular manifestations. In the case of a 26-year-old female with presumed TTP receiving daily plasma exchange, the patient developed loss of vision. Hemorrhage of the optic disc, macula, and retina was identified bilaterally on funduscopic exam. The patient was referred to a retinal specialist for follow-up. There was no clinical improvement despite plasma exchange therapy every other day and dialysis three times a week. Subsequent testing revealed an ADAMTS13 activity level of 61%, and a diagnosis of aHUS was proposed. Eculizumab therapy was started, and renal function subsequently improved. Follow-up ophthalmologic exam showed the patient’s eyes clearing of ocular occlusions and vitreous hemorrhage; no further intervention was needed.Citation49
In the case of a 23-year-old female patient with bilateral serous retinal detachment (SRD), improvement was remarkable with eculizumab treatment. The patient initially presented during the third trimester of pregnancy with preeclampsia and HELLP syndrome and underwent a cesarean section. Following delivery, the patient’s thrombocytopenia and elevated liver enzymes failed to normalize and progressive renal failure set in. She was diagnosed with aHUS via kidney biopsy findings. Treatment with plasmapheresis, HD, and systemic steroids was initiated, but the patient still developed bilateral blurry vision. Fundoscopy revealed bilateral SRD, small retinal hemorrhages, and cotton wool spots. Treatment of plasma exchange, HD, antihypertensive medications, and steroids was reinitiated. Eculizumab was added to the treatment, resulting in improved renal function and total resolution of SRD. The patient’s vision recovered and remained stable at a two-month follow-up.Citation50
Treatment Modalities
Plasma Exchange
The cornerstones of aHUS management include therapeutic plasma exchange and eculizumab. Eculizumab was approved by the US Food and Drug Administration in 2011 for application in aHUS; it now serves as the first-line treatment. Therapeutic plasma exchange is the second-line intervention. depicts plasma exchange divided into its three steps: membrane and centrifuge, plasmapheresis, and immunoadsorption. Plasmapheresis is a long-standing extracorporeal therapy for non-specific removal of serum proteins (and presumably pathogenic autoantibodies) in autoimmune conditions. Unfortunately, it requires plasma reconstitution and puts patients at risk for transfusion reactions.
Figure 5 Plasma exchange filters blood in a three-step process: (1) membrane centrifugation, (2) plasmapheresis, and (3) immunoadsorption.
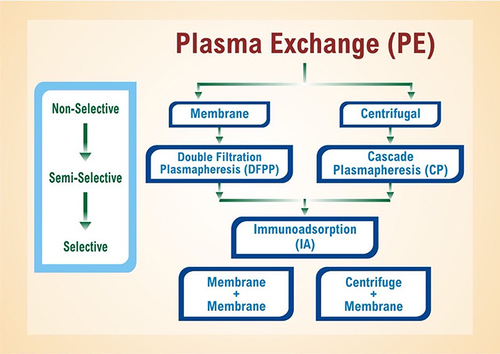
Immunoadsorption is another apheresis modality that employs high-affinity adsorbers to selectively remove immunoglobulins without affecting other plasma components.Citation51 It is commonly applied towards autoimmune neurologic disorders (eg, multiple sclerosis) and antibody-mediated transplant rejection.Citation52 In aHUS, immunoadsorption can remove FH autoantibodies and hyper-functional complement components. Treatment is often initiated in TMA cases prior to a final diagnosis due to the need for acute intervention for suspected TTP. For aHUS, standard practice is to initiate plasma therapy within 24 hours. Starting dosage is 60–75 mL/kg/day for adults and broader to 50–100 mL/kg/day for children.Citation53 Dosage and duration may be individualized based on disease severity.
There is considerable variance in the literature regarding the effectiveness of plasma exchange in managing aHUS, and treatment potency may vary based on the mutations involved. Bresin et al investigated the response of 273 aHUS patients with different mutations to PE. Ninety-seven percent of those with MCP mutations achieved partial or complete remission, 63% of those with CFH mutations, 57% with C3 mutations, and only 25% with IF mutations. The remaining progressed to ESRD. Such results suggest a role for genetic testing in guiding appropriate management of aHUS.Citation54
The safety of plasma exchange is controversial, and reports of adverse events are variable across the literature. Khandelwal et al showed all-grade toxicities in only 12% of 2024 plasma exchange sessions. Mild adverse events included fever and chills, vomiting, abdominal pain, hypotension, hypocalcemia, and urticaria; chills were the most common and occurred in 9% of the sessions. Catheter-associated infections occurred at a rate of 1.45 per 1000 catheter-days. Severe toxicities such as hypocalcemic seizures, life-threatening hemorrhage, and sepsis occurred in 22 sessions.Citation55
Eculizumab
Eculizumab, a recombinant monoclonal antibody, is the current cornerstone for management of aHUS. Eculizumab targets the C5 component of complement activation. Binding to C5 blocks the cleavage of C5 into its effector components C5a and C5b, preventing the subsequent formation of MAC.Citation56 Eculizumab thus directly attenuates the disease mechanism of aHUS and reduces inflammation, endothelial damage, and renal injury. The earlier eculizumab therapy is commenced, the greater the preservation of renal function. However, treatment is often delayed until aHUS is confirmed by genetic testing.Citation57 Dosing is weight-based for pediatric patients but consistent in adults at 900 mg per week for four weeks of induction, 1200 mg for week five, and 1000 mg bi-weekly after.Citation58
The effectiveness of eculizumab has been demonstrated in several studies. A Phase II trial by Licht et al evaluated the efficacy of eculizumab after two years. Twenty patients with aHUS and chronic kidney disease (CKD) were selected. Inclusion criteria included (1) plasma exchange more than once every two weeks but no more than three treatments per week and (2) no decreases in platelet count greater than 25% at least eight weeks prior to the initial dose of eculizumab. Study results were encouraging: 90% of patients achieved hematologic normalization at the two-year cutoff, and all experienced improvements in quality-of-life. No patient required renal transplantation during the study duration or lost an existing graft. Only one participant had a critical TMA-induced event.Citation59 A separate open-label, phase II trial investigated the efficacy of eculizumab in 41 adult patients over a 26-week course. Kidney function was responsive to eculizumab within one week for 22 patients, and more significant GFR improvements were noted in those previously on dialysis. The study demonstrated significant improvement in health status of most patients after the eculizumab course.Citation60
Ideal duration of eculizumab treatment is a topic of ongoing research; adverse effects must be weighed against the risk of relapse. Duration of therapy is typically shorter for secondary aHUS. In a trial of 29 patients with secondary causes (eg, drug reactions, systemic diseases, pregnancy, cancer), rapid recovery of renal function and resolution of MAHA was observed in 20 (68%). Eculizumab was discontinued after a median of 8 weeks with no reported incidents of relapse.Citation61 Duration of therapy is typically longer in patients with complement genetic variations due to increased risk of relapse. In a long-term observational study of 93 aHUS patients on eculizumab, patients with genetic or autoimmune complement abnormalities had a significantly higher rate of reinitiating or never discontinuing eculizumab (67% versus 48%). Variations of particularly high risk included mutations to CFH, CFB, and C3 as well as Factor H autoantibodies.Citation62
Increased levels of C5b-9 at the time of eculizumab discontinuation are associated with an elevated risk of aHUS relapse. In a multi-center trial by Fakhouri et al, 13 out of 55 patients relapsed after eculizumab treatment for at least six months. Eleven (85%) of those who relapsed had elevated C5b-9 levels versus only 23 (55%) of the non-relapsing patients. Multivariable analyses confirmed C5b-9 levels and complement gene variants as independent predictors of aHUS relapse. Fakhouri et al confirm that factoring complement genetics into discontinuation of eculizumab is safe and reasonable.Citation57
Ravulizumab
Ravulizumab is a new C5 inhibitor with immediate and sustained effect across an extended, eight-week dosing interval. It was first introduced in 2018 as a therapeutic candidate for aHUS (and paroxysmal nocturnal hemoglobinuria) and approved for use in the USA in 2019 and EU in 2020. Ravulizumab demonstrates similar efficacy to eculizumab while maintaining a terminal half-life four times longer.Citation63 Safety profiles are also consistent; in a study of 195 patients, no discontinuations due to meningococcal infections or other serious adverse events were reported.Citation64
Similar to eculizumab, ravulizumab binds to C5 and prevents its dissociation into C5a and C5b, thereby interrupting the complement cascade. It employs four amino acid substitutions in the complementarity-determining and Fc regions of eculizumab. Two histidine substitutions in the complementarity-determining regions enhance dissociation of the monoclonal antibody and C5 in the early endosome, attenuating target-mediated drug disposition and improving pharmacokinetics. Two further substitutions lead to increased recycling into the vasculature via the neonatal Fc receptor pathway.Citation65
Renal Transplant
Renal transplantation has poor outcomes in aHUS due to graft failure and disease recurrence. Approximately 50% of patients progress to ESRD after transplantation while maintaining a high rate of relapse.Citation54 Relapse is more common in patients with mutations against CFH, CFI, and C3. Post-transplant prophylaxis with eculizumab has been shown to reduce rates of recurrence in this high-risk population.Citation66 Renal transplant can also lead to de novo aHUS; known causes include immunosuppressive drugs, antibody-mediated rejection, and viral infection. In an analysis of 36 aHUS cases associated with transplant, 14 patients had prior aHUS with high-risk genetic variants, while 22 had de novo aHUS. Prophylactic eculizumab effectively prevented relapse in all 14 high-risk patients. Of note, no pathogenic mutations were identified in the patients with de novo disease.Citation67
Comprehensive Care
Initial diagnostic evaluation of patients with suspected aHUS includes lab work with a complete blood count, lactate dehydrogenase level, and serum creatinine to assess for the pathognomonic findings of hemolytic anemia, thrombocytopenia, and AKI. Urgent measurement of ADAMTS13 activity is indicated to assess for TTP. Therapeutic plasma exchange is typically initiated first-line in adult TMA patients as an acute intervention for potential TTP. If aHUS is suspected, eculizumab should also be started within 24 to 48 hours; early intervention helps preserve renal function. Eculizumab may be administered before plasma exchange if central venous catheterization is not available or complicated by infection or transfusion reactions. Meningococcal vaccine is ideally given two weeks prior to eculizumab, but in the case of emergent treatment, prophylactic antibiotics can provide coverage for two weeks until the vaccine becomes effective.
In addition to ADAMTS13 activity, urgent testing for FH autoantibodies is recommended for additional management options. Prior to eculizumab, standard-of-care for autoantibody-positive aHUS patients included plasma exchange and immunosuppression with oral steroids and intravenous pulses of cyclophosphamide or rituximab.Citation68 Boyer et al reported three cases of children achieving sustained remission and decreases in autoantibody titers with concurrent plasma exchange, prednisone, and cyclophosphamide pulses. In one of the patients, aHUS had previously relapsed after six cycles of PE and four rituximab infusions, suggesting cyclophosphamide as the superior option.Citation69 Eculizumab is now gaining momentum as an alternative to immunosuppressants due to a favorable toxicity profile. Severe toxicities are few and far between; most common are infections such as meningitis, peritonitis, and bacteremia. In a study of 17 children with FH autoantibody-positive aHUS, all six treated with eculizumab completely recovered kidney function.Citation59 Further research comparing eculizumab to immunosuppressants may guide development of standardized protocols.
For all patients with suspected aHUS, early hematology and nephrology consultation is recommended with a multidisciplinary management approach as illustrated in . All nephrotoxic agents (eg, NSAIDs) should be stopped, including those exacerbating aHUS such as bleomycin, cisplatin, gemcitabine, mitomycin C, and calcineurin inhibitors (tacrolimus, cyclosporine). These drugs are commonly employed for chemotherapy and post-transplant immunosuppression.Citation70 Supportive needs should also be monitored including:
Red blood cell transfusion for hemoglobin <7 g/dL in adults and <6 g/dL in children
Platelet transfusion for platelet count <50k/μL
Fluids for hydration and electrolyte imbalances
Figure 6 A multidisciplinary approach involving nephrologists, hematologists, community advocates, and potentially other sub-specialists (eg, gynecologists, dermatologists) is required for comprehensive management of hemolytic-uremic syndrome.
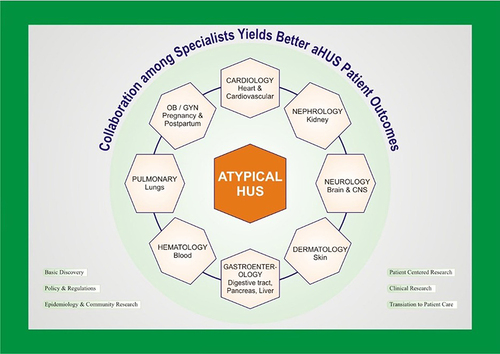
Decisions on plasma exchange and dialysis should be made in partnership with the nephrology service. Dialysis should be reserved for the standard indications of renal replacement therapy in AKI – refractory fluid overload, hyperkalemia, uremia, and metabolic acidosis – but is often applied in conjunction with plasma exchange. Case reports include both HD and peritoneal dialysis in interventions for aHUS. The latter was used to stabilize gangrene in the fingertips of an infant with kidney failure.Citation31
In the setting of pregnancy (or postpartum), aHUS may be less common than preeclampsia and HELLP syndrome.Citation41 However, it is critical to keep aHUS on the differential for cases of MAHA and AKI due to differing management approaches. TMA may be differentiated from the other conditions by normal liver function tests, progressive AKI requiring plasma exchange or dialysis, and severe neurologic manifestations.Citation42,Citation43 When TMA is suspected, nephrology and hematology ought to be consulted and the patient started on standard-of-care anti-complement therapy (eculizumab). Care coordination with gynecology is critical for safe and timely intervention.
Eculizumab is particularly important in aHUS cases with cardiac or neurologic manifestations. Complement hyperactivity can cause steno-occlusive lesions in macro-vasculature such as the carotid arteries, precipitating ischemic changes to the heart, brain, and extremities.Citation27 Even a single dose of eculizumab has been shown to resolve cardiac dysfunction (eg, dilated cardiomyopathy) refractory to PE and dialysis.Citation26 Case reports also describe rapid improvements in digital ischemia in response to eculizumab.Citation31 Early infusion is therefore indicated in cases with cardiac or skin findings to minimize complications and prevent gangrene. Preemptive interventional cardiology consultation may aid in rapid surgical intervention for severe cardiac manifestations such as myocardial infarction and cardiac tamponade.Citation25 Data is currently limited for the benefits of eculizumab infusion in such acute crises.
As previously detailed, varied pathophysiologic mechanisms can lead to diverse amalgams of neurologic symptoms in aHUS. Patients may initially present with non-specific findings such as headache, altered mental status, drowsiness, seizures, and/or visual disturbances.Citation13 Suspicion for occlusive lesions in the cerebral and vertebral arteries should be high in the setting of TMA, and early consultation of neurology and neurosurgery is recommended for management. First-line imaging is typically diffusion-weighted MRI, but the modality is often negative early in the disease course.Citation29 Hyperintense lesions are also difficult to separate from other possible etiologies such as PRES. MRA of the brain is critical to assess for occlusions and guide neurosurgical intervention.
Visual disturbances derived from aHUS can present as sudden-onset blurry vision, diplopia, scotomas, and/or ocular pain.Citation46 Ophthalmology ought to be consulted for comprehensive physical examination of the eyes; concerning findings include flame hemorrhages, vitreous bleeding, retinal vein tortuosity, optic disc edema, and retinal detachment. Intraocular pressure should also be measured. While ocular hypertension does not typically require emergent intervention, persistent elevations over 25 mmHg suggest treatment with topical prostaglandins (eg, bimatoprost), beta-blockers, or laser therapy.Citation71 One case report of an infant with FH deficiency described resolution of ocular symptoms five weeks after treatment with plasma exchange and fresh frozen plasma (FFP); another pediatric case mentioned resolution with PE and steroids.Citation48,Citation49 Adult cases appear less responsive to plasma exchange, dialysis, and steroids, but eculizumab has been shown to induce full recovery of visual function.Citation50 Further research in adult and pediatric populations on the effectiveness of eculizumab as a first-line treatment may guide future management.
Pulmonary complications in aHUS are rare but carry high risk of morbidity and mortality. ICU level-of-care is indicated due to the risk of ARF secondary to pulmonary edema, hemorrhage, or embolism. The risk of ARF is as high as 21% in pediatric aHUS patients.Citation35 Patients with hypertension or fluid overload refractory to medication management (antihypertensives and diuretics, respectively) and dialysis are at the highest risk.Citation32 Threshold for mechanical ventilation should be low for these patients, and preemptive transfer to ICU settings would be appropriate.
Community Advocacy
Patients with aHUS share challenges with other rare disease populations including complex care, difficult diagnosis, and delays in appropriate identification and treatment. There remains a lack of expert consensus regarding the spectrum of complement-mediated renal TMAs as well as the specific clinical and genetic subtypes of aHUS.Citation72 Wide variation in organ systems affected as well as duration, severity, and frequency of aHUS activity leaves patient care unpredictable and multi-faceted. Even within nuclear families sharing genetic predispositions for aHUS, disease presentations may vary from episodic to chronic and early to late-onset. A central difficulty to outreach and education is creation of learning materials for patients and medical professionals that encompass this far-ranging spectrum.Citation73
Patients and physicians are likely to find research and resources tagged with a variety of keywords including complement-mediated renal disease, thrombotic microangiopathy, primary aHUS, primary TMA, familial HUS, and others. The lack of consistency and fragmented information creates a barrier to understanding. In 2021, the National Kidney Foundation created a multi-national interdisciplinary working group to reevaluate the current classification of aHUS and examine options to revise its nomenclature.Citation74 Rare disease patients and families need disease information that is clear, factual, and presented in formats that are both understandable and meaningful. These needs remain largely unmet. As noted in a 2016 Rare Disease UK report, “Nearly 70% of respondents did not feel they were provided with sufficient information on their condition following diagnosis”. The report illustrated disparities between current educational materials and preferred formats such as websites and leaflets.Citation75
Establishing multidisciplinary teams (MDTs) at centers of excellence may provide avenues to streamline treatment and optimize patient care outcomes.Citation76 A 2018 study showed that an MDT approach for TMA cases reduced length of ICU stay by 31% and hastened identification of affected organs.Citation77 The approach also aids in early identification of treatment-naïve patients for enrollment in clinical trials; lack of such support leaves study creation challenging for academics and pharmaceuticals. In a 2021 poll of the international aHUS community by the Alliance Global Action team, only 31 of 122 patients (25%) reported involvement of a multidisciplinary treatment team. However, 40% reported continuity of care with sub-specialists after care establishment in an inpatient setting. Nephrologists and hematology were the most common specialists for overlooking aHUS care. MDTs should also involve the primary care physician for appropriate health maintenance after discharge.Citation78 Further exploration of patient experiences is needed to assess outpatient management of extra-renal manifestations.
Conclusion
Atypical hemolytic-uremic syndrome is an uncommon TMA variant mediated by complement cascade hyperactivity. Patients characteristically present with microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. Point-of-care coordination between primary services, nephrology, and hematology is encouraged for timely intervention with plasma and anti-complement therapies, supportive care measures, and possibly dialysis. Management of extra-renal manifestations also requires collaboration among pertinent specialties. Case reports across sub-specialties suggest excellent responsiveness of extra-renal findings to eculizumab. Advocacy groups may also aid in patient and caregiver understanding and access to multidisciplinary care. Further research into the genetic etiology of aHUS and varied responsiveness to plasma exchange, dialysis, and eculizumab may guide future directed therapy.
Disclosure
Prof. Dr. Olivia Boyer reports personal fees from Alexion, during the conduct of the study; personal fees from Alnylam, CSL/Vifor, and Purespring, outside the submitted work. The authors report no other conflicts of interest in this work.
References
- Loirat C, Fakhouri F, Ariceta G, et al. HUS International. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31(1):15–39. doi:10.1007/s00467-015-3076-8
- Afshar-Kharghan V. Atypical hemolytic uremic syndrome. Hematol Am Soc Hematol Educ Prog. 2016;2016(1):217–225. doi:10.1182/asheducation-2016.1.217
- Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(17):1676–1687. doi:10.1056/NEJMra0902814
- Janeway CA, Travers P, Walport M, et al. Immunobiology: the immune system in health and disease. In: The Complement System and Innate Immunity. 5th ed. New York: Garland Science;2001. Available from: www.ncbi.nlm.nih.gov/books/NBK27100/.
- Morgan BP. The complement system: an overview. Methods Mol Biol. 2000;150:1–13. doi:10.1385/1-59259-056-X:1
- Barnum SR, Bubeck D, Schein TN. Soluble membrane attack complex: biochemistry and immunobiology. Front Immunol. 2020;11:585108. doi:10.3389/fimmu.2020.585108
- Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267–1279. doi:10.1182/blood-2005-10-007252
- Yan K, Desai K, Gullapalli L, Druyts E, Balijepalli C. Epidemiology of atypical hemolytic uremic syndrome: a systematic literature review. Clin Epidemiol. 2020;12:295–305. doi:10.2147/CLEP.S245642
- Parente R, Clark SJ, Inforzato A, Day AJ. Complement factor H in host defense and immune evasion. Cell Mol Life Sci. 2017;74(9):1605–1624. doi:10.1007/s00018-016-2418-4
- Pangburn MK. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunopharmacology. 2000;49(1–2):149–157. doi:10.1016/S0162-3109(00)80300-8
- Bernabeu-Herrero ME, Jimenez-Alcazar M, Anter J, et al. Complement factor H, FHR-3 and FHR-1 variants associate in an extended haplotype conferring increased risk of atypical hemolytic uremic syndrome. Mol Immunol. 2015;67(2 pt B):276–286. doi:10.1016/j.molimm.2015.06.021
- Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT. Complement factor H related proteins (CFHRs). Mol Immunol. 2013;56(3):170–180. doi:10.1016/j.molimm.2013.06.001
- Dragon-Durey MA, Loirat C, Cloarec S, et al. Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:555–563. doi:10.1681/ASN.2004050380
- Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. 2009;104:115–149.
- Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2007;104:240–245. doi:10.1073/pnas.0603420103
- Fremeaux-Bacchi V, Miller EC, Liszewski MK, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112:4948–4952. doi:10.1182/blood-2008-01-133702
- Loirat C, Fremeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. 2011;6:60. doi:10.1186/1750-1172-6-60
- Nester CM, Barbour T, de Cordoba SR, et al. Atypical aHUS: state of the art. Mol Immunol. 2015;67(1):31–42. doi:10.1016/j.molimm.2015.03.246
- Nester CM, Thomas CP. Atypical hemolytic uremic syndrome: what is it, how is it diagnosed, and how is it treated? Hematol Am Soc Hematol Educ Prog. 2012;2012:617–625. doi:10.1182/asheducation-2012.1.617
- Mohammed SK, Mubarik A, Nadeem B, et al. Atypical hemolytic uremic syndrome: a case report. Cureus. 2019;11(5):e4634. doi:10.7759/cureus.4634
- Wu H, Su S, Li L, Zhang L. Atypical hemolytic uremic syndrome and acute tubular necrosis induced by complement factor B gene (CFB) mutation: a case report. Medicine. 2021;100:e25069.
- Basnayake B, Wazil AWM, Nanayakkara N, et al. Atypical hemolytic uremic syndrome: a case report. J Med Case Rep. 2020;14(11). doi:10.1186/s13256-019-2334-y
- Krishnappa V, Gupta M, Elrifai M, et al. Atypical hemolytic uremic syndrome: a meta-analysis of case reports confirms the prevalence of genetic mutations and the shift of treatment regimens. Ther Apher Dial. 2018;22(2):178–188. doi:10.1111/1744-9987.12641
- Jamme M, Raimbourg Q, Chauveau D, et al; French Thrombotic Microangiopathies Reference Centre. Predictive features of chronic kidney disease in atypical haemolytic uremic syndrome. PLoS One. 2017;12(5):e0177894. doi:10.1371/journal.pone.0177894
- Sallée M, Daniel L, Piercecchi MD, et al. Myocardial infarction is a complication of factor H-associated atypical HUS. Nephrol Dial Transplant. 2010;25(6):2028–2032. doi:10.1093/ndt/gfq160
- Vilalta R, Lara E, Madrid A, et al. Long-term eculizumab improves clinical outcomes in atypical hemolytic uremic syndrome. Pediatr Nephrol. 2012;27(12):2323–2326. doi:10.1007/s00467-012-2276-8
- Loirat C, Macher MA, Elmaleh-Berges M, et al. Non-atheromatous arterial stenoses in atypical haemolytic uraemic syndrome associated with complement dysregulation. Nephrol Dial Transplant. 2010;25(10):3421–3425. doi:10.1093/ndt/gfq319
- Gomez-Lado C, Martinon-Torres F, Alvarez-Moreno A, et al. Reversible posterior leukoencephalopathy syndrome: an infrequent complication in the course of haemolytic-uremic syndrome. Rev Neurol. 2007;44(8):475–478.
- Koehl B, Boyer O, Biebuyck-Gouge N, et al. Neurological involvement in a child with atypical hemolytic uremic syndrome. Pediatr Nephrol. 2010;25(12):2539–2542. doi:10.1007/s00467010-1606-y
- Ažukaitis K, Loirat C, Malina M, et al. Macrovascular involvement in a child with atypical hemolytic uremic syndrome. Pediatr Nephrol. 2013;29:1273–1277. doi:10.1007/s00467-013-2713-3
- Malina M, Gulati A, Bagga A, Majid MA, Simkova E, Schaefer F. Peripheral gangrene in children with atypical hemolytic uremic syndrome. Pediatrics. 2013;131(1):e331–5. doi:10.1542/peds.2012-0903
- Roman-Ortiz E, Mendizabal Oteiza S, Pinto S, Lopez-Trascasa M, Sanchez-Corral P, Rodriguez de Cordoba S. Eculizumab long-term therapy for pediatric renal transplant in aHUS with CFH/CFHR1 hybrid gene. Pediatr Nephrol. 2014;29:149–153. doi:10.1007/s00467-013-2591-8
- Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, et al; French Society of Pediatric Network. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007;18:2392–2400. doi:10.1681/ASN.2006080811
- Dragon-Durey MA, Sethi SK, Bagga A, et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. 2010;21:2180–2187. doi:10.1681/ASN.2010030315
- Formeck C, Swiatecka-Urban A. Extra-renal manifestations of atypical hemolytic uremic syndrome. Pediatr Nephrol. 2019;34:1337–1348. doi:10.1007/s00467-018-4039-7
- Berger BE. The alternative pathway of complement and the evolving clinical-pathophysiological spectrum of atypical hemolytic uremic syndrome. Am J Med Sci. 2016;352:177–190. doi:10.1016/j.amjms.2016.05.003
- Brocklebank V, Johnson S, Sheerin TP, et al. Factor H autoantibody is associated with atypical hemolytic uremic syndrome in children in the United Kingdom and Ireland. Kidney Int. 2017;92:1261–1271. doi:10.1016/j.kint.2017.04.028
- Besbas N, Gulhan B, Soylemezoglu O, et al. Turkish pediatric atypical hemolytic uremic syndrome registry: initial analysis of 146 patients. BMC Nephrol. 2017;18:6. doi:10.1186/s12882-016-0420-6
- Webb TN, Griffiths H, Miyashita Y, et al. Atypical hemolytic uremic syndrome and chronic ulcerative colitis treated with eculizumab. Int J Med Pharm Case Rep. 2015;4(5):105–112. doi:10.9734/IJMPCR/2015/18771
- Green H, Harari E, Davidovits M, et al. Atypical HUS due to factor H antibodies in an adult patient successfully treated with eculizumab. Ren Fail. 2014;36(7):1119–1121. doi:10.3109/0886022X.2014.917574
- Fakhouri F, Roumenina L, Provot F, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010;21(5):859–867. doi:10.1681/ASN.2009070706
- Meibody F, Jamme M, Tsatsaris V, et al. Post-partum acute kidney injury: sorting placental and non-placental thrombotic microangiopathies using the trajectory of biomarkers. Nephrol Dial Transplant. 2020;35(9):1538–1546. doi:10.1093/ndt/gfz025
- George JN, Nester CM, McIntosh JJ. Syndromes of thrombotic microangiopathy associated with pregnancy. Hematol Am Soc Hematol Educ Prog. 2015;2015:644–648. doi:10.1182/asheducation-2015.1.644
- Fakhouri F, Scully M, Ardissino G, Al-Dakkak I, Miller B, Rondeau E. Pregnancy-triggered atypical hemolytic uremic syndrome (aHUS): a Global aHUS Registry analysis. J Nephrol. 2021;34(5):1581–1590. doi:10.1007/s40620-021-01025-x
- Huerta A, Arjona E, Portoles J, et al. A retrospective study of pregnancy-associated atypical hemolytic uremic syndrome. Kidney Int. 2018;93(2):450–459. doi:10.1016/j.kint.2017.06.022
- Loeven MA, Rops AL, Lehtinen MJ, et al. Mutations in complement factor h impair alternative pathway regulation on mouse glomerular endothelial cells in vitro. J Biol Chem. 2016;291(10):4974–4981. doi:10.1074/jbc.M115.702506
- Larakeb A, Leroy S, Frémeaux-Bacchi V, et al. Ocular involvement in hemolytic uremic syndrome due to factor H deficiency--are there therapeutic consequences? Pediatr Nephrol. 2007;22(11):1967–1970. doi:10.1007/s00467-007-0540-0
- Zheng X, Gorovoy IR, Mao J, Jin J, Chen X, Cui QN. Recurrent ocular involvement in pediatric atypical hemolytic uremic syndrome. J Pediatr Ophthalmol Strabismus. 2014;51:e62–e65. doi:10.3928/01913913-20140923-03
- Greenwood GT. Case report of atypical hemolytic uremic syndrome with retinal arterial and venous occlusion treated with eculizumab. Int Med Case Rep J. 2015;8:235–239. doi:10.2147/IMCRJ.S90640
- David R, Hochberg-Klein S, Amer R. Resolution of ocular involvement with systemic eculizumab therapy in atypical hemolytic-uremic syndrome. Eye. 2013;27(8):997–998. doi:10.1038/eye.2013.111
- Loo CY, Mohamed Said MS, Mohd R, et al. Immunoadsorption and plasmapheresis are equally efficacious as adjunctive therapies for severe lupus nephritis. Transfus Apher Sci. 2010;43(3):335–340. doi:10.1016/j.transci.2010.10.003
- Koziolek MJ, Tampe D, Bähr M, et al. Immunoadsorption therapy in patients with multiple sclerosis with steroid-refractory optical neuritis. J Neuroinflammation. 2012;9:80. doi:10.1186/1742-2094-9-80
- Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33(6):508–530. doi:10.1016/j.semnephrol.2013.08.003
- Bresin E, Rurali E, Caprioli J, et al; European Working Party on Complement Genetics in Renal Diseases. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. 2013;24(3):475–486. doi:10.1681/ASN.2012090884
- Khandelwal P, Thomas CC, Rathi BS, et al. Membrane-filtration based plasma exchanges for atypical hemolytic uremic syndrome: audit of efficacy and safety. J Clin Apher. 2019;34(5):555–562. doi:10.1002/jca.21711
- Lee H, Kang E, Kang HG, et al. Consensus regarding diagnosis and management of atypical hemolytic uremic syndrome. Korean J Intern Med. 2020;35(1):25–40. doi:10.3904/kjim.2019.388
- Fakhouri F, Fila M, Hummel A, et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: a prospective multicenter study. Blood. 2021;137(18):2438–2449. doi:10.1182/blood.2020009280
- Cheong HI, Jo SK, Yoon SS, et al. Clinical practice guidelines for the management of atypical hemolytic uremic syndrome in Korea. J Korean Med Sci. 2016;31(10):1516–1528. doi:10.3346/jkms.2016.31.10.1516
- Licht C, Greenbaum LA, Muus P, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of Phase 2 studies. Kidney Int. 2015;87(5):1061–1073. doi:10.1038/ki.2014.423
- Fakhouri F, Hourmant M, Campistol JM, et al. Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single-arm, open-label trial. Am J Kidney Dis. 2016;68(1):84–93. doi:10.1053/j.ajkd.2015.12.034
- Cavero T, Rabasco C, López A, et al. Eculizumab in secondary atypical haemolytic uraemic syndrome. Nephrol Dial Transplant. 2017;32(3):466–474. doi:10.1093/ndt/gfw453
- Menne J, Delmas Y, Fakhouri F, et al. Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study. BMC Nephrol. 2019;20(1):125. doi:10.1186/s12882-019-1314-1
- Dixon BP, Madris-Aris AD, Adams B. Two-year efficacy and safety of ravulizumab in adults and children with Atypical Hemolytic Uremic Syndrome (aHUS): analysis of two phase 3 studies. Blood. 2021;138(1):769. doi:10.1182/blood-2021-145040
- Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540–549. doi:10.1182/blood-2018-09-876805
- Sheridan D, Yu ZX, Zhang Y, et al. Design and preclinical characterization of ALXN1210: a novel anti-C5 antibody with extended duration of action. PLoS One. 2018;13(4):e0195909. doi:10.1371/journal.pone.0195909
- Zuber J, Le Quintrec M, Krid S, et al.; French Study Group for Atypical HUS. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant. 2012;12(12):3337–3354. doi:10.1111/j.1600-6143.2012.04252.x
- Portoles J. Characteristics, management and outcomes of atypical haemolytic uraemic syndrome in kidney transplant patients: a retrospective national study. Clin Kidney J. 2021;14(4):1173–1180. doi:10.1093/ckj/sfaa096
- Durey MA, Sinha A, Togarsimalemath SK, Bagga A. Anti-complement-factor H-associated glomerulopathies. Nat Rev Nephrol. 2016;12(9):563–578. doi:10.1038/nrneph.2016.99
- Boyer O, Balzamo E, Charbit M, et al. Pulse cyclophosphamide therapy and clinical remission in atypical hemolytic uremic syndrome with anti-complement factor H autoantibodies. Am J Kidney Dis. 2010;55(5):923–927. doi:10.1053/j.ajkd.2009.12.026
- Raina R, Chauvin A, Fox K, et al. Effect of immunosuppressive therapy on the occurrence of atypical hemolytic uremic syndrome in renal transplant recipients. Ann Transplant. 2018;23:631–638. doi:10.12659/AOT.909781
- Law SK. First-line treatment for elevated intraocular pressure (IOP) associated with open-angle glaucoma or ocular hypertension: focus on bimatoprost. Clin Ophthalmol. 2007;1(3):225–232.
- Fakhouri F, Schwotzer N, Frémeaux-Bacchi V. How I diagnose and treat atypical hemolytic uremic syndrome. Blood. 2023;141(9):984–995. doi:10.1182/blood.2022017860
- Raina R, Grewal MK, Radhakrishnan Y, et al. Optimal management of atypical hemolytic uremic disease: challenges and solutions. Int J Nephrol Renovasc Dis. 2019;12:183–204. doi:10.2147/IJNRD.S215370
- Ritter C. Dr. Anuja Java Co-chairs Working Group in an International Committee for Revising Ahus Nomenclature; 2021. Division of Nephrology. Available from: https://nephrology.wustl.edu/dr-anuja-java-co-chairs-working-group-for-revising-ahus-nomenclature/. Accessed July 31, 2023.
- Muir E. The rare reality - an insight into the patient and family experience of rare disease: report launch. Rare Disease UK; 2016. Available from: https://www.raredisease.org.uk/rduk-news/the-rare-reality-an-insight-into-The-patient-and-family-experience-of-rare-disease-report-launch/. Accessed July 31, 2023.
- Marsden S, Dunbar L, Sandiford N. Do multidisciplinary teams make a difference to the quality of medical care? Br J Hosp Med. 2019;80(12):696–698. doi:10.12968/hmed.2019.80.12.696
- Uriol Rivera MG, Cabello Pelegrin S, Ballester Ruiz C, et al. Impact of a multidisciplinary team for the management of thrombotic microangiopathy. PLoS One. 2018;13(11):e0206558. doi:10.1371/journal.pone.0206558
- Azoulay E, Knoebl P, Garnacho-Montero J, et al. Expert statements on the standard of care in critically ill adult patients with atypical hemolytic uremic syndrome. Chest. 2017;152(2):424–434. doi:10.1016/j.chest.2017.03.055