
Abstract
Costello syndrome (CS) is a rare neurodevelopmental disorder caused by germline mutations in HRAS. It belongs among the RASopathies, a group of syndromes characterized by alterations in components of the RAS/MAPK signaling pathway and sharing overlapping phenotypes. Its typical features include a distinctive facial appearance, growth delay, intellectual disability, ectodermal, cardiac, and musculoskeletal abnormalities, and cancer predisposition. Due to the several comorbidities having a strong impact on the quality of life, a multidisciplinary team is essential in the management of such a condition from infancy to adult age, to promptly address any detected issue and to develop appropriate personalized follow-up protocols and treatment strategies. With the present paper we aim to highlight the core and ancillary medical disciplines involved in managing the health challenges characterizing CS from pediatric to adult age, according to literature and to our large clinical experience.
Introduction
Originally described in 1971, Costello syndrome (CS, OMIM #218040) is a rare multisystemic disorder with a strong impact on quality of life of caregivers and affected individuals.Citation1,Citation2 In 2005, germline missense mutations in the HRAS gene were identified as responsible for the CS phenotype by Aoki et al, with p.Gly12Ser being the most frequently reported variant.Citation3 The HRAS gene encodes a GTPase functioning as a signal hub controlling multiple signaling pathways, including the mitogen activated protein kinase (MAPK) cascade, a major pathway regulating fundamental biological functions such as cell proliferation, differentiation, and survival.Citation4,Citation5 CS is included among the RASopathies, a group of rare multisystem disorders that share the dysregulation of the RAS/MAPK pathway as the underlying pathogenetic mechanism and are characterized by an overlapping clinical phenotype.Citation6 While individually rare, collectively RASopathies represent the most common family of non-chromosomal disorders affecting development and growth.Citation7 Besides CS, this group of conditions includes neurofibromatosis type 1 (NF1, OMIM #162200), Noonan syndrome (NS, OMIM #163950), cardio-facio-cutaneous syndrome (CFCS, OMIM #115150), Mazzanti syndrome (also known as Noonan-like syndrome with loose anagen hair, NS/LAH, OMIM #607721), Noonan syndrome with multiple lentigines, previously known as LEOPARD syndrome (NSML, OMIM PS151100), CBL mutation-associated syndrome (CBLS, OMIM #613563), Legius syndrome (OMIM #611431), and other emerging disorders.Citation6,Citation8,Citation9
CS is characterized by a distinctive facial gestalt, closely resembling storage diseases, failure to thrive, ectodermal anomalies, cardiopathies, musculoskeletal problems, developmental delay (DD)/intellectual disability (ID), friendly personality, and predisposition to certain cancers.Citation10–Citation12
In the light of the multiple and diverse comorbidities affecting individuals with CS, and given the increased life expectancy associated with a better knowledge of this condition, a multidisciplinary and personalized approach has emerged as a key aspect for more effective care of these patients.
This paper provides an overview of the workflow that we routinely apply for the multidisciplinary management of individuals affected by this rare disorder.
From Clinical Suspicion to Molecular Diagnosis: HRAS Variants – from 2005 to Date
In 2005, using a candidate gene analysis, Aoki et al identified four heterozygous missense mutations of HRAS, including p.Gly12Ser, p.Gly12Val, and p.Gly13Asp, in 12 patients with Costello syndrome, all of which had previously been reported as somatic lesions in various tumors ().Citation3
Figure 1 HRAS domain structure and pathogenic variants of HRAS identified in patients with Costello syndrome. Upper panel shows missense mutations, and lower panel shows intragenic duplications. More than 90% of pathogenic variants are clustered in codons 12 and 13. It has been shown that mutations at these codons impair the intrinsic GTPase activity, resulting in constitutive activations of downstream effectors.Citation3,Citation9,Citation136 Reproduced from Aoki Y, Niihori T, Banjo T, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93(1):173–180. Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.Citation137 The de novo 10-nucleotide-long deletion within the intron-D-exon (IDX) is not shown in the figure.
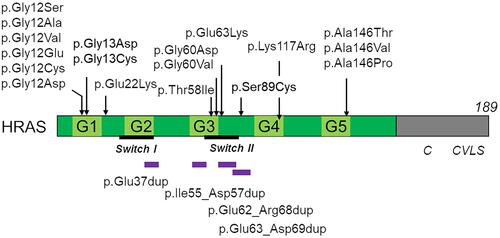
So far, germline HRAS mutations have been identified in more than 90% of patients with Costello syndrome.Citation3,Citation13–Citation17 These mutations are generally de novo events, indicating autosomal dominant inheritance. The p.Gly12Ser substitution has been identified in approximately 80% of patients. It has been suggested that patients with p.Gly12Ala have higher risk of malignancy than those with p.Gly12Ser ().Citation18 p.Gly12Asp, p.Gly12Glu, and p.Gly12Cys are associated with severe neonatal phenotypes with pleural/pericardial effusion, congenital lung and airway abnormalities, and cardiomegaly ().Citation15,Citation17,Citation19–Citation21, Recently, the p.Gly12Asp variant was described in a 31-year-old patient with special but milder manifestations.Citation22 Only seven patients have been reported with p.Gly12Val. This amino acid change is associated with a severe lethal phenotype, including severe hypertrophic cardiomyopathy (HCM), fetal hydrops, and hepatomegaly.Citation3,Citation23–Citation25 Excessive amounts of neuromuscular spindles have been identified in two patients carrying the p.Gly12Val substitution.Citation24,Citation26 p.Gly13Cys is associated with distinctive phenotypes, including dolichocilia (extremely long eye lashes) and loose anagen hair without multifocal atrial tachycardia.Citation27 p.Gly13Asp is associated with less coarse facial features and slow-growing sparse hair, resembling loose anagen hair.Citation28 Individuals heterozygous for the p.Gly13Cys or p.Gly13Asp changes have been reported to develop papillomata or vascular proliferation lesions, but not malignant tumors.Citation28,Citation29 A less evident phenotype has been suggested in patients with rare mutations at codon 58 (p.Thr58Ile),Citation30 60 (p.Gly60Asp),Citation31 117 (p.Lys117Arg),Citation32 and 146 (p.Ala146Thr/Val/Pro).Citation29,Citation33,Citation34 The p.Thr58Ile variant was observed in a girl with no intellectual disability but with severe HCM;Citation30 the rare p.Gly60Val variant was associated to subtle dysmorphic features and early death, highlighting how individuals with less evident CS characteristics may not have a more benign development.Citation35
Table 1 Genotype-phenotype relationships in Costello syndrome with missense HRAS variants, duplications, and deletions
The c.64C>A (p.Glu22Lys) mutation has been identified in patients with fatal congenital HCM and pancreatic nodule and in patients with congenital myopathy and excessive amount of muscle spindles.Citation26,Citation36
As anticipated, the vast majority of pathogenic variants occur as de novo events.Citation17 Anyway, in one family, the p.Thr58Ile substitution was reported in a son–father pair, the former showing macrocephaly and HCM and the latter apparently without any evidence of cognitive and cardiac involvement.Citation37 Also a case in which p.Gly60Asp mutation was transmitted from a mother with subtle dysmorphic features to the son with a mild phenotype was reported.Citation31 Patients who were suspected to have somatic mosaic mutation or germ cell mosaicism in HRAS were reported.Citation38–Citation41 Indeed cases of brothers affected by CS most probably due to germline mosaicism were described.Citation42 The p.Ser89Cys variant was described in two siblings with a milder phenotype and their asymptomatic father.Citation43
Other than missense mutations in HRAS, intragenic duplications/deletions were also identified in a small proportion of patients with clinical features fitting or suggestive of CS. p.Glu37dup has been identified in two patients with mental retardation, short stature, sparse hair, and mild musculoskeletal manifestations.Citation44 p.Ile55_Asp57dup, p.Glu62_Arg68dup, and p.Glu63_Asp69dup have been identified in six patients with a milder or attenuated phenotype.Citation45–Citation48 Finally, a de novo 10-nucleotide-long deletion within the intron-D-exon (IDX) exon of the gene was identified in a subject with DD/ID, autistic features, distinctive coarse facies, reduced growth, and ectodermal anomalies.Citation49 Of note, this deletion was demonstrated to affect HRAS transcript processing, promoting constitutive retention of exon IDX, which is generally skipped during HRAS transcript processing, resulting in a stable and mildly hyperactive GDP/GTP-bound protein constitutively targeted to the plasma membrane.Citation49
Early Diagnosis of Costello Syndrome: Prenatal Findings and Perinatal Features
To schedule a proper monitoring and treatment program, prompt diagnosis at birth or even prenatally has a decisive value. Anticipatory understanding of the possible complications is beneficial for a more effective management, helping physicians to apply a multidisciplinary health approach since birth and to adequately support the family. Furthermore, issues that may develop during pregnancy, such as arrhythmias, macrosomia, relative macrocephaly, and risks of preterm birth, could be more easily addressed if both the medical team and the parents are aware of them, drastically reducing labor-related complications.
Key diagnostic elements that may raise the suspicion of CS in utero are polyhydramnios (which is reported in most pregnancies), supraventricular tachycardia, increased nuchal translucency, macrosomia and macrocephaly (usually after the 20th week of gestation), and peculiar fetal posture; reduced length of long bones has also been reported.Citation50 Fetal arrhythmias are rare and usually responsive to therapy.Citation50,Citation51 The most common cause of polyhydramnios in association to fetal macrosomia is maternal diabetes, but once this condition is ruled out other causes of increased fetal size such as CS should be considered.Citation52
A feature that may help gynecologists in the differential diagnosis is that fetuses with CS have increased head size and body weight, but length is within the reference range. This is probably because macrosomia is mostly related to subcutaneous edema rather than a real overgrowth.Citation25
Most cases with a prenatal diagnosis reported in literature showed a severe phenotype associated to rare variants (eg, p.Gly12Val), often resulting in intrauterine or early postnatal death.Citation21,Citation25
Typical features of newborns with CS include a characteristic facial gestalt with hypertelorism, low-set and posteriorly angulated ears, short nose with bulbous tip, full lips, and macrostomia. Hand posture is also distinctive, with clenched fists and overlapping of fingers.Citation51 Hepatomegaly, small thorax, camptodactyly, and cryptorchidism are often associated to the neonatal lethal CS phenotype.Citation25 When all these elements are present, they should be suggestive of CS, and prompt genetic counseling should be scheduled.
Gynecologists should be aware of the main prenatal features that may raise suspicion for CS (prenatal polyhydramnios, increased nuchal translucency, fetal macrosomia, macrocephaly, and fetal arrhythmias) in order to inform neonatologists on possible medical comorbidities soon after birth and reduce the risk for the affected baby.
Core and Ancillary Medical Specialties in Pediatric Age: Combining Medical Management to Habilitative Therapies
Medical assistance in specialized hospitals is mostly recommended in infancy and childhood; during these periods, the most concerning issues to address are related to growth and neurodevelopment. Cardiorespiratory functions also need to be accurately monitored.
A relevant matter is also the evaluation of visual abilities since many patients with CS may present nystagmus and refractive errors impairing their everyday living and neurodevelopmental skills acquisition.Citation53
Looking at the different anecdotal reports about cancer in pediatric patients with CS, screening protocols should be applied in affected individuals from infancy.Citation12
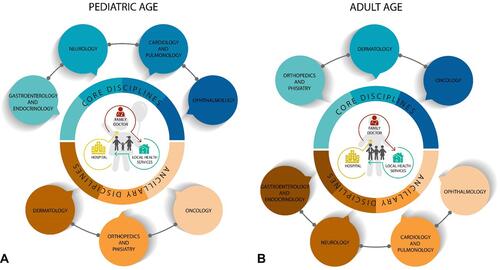
Considering all these issues, as soon as a diagnosis of CS is prompted, it is of utmost importance to lay the foundations to establish personalized follow-up schedules, therapeutic strategies, and habilitative protocols to ensure a good quality of life and to support families/caregivers. At the same time, a virtuous collaboration network between hospital and local health services providing physical, speech, and occupational therapies needs to be established in order to achieve the best clinical outcome ().
Figure 2 Core and ancillary disciplines involved in the management of pediatric and adult patients with CS. The impact of medical issues in the daily lives of patients with CS requires careful management throughout life. A comprehensive multidisciplinary assessment to be performed by physicians and therapists is needed from infancy to adulthood to promptly treat and monitor comorbidities. The latter change according to patients’ age. In particular, the most concerning problem to manage in children is related to failure to thrive. The cardiorespiratory system needs to be accurately evaluated to exclude severe morphological or rhythmic cardiac anomalies. Visual and global neurological functions need to be assessed in order to plan a personalized protocol of habilitative therapies (A). In adults, orthopedic manifestations often require treatment; a comprehensive dermatological evaluation is important to improve skin findings, and a surveillance protocol for cancer risk needs to be followed (B).
Core Disciplines in Pediatric CS Population
Growth and Nutrition
Soon after birth till first years of life, growth delay and severe failure to thrive are two of the most remarkable features characterizing CS, causing significant concern for parents and representing a real challenge for physicians.
Even though weight at birth is within the normal range, according to standardized growth charts for the general population, the weight-for-age is dramatically decreased below normal within the first three years.Citation2,Citation42,Citation54 In previous reports, this first phase of development was indeed described as “marasmic”.Citation54 Based on these observations, normative growth charts for CS have been created.Citation55 Also height is significantly below the reference value for age, with a consequently normal BMI. Poor weight is probably due to a plethora of factors, among which feeding difficulties worsened by global hypotonia have a substantial role. The oro-motor dysfunction with impaired suckling and swallowing movements and oral hypersensitivity are other major contributors to such a phenotype. Severe gastroesophageal reflux disease is also common.Citation42,Citation54,Citation56,Citation57 Hypertrophic pyloric stenosis has also been reported with increased frequency compared to the general population.Citation34 Enteral feeding is most often required during the first three or four years of life through either nasogastric (ng-tube) tube or more commonly gastrostomy (g-tube).Citation29,Citation54,Citation58 The use of g-tube would be preferable compared to ng-tube (when required over 3 months) to improve oral motor skills and dysphagia through personalized speech therapy. A normo-caloric diet has been demonstrated to be as effective as a hypercaloric diet in reaching the expected growth parameters with a better tolerance and fewer gastrointestinal symptoms.Citation54 Children affected by CS usually acquire normal feeding capacities by the age of 4 years; nevertheless growth delays with both short stature and poor weight do persist in adult age.Citation58 Given this complex phenotype, a close follow-up by gastroenterologists and clinical nutritionists (every 3 to 6 months) is necessary in pediatric age groups ().
Table 2 Timing of Clinical and Instrumental Follow-Up According to Literature and Our Experience
It is known that the RAS/MAPK cascade has a role in the regulation of cellular metabolism and growth.Citation56,Citation59 It has recently been demonstrated that an increased resting energy expenditure may play a role in the poor growth pattern in CS. The use of indirect calorimetry may help clinicians provide the right amount of kilocalories per day based on personalized needs. Some factors may be at the base of the increased energy expenditure in CS such as heart defects, lung issues, infections, and endocrine dysfunction, but the intrinsic effect of HRAS mutation has otherwise been primarily considered.Citation56,Citation60
GH and cortisol deficiencies were also observed in CS, sometimes associated to fasting hypoglycemia.Citation61,Citation62 The use of replacement therapy once GH deficiency is diagnosed is controversial due to comorbidities such as HCM, central nervous system (CNS) malformations, and obstructive apneas syndrome.Citation63 Routinely monitoring of growth velocity (every 6–12 months) by an experienced endocrinologist is strongly suggested in infancy, particularly in patients dropping below standard growth charts for CS.Citation29,Citation55,Citation58
Hypoglycemia has also been reported in the absence of pituitary gland and hypothalamus dysfunctions in humans, as well as hypercholesterolemia.Citation56 Similarly, metabolic changes have been reported by Oba et al in HRAS p.Gly12Ser knock-in mice, with growth failure and impaired fatty acid oxidation observed in mice fed with a high rich fat diet. Such findings confirm the roles that the RAS pathway and the HRAS gene have in metabolism regulation.Citation64,Citation65
The risk of endocrinopathy and hypoglycemia should be assessed during prolonged fasting, as also by anesthesiologist when a surgical intervention is planned, and in some cases peri-operative glucose monitoring with steroid coverage may be considered.Citation66
Neurodevelopment and Central Nervous System (CNS) Morphology
Delay in neurodevelopmental milestones is almost universal in CS. A moderate to severe hypotonia is present since birth. Physical therapy (at least 2 to 3 times per week), together with the use of orthosis and postural aids, is crucial to safely feed the baby and to improve the patient’s outcome. Dystonia has also been anecdotally reported contributing to abnormal postures in children with CS;Citation10,Citation67,Citation68 a therapeutic approach with trihexyphenidyl was recently reported in a patient, showing improvement of posture and gait.Citation69
Cognitive impairment is always present, ranging in severity from mild to moderate or severe forms. A comprehensive neurological assessment by clinical neurological evaluation and cognitive tests needs to be performed on a regular basis. Monitoring by a child neurologist and possibly physiatrist is indicated every 3 to 6 months in small children (till 4 to 5 years) and every 6 to 12 months later on. Annual evaluation of cognitive performances and adaptive behavior profile is also recommended in pediatric age (). A high rate of individuals affected by CS show autism spectrum disorder (ASD) traits especially during early infancy, with restlessness, extreme shyness, speech delays, and aversion to touch and auditory stimuli often reported.Citation70,Citation71
Another striking feature of CS is the relative macrocephaly, probably due to megalencephaly. Hyperplasia and increased differentiation towards astroglial cells were observed in induced pluripotent stem cells (iPSCs) carrying HRAS G12S, while in mouse models homozygous for HRAS G12V variant increased brain weight, surface area of cortex, striatum, and corpus callosum were reported.Citation72,Citation73 These models partly explain the CNS abnormalities reported in individuals with CS. It has been hypothesized that the posterior cranial fossa crowding found in patients with CS might be due either to an increased cerebellar volume in a relatively normal cranial fossa,Citation74 or to a hypoplastic cranial fossa, reduced cerebrospinal fluid (CSF) spaces, and an altered anatomy of the foramen magnum with relatively normal cerebellar dimensions.Citation75 Either way the result is a posterior fossa crowding with consequent cerebellar tonsillar herniation, often configuring as a true Chiari type I malformation. Such tonsillar herniation sometimes results in medullary compression, consequent hydrocephalus, syrinx, or hydromyelia.
CNS malformations reported are mostly asymptomatic, but feeding difficulties, respiratory distress and/or apnea, ocular palsy, arching, and headache, consistent with compression of medulla oblongata and upper cervical spine, need to be carefully monitored ().
Surgical interventions with shunt placements or ventriculostomy are the most reported neurosurgical procedures. Posterior fossa decompression is also often performed to improve severe symptoms when present.
For all the above-mentioned reasons, magnetic resonance imaging (MRI) of both brain and spine needs to be performed soon after diagnosis of CS, and personalized follow-up strategies based on major findings are mandatory. If the first MRI screening exam results are negative for CNS involvement, a subsequent MRI after two to three years, or as soon as symptoms are reported, is kindly suggested. In the presence of CNS abnormalities, follow-up schedules are personalized according to individual features (). Noteworthily, reduced prevalence of neurological abnormalities has been observed in individuals carrying the p.Gly13Cys variant ().Citation27
Cardio-Respiratory Issues
Individuals affected by CS, as with other RASopathies, require a careful cardiological evaluation at birth and/or at diagnosis. Congenital heart defects (CHD) and HCM are frequently found in CS and represent major contributors to morbidity and may be a cause of early death.Citation29,Citation66,Citation76–Citation79 Heart failure and cardiocirculatory collapse have also been reported as cause of death in some infants.Citation21 Pulmonary valve stenosis (PVS), dysplasia, mitral valve defects, and other valve abnormalities were also described.Citation15,Citation19,Citation20,Citation23,Citation51,Citation80,Citation81
Electrocardiographic (ECG) anomalies such as arrhythmias (usually supraventricular tachycardia) may occur both during fetal and postnatal life; HCM is not usually present prenatally or at birth, but it subsequently develops.Citation51 Therefore, a comprehensive cardiac evaluation with echocardiography, ECG, and cardiological evaluation needs to be routinely performed since birth, to carefully monitor HCM development and rhythmic anomalies (). HCM can be heterogeneous: either reversal, resolution, or progression has been reported. Pharmacological treatment is usually recommended as first-line therapy in cases of progression of HCM, but sometimes it is not sufficient to prevent its evolution; in such cases, surgical treatment (septal myectomy) is necessary to prevent congestive heart failure (CHF) that contributes to the worsening of clinical symptoms.Citation78,Citation79,Citation82–Citation84
A subset of rarely occurring variants in HRAS have been associated with more severe phenotypes characterized by premature deaths often due to either cardiocirculatory or respiratory causes or multi-organ failure. Structural abnormalities, such as cardiomegaly, PVS, biventricular and septal hypertrophy, and patent foramen ovale, and conduction disturbances, such as atrial and ventricular tachyarrhythmias, have been reported in patients heterozygous for the p.Gly12Glu, p.Gly12Cys, p.Gly12Asp, and p.Gly12Val substitutions ().Citation21,Citation25
In case of surgical intervention, preoperative ECG and echocardiogram should always be performed to screen for HCM and other complications.
Disorders concerning the respiratory system have not been frequently reported in literature, but they are commonly referred to by caregivers, especially in children. Abnormalities of both upper and lower airway tract are frequent findings, as is also a high incidence of postnatal respiratory distress, which is not necessarily associated with prematurity. As for cardiac involvement, it was shown that rare variants, such as p.Gly12Cys, p.Gly12Asp, p.Gly12Glu, and p.Gly12Val, are associated to increased morbidity and mortality linked to respiratory (and cardiac) impairment, with death within the first months of life often due to respiratory/heart failure (). In case of prenatal diagnosis of CS, especially when due to such specific variants, respiratory complications should be expected, and, after a careful evaluation, a management strategy should be planned to face a severe cardio-respiratory phenotype.
Adenoid/tonsillar hypertrophy, upper airway obstruction leading to obstructive sleep apnea (OSA), pharyngomalacia, laryngomalacia, tracheo-bronchomalacia, bronchiectasis, bronchopulmonary dysplasia, lung hypoplasia, and pulmonary vascular dysplasia are some of the reported respiratory abnormalities.Citation85,Citation86 Screening of sleep quality is often necessary, sometimes with the aid of a sleep diary, to investigate the presence of sleep disorders; if positive, ENT evaluation combined with overnight polysomnography to search for OSA is suggested ().
When planning surgery, anesthesiologists should be aware of the difficult airway management in patients with CS. The abovementioned abnormalities along with macroglossia, small jaw, airway papillomata, and scoliosis should always be assessed to choose the safest intubation.Citation66,Citation77
Ocular Findings
Ophthalmologists are among the most visited medical specialists by CS patients in infancy since vision problems and other ocular issues are common findings.Citation87 Refractive errors are present in most individuals, but strabismus, nystagmus, and optic nerve anomalies were also observed. As in other RASopathies, palpebral ptosis is common.Citation53,Citation88
Retinal dystrophy was observed in two boys with p.Gly13Cys, both having nystagmus, photophobia, and abnormal findings at electroretinogram examination ().Citation89
Recently, bilateral anterior capsular plaque and anterior lenticular opacities were described in a CS patient, suggesting the necessity of accurate anterior chamber examination.Citation90
The large number of ocular manifestations in CS underline the importance of early and periodic ophthalmological assessment. A first evaluation should be performed as soon as a diagnosis of CS is established. During infancy, an eye exam with fundus oculi every 6 months is recommended, at least till 3 years of life, to prevent a long-term vision impairment, and subsequently annually or based on major findings (). Precocious visual function rehabilitation should be performed in highly qualified centers together with physical therapy; this is crucial to improve visual impairment and overall quality of life in CS patients.
Ancillary Disciplines in Pediatric CS Population
Orthopedic Problems and Bone Impairment
The musculoskeletal system is deeply compromised in CS, with individuals carrying the common p.Gly12Ser variant having a more severe and impacting phenotype, and the less recurrent p.Gly13Cys variant associated to milder manifestations ().Citation27
Most characteristic findings in pediatric age are ulnar deviation of fingers, anterior chest wall abnormalities, tight Achilles tendons, and pes planus. Most of these manifestations do not require surgical treatment in pediatric age with the exception of tight heel-cords. In fact, the use of orthosis from walking age may both prevent the evolution towards severe tight heel-cords, and may be useful to improve the outcome of surgical treatment, when required, together with adequate physical therapy (several times per week). Therefore, proper orthopedic assessment and treatment from young age are necessary to avoid worsening of the phenotype and loss of autonomy.
Spine is commonly affected in CS with kyphosis and/or scoliosis. Spine X-ray is necessary when a clinical diagnosis of spine abnormalities is present, to monitor the evolution over time of deformities widely ranging from mild to severe (dystrophic scoliosis). Given the high prevalence of hip dysplasia reported, hip X-ray as a screening method during infancy is fundamental, also to exclude a coxa valga subluxans ().Citation10,Citation91
Bone assessment by the age of 5 years with dual-energy X-ray absorptiometry exam (DEXA-scan) and biomarkers of bone metabolism dosage every two years may help clinicians to prevent and to timely treat a vitamin D insufficiency and bone mineral density reduction ().Citation92
Dermatologic Evaluation
As in most RASopathies, the integumentary system in CS is affected. Some ectodermal features do not have any functional implication and only contribute to the recognizable phenotype of individuals with CS, such as sparse and curly hair, deep palmar and plantar creases and pachydermatoglyphia. Papillomata, which usually develop in the nasal and perianal regions, are not present during early infancy but mostly occur in childhood. They have no malignant potential, and they are most often removed for aesthetic reasons or due to frequent itching.
Other features, such as cutis laxa, palmoplantar keratoderma, hyperkeratosis, and eczema, may appear as secondary issues, but taken together they may have a significant effect on patients'/families’ quality of life. Some parents also reported an unusual body odor.Citation2,Citation93,Citation94 Hyper-pigmented lesions and melanocytic nevi have been reported in CS, sometimes with potential malignant evolution (see below). For this reason, an annual clinical and dermatoscopic evaluation by a dermatologist is strongly suggested ().
Tumor Surveillance
As reported by Kratz et al in 2015, CS is a tumor-predisposing syndrome, with affected children having a 42.4-times increased risk to develop cancer compared to the general population of the same age.Citation11,Citation12 The most frequently reported solid tumors in childhood are neuroblastoma (NBS) and rhabdomyosarcoma (RMS), while bladder carcinoma has generally been observed during adolescence and adulthood.Citation95 As recommended by the consensus guidelines for CS, an abdominal ultrasound (US) every 3–6 months, or according to symptomatology, is recommended to check for internal organ tumors ().
Routine clinical surveillance with a general physical examination is the only way to detect the presence of RMS.Citation29
Since transitional cell carcinoma of the bladder has been reported from the second decade, screening protocols should start at the age of 10, with physical-chemical urine examination, urine cytology, and cystoscopy. The last-mentioned examination, even though more invasive, may allow all bladder lesions to be detected and histologically characterized ().Citation96
Based on available data in literature, most cases of patients with CS who developed tumors (RMS, ganglioneuroblastoma, or bladder carcinoma) carry the p.Gly12Ser or p.Gly12Ala variants. Two patients with p.Gly12Cys developed RMS ().Citation15,Citation97 No data about tumors in patients with rarer variants or with intragenic duplications have been reported to date, therefore a clear-cut genotype–phenotype correlation concerning tumor risk according to specific HRAS variants has not been performed yet. This bias could be related to the rarity of CS, the lack of long-term follow-up, and the paucity of variants different from the more prevalent ones.Citation28
Core and Ancillary Medical Specialties in Adult Age: Changing Medical Specialties According to Phenotype Evolution Over Time
The more accurate management of CS from young ages has led to an increased life expectancy; therefore, it has become necessary to improve assessment strategies according to the natural history of the disorder. During adult age, centers specialized in rare disorders acquire a supportive role, guiding major clinical decisions but having a minor function in everyday issues. Unfortunately, difficulties in finding effective management plans have been reported by most adult individuals affected by CS when they had to find a general practitioner or an occupational therapist,Citation87 underlining a significant healthcare gap in the transition process from infancy to adulthood and a more challenging management of medical problems in adult age. The multidisciplinary approach is still necessary to take care of overall comorbidities. Within them musculoskeletal and bone issues surely require a primary attention ().
Core Disciplines in Adult CS Population
Orthopedics Problems and Bone Impairment
Since orthopedic manifestations have a huge effect on quality of life and functional abilities, proper management of such issues is crucial in adult age.Citation98 Therefore, the multidisciplinary team should always include orthopedists, physical therapists, and an expert on bone metabolism (). During follow-up visits, the 6 minutes walking test (6MWT), Pediatrics Outcomes Data Collection Instrument (PODCI), and other functional tests might be useful tools to evaluate both progression and functional implications of skeletal abnormalities.Citation10,Citation98 Hip X-ray to monitor a coxa valga subluxans is suggested also during adolescence ().Citation99 Both clinical and radiological evaluation of spinal abnormalities is essential to monitor possible worsening in adult age due to the intrinsic role played by RAS/MAPK pathway on bone cells.Citation10,Citation87,Citation91,Citation99–Citation101
All bone deformities evolve over time in CS, resulting in more difficult surgical interventions;Citation10 therefore, the prompt recognition and treatment of such problems leads to easier and quicker post-surgery clinical outcome. The most commonly performed surgeries are tendon releases (performed in infancy and/or adult age), hip osteotomy, and spinal fusion.Citation87
Another issue to take into consideration in adults concerns the reduced bone mineral density that occurs in individuals with CS, generally in association with a constitutional decrease of 25-OH vitamin D.Citation92,Citation102 Observed osteopenia/osteoporosis has also been reported in some anecdotal cases of vertebral crush fractures and bone pain.Citation100 As also suggested in individuals affected by CFCS,Citation103 the abovementioned findings highlight the importance of monitoring bone homeostasis in all individuals with CS with DEXA-scan and bone biomarkers of bone metabolism every 24 months based on basal findings and personalized needs (). As it was often reported to be constitutionally low, high-dose vitamin D supplementation therapy according to patients’ basal levels and needs is strongly suggested from pediatric to adult age to at least maintain stable vitamin D levels and bone density.Citation92,Citation102
Given their peculiar phenotype, individuals with CS acquire an atypical posture, with an anteriorly flexed trunk, flexed elbows and wrists. Also small and large joint contractures are reported.Citation100 Muscles are severely impaired too, with generalized hypotrophy that may be due to alterations during myoblast differentiation.Citation10 Such reported skeletal abnormalities, together with small and large joint contractures, are most probably involved in the chronic pain commonly reported in CS.Citation98,Citation104 This is further worsened by hypo-/hypertonia, which alters both static and dynamic posture.Citation104 Given all these issues, it is of utmost importance for orthopedics and physical therapists to share decision-making and plan together a timely monitoring of muscle-skeletal assessment in adults with CS.Citation29,Citation91
Dermatologic Evaluation
In adults, dermatological findings may progress over time, with severe calluses, hyperpigmented and xerotic skin, and acanthosis nigricans being more frequently present. Antihistaminic oral therapy and topical treatment with moisturizing to improve patient’ discomfort due to continuous itching are often necessary. Periorificial papillomata do persist in adult age, and they can be removed only surgically or with local treatment.
In all RASopathies a higher number of nevi compared to the general population was reported, and an increased risk to develop atypical lesions when the number of nevi was >30 was observed. It was also observed that mutations in downstream components of the RAS pathway, such as those resulting in hyperactivation of the BRAF, MAP2K1, and MAP2K2 kinases, may predispose to an increased number of nevi.Citation105
Since melanoma in situ and melanocytoma were anecdotally reported in CS,Citation106 a careful skin assessment through clinical and dermatoscopic examination by an expert dermatologist should be performed every 12 months in the adult population according to the patients’ needs, to promptly remove atypical lesions and to provide a histopathological analysis ().
Tumor Surveillance in Adult Age
Data from literature provide evidence of reduced risk to develop RMS and NBS in adult life, whereas transitional cell carcinoma of the bladder seems to be particularly frequent both in adolescence and young adults with CS.Citation100 Bladder lesions are often asymptomatic, therefore abdominal US and urinalysis may not be sufficient to detect low-grade lesions. Cystoscopy should therefore be implemented as screening, diagnostic, and therapeutic intervention to remove any detected bladder lesion before its evolution towards malignancy ().Citation96
Ancillary Disciplines in Adult CS Population
Nutrition
After infancy/early childhood, feeding problems are no longer an issue. Adult individuals with CS usually do not present anymore oral motor dysfunction with swallowing difficulties, and neither aversion to food nor vomiting. Their families often report that they have an appropriate and balanced diet. Anyway, since some disturbances persist, follow-up evaluation by gastroenterologists is still recommended.
Constipation and abdominal pain, consistently to what was found in other RASopathies, are the issues that are most often reported.Citation104 Such pain may be attributed to a visceral hypersensitivity and therefore has a functional origin. Sometimes also gastroesophageal reflux disease (GERD) may persist in adult age, and therefore adequate pharmacological therapy is required.
Neuro-Psychological Issues
Besides reported anatomical and functional neurological abnormalities, consistent with the role played by the RAS pathway in neuronal function and plasticity, cognitive impairment is always present, with ID reported in the vast majority of patients with CS.Citation107–Citation109
While in infancy a shy personality with autism disorder traits is reported, by the age of eight to ten years they are extremely sociable and happy.Citation110 Some authors speculated that such improvement occurs as feeding issues resolve, as it also occurs in patients affected by other neurodevelopmental disorders.Citation111 Socialization was reported as a relative strength in individuals affected by CS, while more issues were encountered in daily living skills.Citation112
Nevertheless, consistently to what was observed in other RASopathies,Citation113 psychiatric disorders such as depression, anxiety, and separation anxiety have been commonly reported, especially in male individuals with CS, who showed a superior number of maladaptive behaviors. To target such specific issues, after a primary neuropsychological evaluation, psychotherapy and behavioral therapy may be useful, and also medications in the most severe cases of anxiety to avoid any functional implication in everyday activities.Citation114
Epileptic seizures are a rare finding in patients with CS, even though different electroencephalographic (EEG) abnormalities have been reported in literature.Citation115
Cardiological Issues
Based on cardiological evaluation performed in pediatric age, a comprehensive cardiological assessment needs to be performed during the entire lifespan of affected patients. Considering the available data reporting either improvement, stabilization, or worsening of HCM during life, yearly cardiac US follow-up is strongly suggested. Moreover, based on the few case reports of sudden death in CS, and the risk to develop fatal arrhythmias, yearly ECG exam is also recommended ().
Ophthalmologic Findings
Ocular disorders reported in CS are a common cause of vision loss, with subsequent decline in the quality of life. Therefore, proper ophthalmologic evaluation with accurate fundus oculi exam should be performed every year throughout the life of individuals with CS, or with a personalized timing according to patients’ needs. Visual rehabilitation therapy in specialized centers is fundamental to avoid further deterioration of visual capacity in the presence of ocular disorders ().
Discussion
In recent years, thanks to the understanding of the genetic basis of diseases, better knowledge of medical comorbidities, and implementation of personalized treatments, life expectancy for individuals affected by CS has significantly increased. Nevertheless, the pathogenetic mechanisms underlying some of the varied medical issues characterizing this disorder are still not clear, making the development of new treatments and follow-up strategies challenging. Overall, a multidisciplinary assessment is undeniably required to effectively take care of affected patients and their families, and to monitor and promptly treat comorbidities as soon as they occur. A close multisystem evaluation especially during infancy, childhood, and young ages dramatically improves the outcome of young patients. In detail, a multidisciplinary team involving pediatricians, gastroenterologists, nutritionists, endocrinologists, child neurologists, and physical therapists is required to address the failure to thrive that characterizes the first years of life of individuals with CS. The recent documentation of increased basal metabolism and resting energy expenditure surely needs to be supported by future studies on larger cohorts to confirm its negative impact on growth and metabolic profile.Citation56,Citation60
A regular overall neurological assessment by a child neurologist and the routine habilitative therapies performed by physical, speech, and occupational therapists are crucial to improve quality of life, to support families, and to reach best outcomes during patients’ lifespan. Changes in behavioral phenotype are frequently found in CS, with irritability during infancy and early childhood, and a more friendly and sociable attitude in adulthood, compared to all other RASopathies,Citation113 requiring specific support by specialized doctors.
Cardiologists need to be included in the multidisciplinary team since birth to help in defining correct timing and strategies of follow-up according to the evolution over time of a cardiac phenotype characterized by HCM and arrhythmias. Pneumological evaluation is also recommended from pediatric age to exclude OSA or other pulmonary involvement mostly associated to rare variants with worse phenotypes.
Ophthalmological evaluation is recommended from birth since vision deterioration could have a negative impact on neurodevelopment, limiting social interactions and the improvement of life skills.
Orthopedists are other fundamental figures in the multidisciplinary team given the worsening musculoskeletal involvement in CS. Furthermore, the recent reports about reduction in BMD both in pediatric and adult individuals with CS, sometimes associated to an increased risk of fractures in adult age, need to be supported by further studies to understand the underlying pathogenetic mechanisms caused by HRAS mutation and to develop proper treatments.
A comprehensive dermatological assessment by specialized dermatologists should be routinely performed at least yearly to improve troublesome symptoms such as itching and sweating, and to monitor the progression of melanocytic nevi.
While cancer risk is now well-recognized, dedicated studies should be performed to define the prevalence and risk of tumor development more accurately as well as to properly address the issue of personalized screening protocols.
RAS GTPases function as molecular switches controlling a major intracellular signaling network that, depending on the cellular context, guides diverse biological functions, including cell fate determination, proliferation, survival, differentiation, migration, and senescence. This multifaced role is attained by the control of a number of signaling pathways mediating different cellular processes. Among these, the MAPK and PI3K/AKT/mTOR pathways are commonly dysregulated in cancer, and components of these cascades have been identified as targets for therapeutic intervention.Citation116,Citation117 Indeed the development of therapies targeting these pathways has opened a way to approach RASopathies by selective inhibition of the dysregulated cascades implicated in pathogenesis.Citation118,Citation119 First evidence of such potential beneficial effects is supported by the recent approval of MEK inhibitors (MEKi) such as selumetinib for the treatment of symptomatic inoperable plexiform neurofibromas in children affected by NF1.Citation120,Citation121 Recent evidences also support the use of trametinib for the treatment of HCM in Noonan syndrome.Citation122 The use of selective MEKi, as hopefully other new targeted molecules, may be promising for early treatment or even the reversal of manifestations associated to enhanced activity of the MAPK pathway. A certain efficacy in preventing the development of RASopathy features was proven in 2015 in animal models by prenatal administration of MEKi.Citation123 Recent preclinical studies proved that inhibition of MEK could improve the myopathy associated to HRAS G12V variant.Citation124 Unfortunately, MEKi have a narrow therapeutic index, but their toxicities may be controlled by intermittent dosing. Furthermore, the combination of such molecules with RAF monomer inhibitors (which instead help ERK signaling pathway in normal cells) may be a promising therapeutic strategy to avoid adverse effects.Citation125
Besides the MAPK signaling cascade, overexpression of CS-causing HRAS mutants has been demonstrated to result in variably enhanced growth factor-dependent stimulation of the PI3K-AKT-mTOR pathway in multiple experimental in vitro systems.Citation44,Citation126 These considerations suggest that targeting of both pathways might be required for an effective managing of the evolutive complications of CS. It should be also noted that the PI3K/AKT/mTOR and MAPK cascades are interconnected with multiple points of convergence, cross-talk, and feedback loops. The presence of compensatory loops able to cross-activate one pathway following the blockade of the other has been demonstrated.Citation126,Citation127 Based on these considerations, the blockade of both pathways with a combined targeted approach should be taken into account.
Another treatment opportunity could be represented by farnesyl transferase inhibitors (FTi) such as tipifarnib, as it suppresses HRAS function, whereas NRAS and KRAS overcome its effects.Citation69,Citation118,Citation125 Future studies considering other RASopathies may be promising, even if the rarity of the disease, the relatively young age of the affected population, interpatient variability, and the possible long-term evolution of such disorders may represent a challenge for clinical trial development.Citation128,Citation129
Similarly to other rare diseases, CS represents a continuous challenge both for patients and their physicians. Starting from being diagnosed, several issues are faced by the family of a patient with CS, such as receiving optimal care and the financial burden of disease-specific treatments. Moreover, physicians struggle to obtain enough knowledge and understanding of the condition, especially in primary centers. This is the reason for which it is of great importance to collect relatively large cohorts of patients to evaluate and follow throughout their lives.Citation130 Primary care physicians’ role is to manage patients’ care together with specialized centers and facilitate communication with families and local health services. Specialized hospitals have a major role during the entire life of patients: the management of complex care needs, especially in pediatric age, the possible inclusion in research initiatives, multicenter studies, and enrollment in clinical trials for the development of new experimental therapies thereafter. It is of utmost importance for patients and families to develop mutual trust and understanding with their primary care physician, the other specialized clinicians, and researchers taking care of their condition.Citation131
The presence of clinical guidelines for CS makes their actual management easier;Citation29 however, many points should still be addressed to improve care and outcomes of patients affected by this syndrome. The diagnosis of a rare disease such as CS, with all its consequences, may represent a big concern for patients and their families, often becoming a cause of loneliness and isolation from society. Not only patients but also caregivers struggle, especially when adapting to a new routine and abruptly changing their everyday life and their perception of normality. In such situations, when families are spending most of their times actively looking for answers, social support may be missing, having a great impact on psychological health of caregivers. Several issues are often associated to the rarity of the disease, and to the nonexistent comparison with peers. For this reason, family support groups represent a significant resource for individuals affected by CS and their families/caregivers. They allow an easier circulation of important information and provide emotional and social support, consequently improving psychological health of caregivers. Support groups allow the patients to encounter other people with the same conditions, with whom they can share similar life experiences, thoughts, concerns, and feelings, acquiring a sense of community, allowing close interpersonal relationships outside the family environment and to develop coping mechanisms to face everyday struggles.Citation131–Citation134 Furthermore, international support communities may allow productive collaborations with clinicians and researchers, allowing a better everyday care, and also provide interesting insights and ideas for further studies and trials.Citation135
To conclude, CS represents a clinically complex rare condition for which good clinical management recommendations are available in literature.Citation29 However, given the advancing knowledge about comorbidities, and the only few data about adult patients and the lack of personalized target therapies, great efforts are required both by clinicians and by basic scientists to better understand the disease mechanisms leading to clinical manifestations. Future works, such as longitudinal natural history studies, are required to provide further knowledge and develop effective treatment strategies.
The comprehensive routine monitoring of patients with CS through a strong multidisciplinary network involving specialized centers, local health services, and family doctors is necessary to achieve the best patient outcomes, to improve assessment protocols on this rare disorder, and to support families during lifetime changes of care needs both based on the natural history of CS and personalized requirements. It is of utmost importance for healthcare providers, especially those who do not routinely care for individuals affected by CS, to follow existing clinical guidelines and recommendations to improve the quality of life and increase life expectancy of their patients.
Abbreviations
CS, Costello syndrome; MAPK, mitogen activated protein kinase; NF1, neurofibromatosis type 1; NS, Noonan syndrome; CFCS, cardio-facio-cutaneous syndrome; NS/LAH, Noonan syndrome with loose anagen hair; NSML, Noonan syndrome with multiple lentigines; CBLS, CBL-mutation associated syndrome; DD, developmental delay; ID, intellectual disability; HCM, hypertrophic cardiomyopathy; ng-tube, nasogastric tube; g-tube, gastrostomy tube; CNS, central nervous system; ASD, autism spectrum disorder; iPSCs, induced pluripotent stem cells; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging; CHD, congenital heart defect; PVS, pulmonary valve stenosis; ECG, electrocardiography; CHF, congestive heart failure; OSA, obstructive sleep apnea; DEXA, dual-energy X-ray absorptiometry; NBS, neuroblastoma; RMS, rhabdomyosarcoma; US, ultrasound; 6MWT, six minutes walking test; PODCI, Pediatrics Outcomes Data Collection Instrument; MEKi, MEK inhibitors; FTi, farnesyl transferase inhibitors.
Author Consent
All authors meet authorship criteria. Nobody who qualified for authorship has been excluded.
Acknowledgments
Our special thanks go to national and international families support groups: Associazione Italiana Sindromi Costello e cardio-facio-cutanea, Costello kids, Costello Syndrome Family Network, and the International Costello Syndrome Support Group.
Disclosure
The authors report no conflict of interest in this work.
Additional information
Funding
References
- Costello JM. A new syndrome: mental subnormality and nasal papillomata. J Paediatr Child Health. 1977;13(2):114–118. doi:10.1111/j.1440-1754.1977.tb01135.x
- Gripp KW, Rauen KA Costello syndrome. 2020:1–29.
- Aoki Y, Niihori T, Kawame H, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37(10):1038–1040. doi:10.1038/ng1641
- Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–531. doi:10.1038/nrm2438
- Mitin N, Rossman KL, Der CJ. Signaling interplay in ras superfamily function. Curr Biol. 2005;15(14):563–574. doi:10.1016/j.cub.2005.07.010
- Aoki Y, Niihori T, Inoue SI, Matsubara Y. Recent advances in RASopathies. J Hum Genet. 2016;61(1):33–39. doi:10.1038/jhg.2015.114
- Tidyman WE, Rauen KA. Pathogenetics of the RASopathies. Hum Mol Genet. 2016;25(R2):R123–R132. doi:10.1093/hmg/ddw191
- Motta M, Pannone L, Pantaleoni F, et al. Enhanced MAPK1 function causes a neurodevelopmental disorder within the RASopathy clinical spectrum. Am J Hum Genet. 2020;107(3):499–513. doi:10.1016/j.ajhg.2020.06.018
- Tajan M, Paccoud R, Branka S, Edouard T, Yart A. The RASopathy family: consequences of germline activation of the RAS/MAPK pathway. Endocr Rev. 2018;39(5):676–700. doi:10.1210/er.2017-00232
- Leoni C, Romeo DM, Pelliccioni M, et al. Musculo-skeletal phenotype of Costello syndrome and cardio-facio-cutaneous syndrome: insights on the functional assessment status. Orphanet J Rare Dis. 2021;16(1):1–11. doi:10.1186/s13023-021-01674-y
- Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet. 2011;157(2):83–89. doi:10.1002/ajmg.c.30300
- Kratz CP, Franke L, Peters H, et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer. 2015;112(8):1392–1397. doi:10.1038/bjc.2015.75
- Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA. HRAS mutations in Costello syndrome: detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A. 2006;140(1):8–16. doi:10.1002/ajmg.a.31078
- Gripp KW, Lin AE, Stabley DL, et al. HRAS mutation analysis in Costello syndrome: genotype and phenotype correlation karen. Am J Hum Genet. 2006;221(3):212–221.
- Kerr B, Delrue M-A, Sigaudy S, et al. Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med Genet. 2006;43(5):401–405. doi:10.1136/jmg.2005.040352
- Schulz AL, Albrecht B, Arici C, et al. Mutation and phenotypic spectrum in patients with cardio-facio-cutaneous and Costello syndrome. Clin Genet. 2008;73(1):62–70. doi:10.1111/j.1399-0004.2007.00931.x
- Niihori T, Aoki Y, Okamoto N, et al. HRAS mutants identified in Costello syndrome patients can induce cellular senescence: possible implications for the pathogenesis of Costello syndrome. J Hum Genet. 2011;56(10):707–715. doi:10.1038/jhg.2011.85
- Aoki Y, Niihori T, Narumi Y, Kure S, Matsubara Y. The RAS/MAPK syndromes: novel roles of the RAS pathway in human genetic disorders. Hum Mutat. 2008;29(8):992–1006. doi:10.1002/humu.20748
- Lo IFM, Brewer C, Shannon N, et al. Severe neonatal manifestations of Costello syndrome. J Med Genet. 2008;45(3):167–171. doi:10.1136/jmg.2007.054411
- Lorenz S, Petersen C, Kordaß U, Seidel H, Zenker M, Kutsche K. Two cases with severe lethal course of Costello syndrome associated with HRAS p.G12C and p.G12D. Eur J Med Genet. 2012;55(11):615–619. doi:10.1016/j.ejmg.2012.07.007
- Weaver KN, Wang D, Cnota J, et al. Early-lethal Costello syndrome due to rare HRAS tandem base substitution (c.35-36GClAA; p.G12E)-associated pulmonary vascular disease. Pediatr Dev Pathol. 2014;17(6):421–430. doi:10.2350/14-05-1488-OA.1
- Qian W, Zhang M, Huang H, et al. Costello syndrome with special cutaneous manifestations and HRAS G12D mutation: a case report and literature review. Mol Genet Genomic Med. 2021;9(6):e1690. doi:10.1002/mgg3.1690
- Burkitt‐Wright EMM, Bradley L, Shorto J, et al. Neonatal lethal Costello syndrome and unusual dinucleotide deletion/insertion mutations in HRAS predicting p.Gly12Val. Am J Med Genet A. 2012;158A(5):1102–1110. doi:10.1002/ajmg.a.35296
- Quélin C, Loget P, Rozel C, et al. Fetal Costello syndrome with neuromuscular spindles excess and p.Gly12Val HRAS mutation. Eur J Med Genet. 2017;60(7):395–398. doi:10.1016/j.ejmg.2017.03.014
- Bend EG, Louie RJ, Stevenson RE. Fetal edema, not overgrowth, is associated with neonatal lethal Costello syndrome due to the HRAS p.Gly12Val mutation. Clin Dysmorphol. 2019;28(2):71–73. doi:10.1097/MCD.0000000000000260
- Van Der Burgt I, Kupsky W, Stassou S, et al. Myopathy caused by HRAS germline mutations: implications for disturbed myogenic differentiation in the presence of constitutive HRas activation. J Med Genet. 2007;44(7):459–462. doi:10.1136/jmg.2007.049270
- Gripp KW, Hopkins E, Sol-church K, et al. Phenotypic analysis of individuals with Costello syndrome due to HRAS p.G13C. Am J Med Genet A. 2011;155(4):706–716. doi:10.1002/ajmg.a.33884.Phenotypic
- Bertola D, Buscarilli M, Stabley DL, et al. Phenotypic spectrum of Costello syndrome individuals harboring the rare HRAS mutation p.Gly13Asp. Am J Med Genet A. 2017;173(5):1309–1318. doi:10.1002/ajmg.a.38178
- Gripp KW, Morse LA, Axelrad M, et al. Costello syndrome: clinical phenotype, genotype, and management guidelines. Am J Med Genet A. 2019;179(9):1725–1744. doi:10.1002/ajmg.a.61270
- Hiippala A, Vasilescu C, Tallila J, et al. The rare Costello variant HRAS c.173C>T (p.T58I) with severe neonatal hypertrophic cardiomyopathy. Am J Med Genet A. 2016;170(6):1433–1438. doi:10.1002/ajmg.a.37596
- Gripp KW, Sol-Church K, Smpokou P, et al. An attenuated phenotype of Costello syndrome in three unrelated individuals with a HRAS c.179G>A (p.Gly60Asp) mutation correlates with uncommon functional consequences. Am J Med Genet A. 2015;167A(9):2085–2097. doi:10.1002/ajmg.a.37128
- Denayer E, Parret A, Chmara M, et al. Mutation analysis in Costello syndrome: functional and structural characterization of the HRAS p.Lys117Arg mutation. Hum Mutat. 2008;29(2):232–239. doi:10.1002/humu.20616
- Chiu ATG, Leung GK-C, Chu -YW-Y, Gripp KW, Chung BH-Y. A novel patient with an attenuated Costello syndrome phenotype due to an HRAS mutation affecting codon 146-Literature review and update. Am J Med Genet A. 2017;173(4):1109–1114. doi:10.1002/ajmg.a.38118
- Gripp KW, Innes AM, Axelrad ME, et al. Costello syndrome associated with novel germline HRAS mutations: an attenuated phenotype? Am J Med Genet A. 2008;146A(6):683–690. doi:10.1002/ajmg.a.32227
- Gripp KW, Kolbe V, Brandenstein LI, Rosenberger G. Attenuated phenotype of Costello syndrome and early death in a patient with an HRAS mutation (c.179G>T; p.Gly60Val) affecting signalling dynamics. Clin Genet. 2017;92(3):332–337. doi:10.1111/cge.12980
- Sheffield BS, Yip S, Ruchelli ED, et al. Fatal congenital hypertrophic cardiomyopathy and a pancreatic nodule morphologically identical to focal lesion of congenital hyperinsulinism in an infant with Costello syndrome: case report and review of the literature. Pediatr Dev Pathol. 2015;18(3):237–244. doi:10.2350/14-07-1525-CR.1
- Gripp KW, Hopkins E, Serrano A, Leonard NJ, Stabley DL, Sol-Church K. Transmission of the rare HRAS mutation (c. 173C > T; p.T58I) further illustrates its attenuated phenotype. Am J Med Genet A. 2012;158A(5):1095–1101. doi:10.1002/ajmg.a.35294
- Girisha KM, Lewis LE, Phadke SR, Kutsche K. Costello syndrome with severe cutis laxa and mosaic HRAS G12S mutation. Am J Med Genet A. 2010;152A(11):2861–2864. doi:10.1002/ajmg.a.33687
- Gripp KW, Stabley DL, Nicholson L, Hoffman JD, Sol-Church K. Somatic mosaicism for an HRAS mutation causes Costello syndrome. Am J Med Genet A. 2006;140(20):2163–2169. doi:10.1002/ajmg.a.31456
- Liang J, Guo Y, Lu Z, Yu H, Wu L, Yao Z. Woolly hair nevus caused by somatic mutation and Costello syndrome caused by germline mutation in HRAS: consider parental mosaicism in prenatal counseling. J Dermatol. 2022;49(1):161–164. doi:10.1111/1346-8138.16177
- Sol-Church K, Stabley DL, Demmer LA, et al. Male-to-male transmission of Costello syndrome: G12S HRAS germline mutation inherited from a father with somatic mosaicism. Am J Med Genet A. 2009;149A(3):315–321. doi:10.1002/ajmg.a.32639
- Zampino G, Pantaleoni F, Carta C, et al. Diversity, parental germline origin, and phenotypic spectrum of de novo HRAS missense changes in Costello syndrome. Hum Mutat. 2007;28(3):265–272. doi:10.1002/humu.20431
- Gripp KW, Bifeld E, Stabley DL, et al. A novel HRAS substitution (c.266C>G; p.S89C) resulting in decreased downstream signaling suggests a new dimension of RAS pathway dysregulation in human development. Am J Med Genet A. 2012;158A(9):2106–2118. doi:10.1002/ajmg.a.35449
- Gremer L, De Luca A, Merbitz-Zahradnik T, et al. Duplication of Glu37 in the switch I region of HRAS impairs effector/GAP binding and underlies Costello syndrome by promoting enhanced growth factor-dependent MAPK and AKT activation. Hum Mol Genet. 2010;19(5):790–802. doi:10.1093/hmg/ddp548
- Gripp KW, Baker L, Robbins KM, et al. The novel duplication HRAS c.186_206dup p. (Glu62_Arg68dup): clinical and functional aspects. Eur J Hum Genet. 2020;28(11):1548–1554. doi:10.1038/s41431-020-0662-4
- Lorenz S, Lissewski C, Simsek-Kiper PO, et al. Functional analysis of a duplication (p.E63_D69dup) in the switch II region of HRAS: new aspects of the molecular pathogenesis underlying Costello syndrome. Hum Mol Genet. 2013;22(8):1643–1653. doi:10.1093/hmg/ddt014
- Nagai K, Niihori T, Okamoto N, et al. Duplications in the G3 domain or switch II region in HRAS identified in patients with Costello syndrome. Hum Mutat. 2022;43(1):3–15. doi:10.1002/humu.24287
- Xu F, Wang HJ, Lin ZM, Yu B. Recurrent duplication mutation in HRAS causing mild Costello syndrome in a Chinese patient. Clin Exp Dermatol. 2015;40(4):404–407. doi:10.1111/ced.12571
- Pantaleoni F, Lev D, Cirstea IC, et al. Aberrant HRAS transcript processing underlies a distinctive phenotype within the RASopathy clinical spectrum. Hum Mutat. 2017;38(7):798–804. doi:10.1002/humu.23224
- Schøler Nørgaard M, Mogra R, Pinner J, et al. Fetal Costello syndrome: description of phenotype of HRAS exon 1 mutations. Ultrasound Obstet Gynecol. 2020;55(2):274–275. doi:10.1002/uog.20281
- Lin AE, O’Brien B, Demmer LA, et al. Prenatal features of Costello syndrome: ultrasonographic findings and atrial tachycardia. Prenat Diagn. 2009;29(7):682–690. doi:10.1002/pd.2276
- Lan L-B, Li D-Z. Idiopathic polyhydramnios and foetal macrosomia in the absence of maternal diabetes: clinical vigilance for Costello syndrome. J Obstet Gynaecol. 2021;1–3. doi:10.1080/01443615.2021.1959533
- Shankar SP, Fallurin R, Watson T, et al. Ophthalmic manifestations in Costello syndrome caused by Ras pathway dysregulation during development. Ophthalmic Genet. 2021:1–10. DOI:10.1080/13816810.2021.1978103.
- Zampino G, Mastroiacovo P, Ricci R, et al. Costello syndrome: further clinical delineation, natural history, genetic definition, and nosology. Am J Med Genet. 1993;47(2):176–183. doi:10.1002/ajmg.1320470210
- Sammon MR, Doyle D, Hopkins E, et al. Normative growth charts for individuals with Costello syndrome. Am J Med Genet Part A. 2012;158 A(11):2692–2699. doi:10.1002/ajmg.a.35534
- Leoni C, Onesimo R, Giorgio V, et al. Understanding growth failure in Costello syndrome: increased resting energy expenditure. J Pediatr. 2016;170:322–324. doi:10.1016/j.jpeds.2015.11.076
- Aftab S, Dattani MT. Pathogenesis of growth failure in Rasopathies. Pediatr Endocrinol Rev. 2019;16(Suppl 2):447–458. doi:10.17458/per.vol16.2019.ad.pathogenesisrasopathies
- Leoni C, Giorgio V, Onesimo R, Kuczynska E, Zampino G. Impact of Costello syndrome on growth patterns. Am J Med Genet Part A. 2020;182(11):2797–2799. doi:10.1002/ajmg.a.61812
- Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320–328. doi:10.1016/j.tibs.2011.03.006
- Carpentieri G, Leoni C, Pietraforte D, et al. Hyperactive HRAS dysregulates energetic metabolism in fibroblasts from patients with Costello syndrome via enhanced production of reactive oxidizing species. Hum Mol Genet. 2021. doi:10.1093/hmg/ddab270
- Gregersen N, Viljoen D. Costello syndrome with growth hormone deficiency and hypoglycemia: a new report and review of the endocrine associations. Am J Med Genet A. 2004;129A(2):171–175. doi:10.1002/ajmg.a.30189
- Gripp KW, Lin AE. Costello syndrome: a Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med. 2012;14(3):285–292. doi:10.1038/gim.0b013e31822dd91f
- Fidan M, Chennappan S, Cirstea IC. Studying metabolic abnormalities in the Costello syndrome HRAS G12V mouse model: isolation of mouse embryonic fibroblasts and their in vitro adipocyte differentiation. Methods Mol Biol. 2021;2262:397–409. doi:10.1007/978-1-0716-1190-6_24
- Leoni C, Flex E. Costello syndrome: the challenge of hypoglycemia and failure to thrive. EBioMedicine. 2018;27:5–6. doi:10.1016/j.ebiom.2017.12.006
- Oba D, Inoue S, Miyagawa-Tomita S, et al. Mice with an oncogenic HRAS mutation are resistant to high-fat diet-induced obesity and exhibit impaired hepatic energy homeostasis. EBioMedicine. 2018;27:138–150. doi:10.1016/j.ebiom.2017.11.029
- Ugata K, Imamachi N, Hashimoto A, Saito Y. Anesthetic management in an adult patient with Costello syndrome: a case report. A&A Pract. 2019;13(2):41–43. doi:10.1213/xaa.0000000000000983
- Dileone M, Zampino G, Profice P, et al. Dystonia in Costello syndrome. Parkinsonism Relat Disord. 2012;18(6):798–800. doi:10.1016/j.parkreldis.2012.03.015
- Alfieri P, Piccini G, Caciolo C, et al. Differential effects of HRAS mutation on LTP-like activity induced by different protocols of repetitive transcranial magnetic stimulation. Am J Med Genet Part A. 2011;9(1):3445–3456. doi:10.1002/ajmg.a.36374
- Romeo DM, Specchia A, Fasano A, et al. Treatment of dystonia using trihexyphenidyl in Costello syndrome. Brain Sci. 2020;10(7):450. doi:10.3390/brainsci10070450
- Kawame H, Matsui M, Kurosawa K, et al. Further delineation of the behavioral and neurologic features in Costello syndrome. Am J Med Genet. 2003;118(1):8–14. doi:10.1002/ajmg.a.10236
- Axelrad ME, Glidden R, Nicholson L, Gripp KW. Adaptive skills, cognitive, and behavioral characteristics of Costello syndrome. Am J Med Genet. 2004;128 A(4):396–400. doi:10.1002/ajmg.a.30140
- Schreiber J, Grimbergen L-A, Overwater I, et al. Mechanisms underlying cognitive deficits in a mouse model for Costello syndrome are distinct from other RASopathy mouse models. Sci Rep. 2017;7(1):1256. doi:10.1038/s41598-017-01218-0
- Kang M, Lee Y-S. The impact of RASopathy-associated mutations on CNS development in mice and humans. Mol Brain. 2019;12(1):96. doi:10.1186/s13041-019-0517-5
- Gripp KW, Hopkins E, Doyle D, Dobyns WB. High incidence of progressive postnatal cerebellar enlargement in Costello syndrome: brain overgrowth associated with HRAS mutations as the likely cause of structural brain and spinal cord abnormalities. Am J Med Genet Part A. 2010;152(5):1161–1168. doi:10.1002/ajmg.a.33391
- Calandrelli R, D’Apolito G, Marco P, Zampino G, Tartaglione T, Colosimo C. Costello syndrome: analysis of the posterior cranial fossa in children with posterior fossa crowding. Neuroradiol J. 2015;28(3):254–258. doi:10.1177/1971400915592549
- Lin AE, Alexander ME, Colan SD, et al. Clinical, pathological, and molecular analyses of cardiovascular abnormalities in Costello syndrome: a Ras/MAPK pathway syndrome. Am J Med Genet Part A. 2011;155(3):486–507. doi:10.1002/ajmg.a.33857
- Gelb BD, Roberts AE, Tartaglia M. Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog Pediatr Cardiol. 2015;39(1):13–19. doi:10.1016/j.ppedcard.2015.01.002
- Leoni C, Blandino R, Delogu AB, et al. Genotype-cardiac phenotype correlations in a large single-center cohort of patients affected by RASopathies: clinical implications and literature review. Am J Med Genet. 2021. doi:10.1002/ajmg.a.62529
- Lioncino M, Monda E, Verrillo F, et al. Hypertrophic cardiomyopathy in rasopathies: diagnosis, clinical characteristics, prognostic implications, and management. Heart Fail Clin. 2022;18(1):19–29. doi:10.1016/j.hfc.2021.07.004
- Kuniba H, Pooh RK, Sasaki K, et al. Prenatal diagnosis of Costello syndrome using 3D ultrasonography amniocentesis confirmation of the rare HRAS mutation G12D. Am J Med Genet A. 2009;149A(4):785–787. doi:10.1002/ajmg.a.32335
- Mori M, Yamagata T, Mori Y, et al. Elastic fiber degeneration in Costello syndrome. Am J Med Genet. 1996;61(4):304–309. doi:10.1002/(SICI)1096-8628(19960202)61:4<304::AID-AJMG2>3.0.CO;2-U
- Calcagni G, Gagliostro G, Limongelli G, et al. Atypical cardiac defects in patients with RASopathies: updated data on CARNET study. Birth Defects Res. 2020;112(10):725–731. doi:10.1002/bdr2.1670
- Calcagni G, Limongelli G, D’Ambrosio A, et al. Data on cardiac defects, morbidity and mortality in patients affected by RASopathies. CARNET study results. Data Br. 2018;16:649–654. doi:10.1016/j.dib.2017.11.085
- Calcagni G, Limongelli G, D’Ambrosio A, et al. Cardiac defects, morbidity and mortality in patients affected by RASopathies. CARNET study results. Int J Cardiol. 2017;245:92–98. doi:10.1016/j.ijcard.2017.07.068
- Gomez-Ospina N, Kuo C, Ananth AL, et al. Respiratory system involvement in Costello syndrome. Am J Med Genet Part A. 2016;170(7):1849–1857. doi:10.1002/ajmg.a.37655
- Vasta I, Scarano E, Rigante M, et al. Obstructive sleep apnea in Costello syndrome. Am J Hum Genet. 2006;221(3):212–221.
- Shikany AR, Baker L, Stabley DL, et al. Medically actionable comorbidities in adults with Costello syndrome. Am J Med Genet Part A. 2020;182(1):130–136. doi:10.1002/ajmg.a.61394
- Alfieri P, Cesarini L, Zampino G, et al. Visual function in Noonan and LEOPARD syndrome. Neuropediatrics. 2008;39(6):335–340. doi:10.1055/s-0029-1216354
- Pierpont ME, Richards M, Engel WK, Mendelsohn NJ, Summers CG. Retinal dystrophy in two boys with Costello syndrome due to the HRAS p.Gly13Cys mutation. Am J Med Genet A. 2017;173(5):1342–1347. doi:10.1002/ajmg.a.38110
- Thakur A, Thakur V, Choudhary T, Mahajan R, Kulshrestha A, Gupta A. Anterior lenticular opacities in Costello syndrome. Am J Ophthalmol Case Rep. 2021;22:101036. doi:10.1016/j.ajoc.2021.101036
- Stevenson DA, Yang FC. The musculoskeletal phenotype of the RASopathies. Am J Med Genet C Semin Med Genet. 2011;157(2):90–103. doi:10.1002/ajmg.c.30296
- Leoni C, Bisanti C, Viscogliosi G, et al. Bone tissue homeostasis and risk of fractures in Costello syndrome: a 4-year follow-up study. Am J Med Genet A. 2021. doi:10.1002/ajmg.a.62615
- Siegel DH, Mann JA, Krol AL, Rauen KA. Dermatological phenotype in Costello syndrome: consequences of Ras dysregulation in development. Br J Dermatol. 2012;166(3):601–607. doi:10.1111/j.1365-2133.2011.10744.x
- Marukian NV, Levinsohn JL, Craiglow BG, Milstone LM, Choate KA. Palmoplantar keratoderma in Costello syndrome responsive to acitretin. Pediatr Dermatol. 2017;34(2):160–162. doi:10.1111/pde.13057
- Gripp KW. Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet. 2005;137 C(1):72–77. doi:10.1002/ajmg.c.30065
- Leoni C, Paradiso FV, Foschi N, et al. Prevalence of bladder cancer in Costello syndrome: new insights to drive clinical decision-making. Clin Genet. 2022;101(4):454–458. doi:10.1111/cge.14111
- Menke J, Pauli S, Sigler M, et al. Uniparental trisomy of a mutated HRAS proto-oncogene in embryonal rhabdomyosarcoma of a patient with Costello syndrome. J Clin Oncol. 2015;33(13):e62–5. doi:10.1200/JCO.2013.49.6539
- Johnson B, Goldberg-Strassler D, Gripp K, Thacker M, Leoni C, Stevenson D. Function and disability in children with Costello syndrome and Cardiofaciocutaneous syndrome. Am J Med Genet A. 2015;167A(1):40–44. doi:10.1002/ajmg.a.36828
- Detweiler S, Thacker MM, Hopkins E, Conway L, Gripp KW. Orthopedic manifestations and implications for individuals with Costello syndrome. Am J Med Genet Part A. 2013;161(8):1940–1949. doi:10.1002/ajmg.a.36047
- White SM, Graham JM, Kerr B, et al. The adult phenotype in Costello syndrome. Am J Med Genet. 2005;136 A(2):128–135. doi:10.1002/ajmg.a.30747
- Reinker KA, Stevenson DA, Tsung A. Orthopaedic conditions in Ras/MAPK related disorders. J Pediatr Orthop. 2011;31(5):599–605. doi:10.1097/BPO.0b013e318220396e
- Leoni C, Stevenson DA, Martini L, et al. Decreased bone mineral density in Costello syndrome. Mol Genet Metab. 2014;111(1):41–45. doi:10.1016/j.ymgme.2013.08.007
- Leoni C, Viscogliosi G, Onesimo R, et al. Characterization of bone homeostasis in individuals affected by cardio-facio-cutaneous syndrome. Am J Med Genet A. 2022;188(2):414–421. doi:10.1002/ajmg.a.62588
- Leoni C, Triumbari EKA, Vollono C, et al. Pain in individuals with RASopathies: prevalence and clinical characterization in a sample of 80 affected patients. Am J Med Genet Part A. 2019;179(6):940–947. doi:10.1002/ajmg.a.61111
- Kiuru M, Urban J, Zhu G, et al. RAS pathway influences the number of melanocytic nevi in cardiofaciocutaneous and Costello syndromes. J Am Acad Dermatol. 2020;82(5):1091–1093. doi:10.1016/j.jaad.2020.01.038
- Leoni C, Guerriero C, Onesimo R, et al. Melanocytic nevi in RASopathies: insights on dermatological diagnostic handles. J Eur Acad Dermatology Venereol. 2021;35(1):e83–e85. doi:10.1111/jdv.16824
- Krab LC, Goorden SMI, Elgersma Y. Oncogenes on my mind: ERK and MTOR signaling in cognitive diseases. Trends Genet. 2008;24(10):498–510. doi:10.1016/j.tig.2008.07.005
- Weeber EJ, Sweatt JD. Molecular neurobiology of human cognition. Neuron. 2002;33(6):845–848. doi:10.1016/s0896-6273(02)00634-7
- Alfieri P, Piccini G, Caciolo C, et al. Behavioral profile in RASopathies. Am J Med Genet Part A. 2014;164(4):934–942. doi:10.1002/ajmg.a.36374
- Bizaoui V, Gage J, Brar R, Rauen KA, Weiss LA. RASopathies are associated with a distinct personality profile HHS Public Access. Am J Med Genet B Neuropsychiatr Genet. 2018;177(4):434–446. doi:10.1002/ajmg.b.32632
- Schwartz DD, Katzenstein JM, Highley EJ, et al. Age-related differences in prevalence of autism spectrum disorder symptoms in children and adolescents with Costello syndrome. Am J Med Genet Part A. 2017;173(5):1294–1300. doi:10.1002/ajmg.a.38174
- Axelrad ME, Schwartz DD, Fehlis JE, et al. Longitudinal course of cognitive, adaptive, and behavioral characteristics in Costello syndrome. Am J Med Genet Part A. 2009;149(12):2666–2672. doi:10.1002/ajmg.a.33126
- Perrino F, Licchelli S, Serra G, et al. Psychopathological features in Noonan syndrome. Eur J Paediatr Neurol. 2018;22(1):170–177. doi:10.1016/j.ejpn.2017.09.009
- Axelrad ME, Schwartz DD, Katzenstein JM, Hopkins E, Gripp KW. Neurocognitive, adaptive, and behavioral functioning of individuals with Costello syndrome: a review. Am J Med Genet C Semin Med Genet. 2011;157(2):115–122. doi:10.1002/ajmg.c.30299
- Della Marca G, Leoni C, Dittoni S, et al. Increased sleep spindle activity in patients with Costello syndrome (HRAS gene mutation). J Clin Neurophysiol. 2011;28(3):314–318. doi:10.1097/WNP.0b013e31821c3ad5
- Leoni C, Gullo G, Resta N, et al. First evidence of a therapeutic effect of miransertib in a teenager with Proteus syndrome and ovarian carcinoma. Am J Med Genet A. 2019;179(7):1319–1324. doi:10.1002/ajmg.a.61160
- De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16(Suppl 2):S17–27. doi:10.1517/14728222.2011.639361
- Gross AM, Frone M, Gripp KW, et al. Advancing RAS/RASopathy therapies (ART): an NCI-sponsored intramural and extramural collaboration for the study of RASopathies. Am J Med Genet A. 2020;182(4):866–876. doi:10.1002/ajmg.a.61485
- Kontaridis MI, Roberts AE, Schill L, et al. The seventh international RASopathies symposium: pathways to a cure-expanding knowledge, enhancing research, and therapeutic discovery. Am J Med Genet A. 2022. doi:10.1002/ajmg.a.62716
- Markham A, Keam SJ. Selumetinib: first Approval. Drugs. 2020;80(9):931–937. doi:10.1007/S40265-020-01331-X
- Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform Neurofibromas [published correction appears in N Engl J Med. 2020 Sep 24;383(13):1290]. N Engl J Med. 2020;382(15):1430–1442. doi:10.1056/NEJMoa1912735
- Andelfinger G, Marquis C, Raboisson MJ, et al. Hypertrophic cardiomyopathy in Noonan syndrome treated by MEK-inhibition. J Am Coll Cardiol. 2019;73(17):2237–2239. doi:10.1016/j.jacc.2019.01.066
- Jindal GA, Goyal Y, Burdine RD, Rauen KA, Shvartsman SY. RASopathies: unraveling mechanisms with animal models. Dis Models Mech. 2015. doi:10.1242/dmm.022442
- Tidyman WE, Goodwin AF, Maeda Y, Klein OD, Rauen KA. MEK-inhibitor-mediated rescue of skeletal myopathy caused by activating Hras mutation in a Costello syndrome mouse model. Dis Model Mech. 2022;15(2). doi:10.1242/dmm.049166
- Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin Cancer Res. 2015;21(8):1819–1827. doi:10.1158/1078-0432.CCR-14-3214
- Rosenberger G, Meien S, Kutsche K. Oncogenic HRAS mutations cause prolonged PI3K signaling in response to epidermal growth factor in fibroblasts of patients with Costello syndrome. Hum Mutat. 2009;30(3):352–362. doi:10.1002/humu.20855
- Saini KS, Loi S, de Azambuja E, et al. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev. 2013;39(8):935–946. doi:10.1016/j.ctrv.2013.03.009
- Bergqvist C, Wolkenstein P. MEK inhibitors in RASopathies. Curr Opin Oncol. 2021;33(2):110–119. doi:10.1097/CCO.0000000000000711
- Stevenson DA, Schill L, Schoyer L, et al. The fourth international symposium on genetic disorders of the Ras/MAPK pathway. Am J Med Genet Part A. 2016;170(8):1959–1966. doi:10.1002/ajmg.a.37723
- Stoller JK. The challenge of rare diseases. Chest. 2018;153(6):1309–1314. doi:10.1016/j.chest.2017.12.018
- Wilson BT, Hughes J. Joining the hidden revolution in rare diseases: working with family support groups. Arch Dis Child. 2020;105(2):107–108. doi:10.1136/archdischild-2019-317227
- Germeni E, Vallini I, Bianchetti MG, Schulz PJ. Reconstructing normality following the diagnosis of a childhood chronic disease: does “rare” make a difference? Eur J Pediatr. 2018;177(4):489–495. doi:10.1007/s00431-017-3085-7
- Delisle VC, Gumuchian ST, Rice DB, et al. Perceived benefits and factors that influence the ability to establish and maintain patient support groups in rare diseases: a scoping review. Patient. 2017;10(3):283–293. doi:10.1007/s40271-016-0213-9
- Anderson M, Elliott EJ, Zurynski YA. Australian families living with rare disease: experiences of diagnosis, health services use and needs for psychosocial support. Orphanet J Rare Dis. 2013;8(1):1. doi:10.1186/1750-1172-8-22
- Baas M, Huisman S, van Heukelingen J, Koekkoek G, Laan HW, Hennekam RC. Building treasures for rare disorders. Eur J Med Genet. 2015;58(1):11–13. doi:10.1016/j.ejmg.2014.10.006
- Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–4689.
- Aoki Y, Niihori T, Banjo T, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93(1):173–180. doi:10.1016/j.ajhg.2013.05.021