Abstract
The discovery of the tetrodotoxin-resistant (TTX-R) Na+ channel in nociceptive neurons has provided a special target for analgesic intervention. In a previous study we found that both morphine tolerance and persistent visceral inflammation resulted in visceral hyperalgesia. It has also been suggested that hyperexcitability of sensory neurons due to altered TTX-R Na+ channel properties and expression contributes to hyperalgesia; however, we do not know if some TTX-R Na+ channel property changes can be triggered by visceral hyperalgesia and morphine tolerance, or whether there are similar molecular or channel mechanisms in both situations. To evaluate the effects of morphine tolerance and visceral inflammation on the channel, we investigated the dorsal root ganglia (DRG) neuronal change following these chronic treatments. Using whole-cell patch clamp recording, we recorded TTX-R Na+ currents in isolated adult rat lumbar and sacral (L6−S2) DRG neurons from normal and pathologic rats with colon inflammatory pain or chronic morphine treatment. We found that the amplitudes of TTX-R Na+ currents were significantly increased in small-diameter DRG neurons with either morphine tolerance or visceral inflammatory pain. Meanwhile, the result also showed that those treatments altered the kinetics properties of the electrical current (ie, the activating and inactivating speed of the channel was accelerated). Our current results suggested that in both models, visceral chronic inflammatory pain and morphine tolerance causes electrophysiological changes in voltage-gated Na channels due to the chronic administration of these medications. For the first time, the present investigation explored the adaptations of this channel, which may contribute to the hyperexcitability of primary afferent nerves and hyperalgesia during these pathologic conditions. The results also suggest that neurophysiologic mechanisms of morphine tolerance and visceral hyperalgesia are related at the TTX-R Na+ channel.
Introduction
Visceral pain, especially the type associated with inflammation of visceral organs, is an important chronic pain syndrome. It is poorly understood and difficult to treat clinically. Compared with somatic pain, visceral pain is more likely to result in hyperalgesia.Citation1 Meanwhile, previous investigations suggested that the repeated use of opioids could, through interactions between opioid and NMDA receptors,Citation2 induce pain hypersensitivity similar to persistent noxious input. It was reported that there was long-lasting increased pain sensitivity in rats following exposure to heroin.Citation3 When conducting an experiment on colorectal distension in the awake rats, we also found that chronic morphine treatment resulted in obvious visceral hyperalgesia, and that the visceral hyperalgesia and morphine tolerance may be interrelated at the level of the dorsal horn of the spinal cord, as they may share common neural mechanisms that modulate the activation of excitatory amino acid receptors, as well as play a role in the fluctuation of intracellular Ca2+ concentration or nitric oxide production in pain-modulating neurons in the dorsal horn.Citation6 Visceral pain/hypersensitivity is a cardinal symptom of functional gastrointestinal disorders. With their peripheral and central projections, sensory neurons in the dorsal root ganglia (DRG) are the “gateway” for painful signals emanating from both somatic and visceral structures. However, there have been no investigations regarding the possible common mechanisms involved with the nociceptive neurons of peripheral DRG during morphine tolerance and visceral hyperalgesia.
The discovery of a tetrodotoxin-resistant voltage-gated Na+ channel (TTX-R; Nav 1.8), which has a specific distribution and role in sensory processing and nociception, has provided a promising target for pharmaceutical pain interventions.Citation5–Citation7 Much evidence supports the importance of the TTX-R Na+ current in the sensitization of primary afferent neurons and inflammatory hyperalgesia.Citation31–Citation37 The effects of inflammatory mediators and antinociceptive agents on the TTX-R Na+ current (TTX-R INa) as well as the test for the reduction of the current density in TTX-R INa by antisense oligonucleotides suggest that the modulation of these currents contributes to inflammatory and neuropathic hyperalgesia, and that blocking Nav1.8 would be an effective treatment of neuropathic pain.Citation8–Citation11 Studies have shown that the TTX-R Na+ channel is crucially involved in the pain processing pathways and that blockade of the expression or function of this channel may produce analgesia without side effects.Citation6 The modulation of TTX-R INa in colonic afferents is an underlying mechanism of hyperalgesia and pain associated with colon inflammation; therefore, this current could constitute a novel target for therapeutic relief of visceral inflammatory pain.Citation12 Selective Nav tetrodotoxin-resistant channels (Nav 1.8 and Nav 1.9) are expressed exclusively in nociceptive neurons in the DRGs where they play a key role in pain sensation, providing an opportunity for the development of novel peripheral analgesics.Citation13,Citation14,Citation36,Citation39 In recent years, the discovery of the highly selective blocker of Nav 1.8 channels (A-803467) and μO-conotoxin MrVIB brought us new hope to properly target the Nav 1.8 channel. It was reported that A-803467 can block the spontaneous activity in small DRG neurons produced by peripheral inflammation.Citation15,Citation16
DRG neurons are the starting-point of the somatic and visceral pain pathway, and following some injuries these cells can produce abnormal spontaneous activity or bursting which can produce chronic pain.Citation17,Citation18 Sodium channels in the DRG neuron play an important role in nociception and could serve as a molecular engine in chronic pain.Citation19,Citation40 It was reported that neuropathic pain can induce spontaneous firing in neuronal cell bodies and retain the intrinsic excitability properties from freshly isolated DRG neurons. Furthermore, the increased excitability resides at least partly in the cell body and is retained in vitro.Citation20–Citation22 Therefore, DRG neurons in an in vitro model can be a useful tool to study cellular mechanisms of nociception; however, the investigation must take the substantial heterogeneity of DRG neurons and their Na+ currents into account. To some extent, the sensory function of a neuron in DRG is related to its size, because small neurons tend to be associated with unmyelinated C fibers, intermediate-sized neurons with Aδ fibers, and large ones with Aβ myelinated fibers.Citation23,Citation24
Since the Na+ channel is the primary inward charge carrier for action potentials in most DRG neurons, changes in Na+ channel properties or density might be responsible for some of the changes in excitability after hyperalgesia.Citation21 It was reported that the development of tolerance to opioids was associated with hyperalgesia.Citation2 The persistent inflammation of the colonic wall also produced visceral hyperalgesia in our previous studies;Citation4 however, the effects of morphine tolerance and visceral hyperalgesia on the TTX-R sodium current of DRG has not been studied. It is not known whether similar changes occur after visceral inflammation or morphine tolerance. We hypothesized that the TTX-R sodium channel in DRG sensory neurons is a specific and effective target for analgesic interventions of visceral hyperalgesia. To evaluate the electrophysiological changes that contribute to this pathologic mechanism, we used a whole-cell patch-clamp model to study the properties of the TTX-R Na+ current from DRG neurons that were freshly isolated from rats that had chronic visceral inflammation and were undergoing multiple morphine treatments.
Materials and methods
Preparation of animals
A total of 80 adult Wistar rats, weighing 120–180 g, were used. All rats were kept under controlled environmental conditions (23°C–24°C, light from 7:00 to 19:00). All procedures were approved by the China Animal Care Committee, and were in accordance with guidelines established for the use of experimental animals by the International Association for the Study of Pain.
Experiment of visceral inflammatory pain
Fifty Wistar rats were anesthetized by isoflurane using a special fixed installation. The local inflammation of the sigmoid colon wall as our previous method,Citation42 was induced by per endoscopic injection of a 0.3 mg/0.1 mL solution of 0.1 mL of bee venom (lyophilized powder from Apis mellifera; Sigma-Aldrich Co, St Louis, MO) to the submucosa under isoflurane anesthesia in rats, and typical visceral pain responses and edema were produced according to the Miampamba method.Citation25 The procedure resulted in behavioral patterns that were significantly different from those in animals injected with saline alone. The rats were divided into two groups: (1) rats treated with brief anesthesia and saline injection twice a day; and (2) rats receiving injection of bee venom solution into the colon wall twice a day. Animals receiving endoscopic injections of the same volume of isotonic saline served as the control group. The dissection and dissociation of rat DRG neurons were carried out after 4 days of visceral stimulation or saline administration, and the last stimulus was given 2 hours prior to dissection.
Experiment of the chronic morphine treatment
A total of 30 Wistar adult rats were divided into two equal groups: (1) rats treated with a subcutaneous injection of saline twice a day; and (2) rats treated with 20 mg/kg of a subcutaneous morphine injection twice a day. The doses of morphine that were administered in this study have been shown to induce the development of resistance to the antinociceptive effects of morphine (). The tests were done after 4–5 days of drug or saline administration. To examine the antinociceptive effects of morphine, the tail-flick test was used. The development of tolerance was determined by measuring tail-flick latencies after drug or saline administration.Citation2
Visceral hyperalgesia test
Colorectal distension (CRD) is a reproducible, reliable, and valid noxious visceral stimulus that can be used in studies performed in awake, unanesthetized, unrestrained rats. CRD produces aversive behavior and cardiovascular and visceromotor responses (VMR). VMR is the abdominal and hind limb muscle contraction in response to CRD, which is the signal of visceral pain. Electrodes were implanted in the external oblique abdominal muscle. After a 1-week recovery period, a 5 cm-long balloon was inserted 7 cm into the descending colon and rectum under 2% isoflurane general anesthesia. CRD was performed by rapidly inflating the balloon to constant pressures: 10, 20, 40, 60, and 80 mmHg followed by 2-minute intervals to check the VMT (the threshold of VMR). We also used VMR induced by CRD to determine the development of morphine tolerance. Furthermore, we combined the CRD method with colorectal inflammation for use as a visceral hyperalgesia model. Before and after we injected bee venom into the sigmoid wall of rats, we measured the contracted threshold of VMR evoked by CRD to detect the extent of the visceral hyperalgesia. For the morphine tolerance group, we also measured VMT evoked by CRD before and after daily administration of morphine to detect whether visceral hyperalgesia occurred.
Cell preparation
DRG lumbar-sacral segments L6–S2 were harvested bilaterally from rats after 3–5 days following treatment (n = 80). Rats were deeply anesthetized with isoflurane. After we exposed and freed the vertebral column, the spinal cord was removed en bloc from L4 to S2 and submerged in a cold dissection solution. The L5–S2 dorsal roots were identified and the L6–S2 ganglia were isolated and minced with iridectomy scissors. The minced ganglia were incubated and digested in a mixture of Dulbecco’s modified Eagle’s medium (Gibco® BRL, Life Technologies Corporation, Carlsbad, CA) with 1 mg/mL collagenase (Type 1; Sigma-Aldrich Co) and 0.4 mg/mL of trypsin (Type 3; Sigma-Aldrich Co) for 35 minutes at 37°C, agitated by gentle bubbling with 95% O2 and 5% CO2, and then washed with both the Dulbecco’s modified Eagle’s medium solution and 10% fetal calf serum to stop the trypsin reaction. The cells were dissociated by gentle blowing and then placed on poly-D-lysine-coated 35 mm culture cover slips. DRG neurons were studied after short-term incubation at 37°C in a humidified 95% O2–5% CO2 incubator. All recordings were done 1–6 hours after cell plating to minimize phenotypic changes that may have been induced by the isolation and culture of the cells. To check the living state of the isolated cells, some of the cells were cultured at 37°C in a humidified 95% air −5% CO2 incubator overnight; it was proven that the isolated DRG neurons of adult rats had a healthy growth state ().
Figure 1 The dissociated and cultured rat DRG neurons and representative Na+ current from individual DRG neurons. (A) The small and medium dissociated DRG neurons were used in the experiment. (B) Dissociated DRG neurons that were cultivated from an adult rat; 3 days after cultivation, neuron and dendrite growth was evident. The scale is 25 μm. (C) All INa+ channels. (D) TTX-RI Na+. (E) TTX-S INa+. In these figures, C–D = E.
Abbreviations: DRG, dorsal root ganglion; TTX-R, tetrodotoxin-resistant; TTX-S, tetrodotoxin-sensitive.
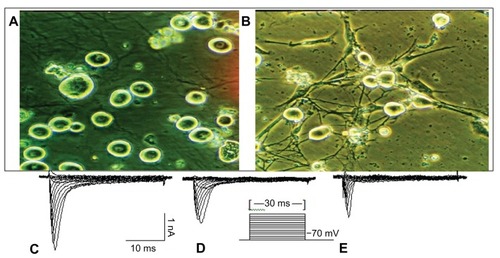
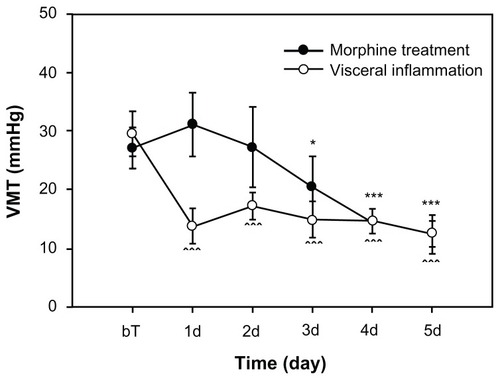
Figure 2 Visceral hyperalgesia phenomena following colon inflammation and morphine treatment.
Abbreviations: VMT, threshold of visceromotor responses; VMR, visceromotor responses; CRD, colorectal distension.
Electrophysiological recording
Sodium currents in small and middle (18–35 μm in diameter) DRG neurons were studied after short-term settling (2–6 hours). The recorded isolated DRG neurons were of a healthy state (). Whole-cell patch-clamp recordings were conducted at room temperature (22°C) using a patch clamp amplifier (Axopatch-1D; Axon Instruments, Union City, CA) and the Clampex program (version 7.0; Axon Instruments). Fire-polished electrodes were fabricated from 1.7 mm capillary glass using the instruments (MF-9; NARISHIGE Group, Tokyo, Japan) and puller (Sachs-Flaming PC-84; Sutter Instrument Company, Novato, CA). Gigaohm seals were achieved with the use of glass microelectrodes with impedances of 2–3 MΩ. Voltage errors were minimized using 80%–85% series resistance compensation, and the capacitance artifact was canceled using the computer-controlled circuitry of the patch-clamp amplifier. Linear leak subtraction, based on resistance estimates from four to five hyperpolarizing pulses applied before the depolarizing test potential, was used for all voltage-clamp recordings. Membrane currents were usually filtered at 2.5 kHz and sampled at 10 kHz.
The pipette solution contained (in mmol/L): 120 CsF, 5 EGTA, 5 NaCl, 5 MgCl2, 1 GTP, and 15 HEPES, pH 7.3. The osmolarity of the solution was adjusted to 300 mOsm. The extracellular bathing solution was (in mmol/L): 45 NaCl, 4 KCl, 4 MgCl2, 0.05 CaCl2, and 10 HEPES, 5 glucose, 20 tetraethylammonium chloride, 65 choline chloride, pH 7.3. pH was adjusted to 7.3 with 1 N CsOH. The osmolarity of the solution was adjusted to 320 mOsm. Reduced extracellular Na+ was required to allow for the recording of large cells that often saturated the voltage clamp amplifier or caused unacceptable serial resistance errors. Extracellular calcium was induced at a concentration low enough to eliminate significant affection from Ca2+ current. Mg2+ was elevated slightly above the baseline physiological level to maintain surface charge screening in the absence of a physiological Ca2+ concentration. Internal Cs+ salts were used to minimize currents through voltage-dependent K+ channels and Ca2+ channels. Tetrodotoxin (TTX) was initially dissolved with a stock concentration of 100 μmol/L and then diluted to a final concentration of 1 μmol/L before application.
First, to examine the DRG neuronal status, membrane potentials and basic properties were studied using current-clamp mode. Recordings of current and voltage were performed using the whole-cell patch-clamp method. Membrane potentials and properties were using the current-clamp mode before TTX was added. Holding potential was −70 mV, and cells showing changes in Na+ current parameters (eg, current decline after a 5-minute recording period), were not used for analysis. Na+ currents can be readily separated according to their sensitivity to TTX (1 μM). Current-voltage relationships of Na+ currents in DRG neurons were elicited by applying depolarizing pulses that were 60 ms in duration over the voltage range of −70 to +60 mV in 5 or 10 mV increments. For the activation, inactivation, and recovery of the channel, double-pulse experiments using conditioning pulses and subsequent test pulses were performed. The pulse width was 60 ms, which was enough to inactivate the sodium channel. The interval between the two pulses was 60 ms. The half-maximal activation and inactivation (V1/2) and the slope factor (k) could be obtained from these curves.
DRG neurons were classified according to their cell diameters in order to detect possible changes in Na+ currents that may be concentrated in a single type of neuron. Based on previous studies correlating fiber type with body size,Citation18 the DRG neurons were identified into the following three groups: small neurons (<25 μm in diameter) largely associated with C fibers; medium neurons (26–30 μm in diameter), typically associated with Aδ fibers; and large neurons (>35 μm in diameter), typical of Aβ fibers. Cell diameter was visually determined using a calibrated reticule in the light path of the microscope during recording. Neurons with a diameter of 18–35 μm that essentially represented peripheral nociceptors in the ganglia were used.
Data analysis
Only DRG neurons that were <35 μm in diameter were included in the data analysis. Data were analyzed with Clampfit 6.05 (Axon Instruments, Union City, CA) and expressed as means ± SE. Origin software 5.5 (University of Oxford, Department of Materials, Oxford, UK) was used for statistical analysis and curve plotting. The Blotzmann function (I/Imax = 1 + exp [(V-V1/2)/k]−1), where V1/2max is the voltage of half activation and inactivation and k is a slope factor, was used to fit activation and inactivation curves.Citation26 The threshold potential of each activation of current was also obtained from the individual fitting curves. Statistical differences were determined and analyzed using two-way analysis of variance and Student’s t-test (two-tailed) among treatment groups. P < 0.05 was considered statistically significant.
Results
Using the whole-cell patch-clamp recording model, we studied some properties of TTX-R Na+ currents from 350 DRG (L6–S2) neurons freshly isolated from rats with visceral inflammatory pain and undergoing a chronic treatment of morphine. The slow TTX-R current was predominant in most small neurons, whereas the TTX-sensitive current was mainly found in large neurons; medium-sized neurons usually exhibited both. showed the representative Na+ current curves from individual DRG neurons. presents whole Na+ current curves. indicates the TTX-R Na+ current curve, and showed the TTX-sensitive current curve.
For some rats, we detected the extent of the visceral hyperalgesia testing the VMT evoked by CRD. For the visceral inflammatory group (n = 10), it was shown that VMT were significantly decreased after colon inflammation (). For the morphine tolerance group (n = 10), we tested VMT values as baseline before morphine injection. We then tested VMT again in 5 days after daily morphine administration. The test was conducted for approximately 10 hours before the last morphine injection was administered. We found that the VMTs from the third to the fifth day exhibited significant differences when compared to baseline values.
Properties of TTX-R voltage-gated sodium channel in DRG (L6–S2) neurons
According to the morphological method, we divided the DRG neurons into small cells (16–24 μm), medium cells (25–35 μm), and large cells (36–50 μm). The average diameter of the small cells (58% of the total number of cells (203/350)), was 22.32 ± 0.14 μm (n = 203), and the average diameter of the medium cells (accounting for 29.4% of all cells, 103/350) was 28.29 ± 0.30 μm (n = 103). In the present experiments, the neurons that were 16–35 μm in diameter represented the sensory neuron relative to nociceptors in the ganglia, and these were recorded.
Na+ currents were separated by their sensitivity to 1 μM TTX. The TTX-R current was recorded in the extracellular solution with 1 μM TTX. Also, the voltage dependence of activation and steady-state inactivation for total TTX-r and TTX-sensitive Na+ currents were measured. The results showed that the TTX-R Na+ current had slow kinetics and slow activating or inactivating currents ( and ). TTX-sensitive currents have faster kinetics and more negative activation and inactivation profiles than TTX-R currents, but we did not show the representative traces of total, TTX-R, and TTX-sensitive Na+ currents here. There are significant differences in the amplitude, rise time, and decay time of TTX-resistant and TTX-sensitive Na+ currents in DRG neurons ().
Table 1 The difference in the amplitude, 10%–90% rise time and decay time of TTX-R and TTX-sensitive Na+ current in DRG neurons
Table 4 Effect of visceral inflammatory pain or morphine tolerance on voltage dependence of activation and steady-state inactivation of TTX-R Na+ current in DRG neurons
Both morphine tolerance and visceral pain enhanced amplitude and excitability of TTX-R sodium currents in the DRG neurons
showed that the average Na+ current amplitude increased when cell size increased. There was no significant difference in cell diameter among the neurons of the control and experiment groups, although there was a decreasing tendency in the visceral pain group (). However, when compared with the control group, visceral inflammation significantly increased the amplitudes of TTX-R currents in small and medium diameter DRG neurons ( and ). For the chronic morphine treatment group, the amplitudes of TTX-R currents in medium cells were also significantly increased, which did not occur in the small diameter neurons in this group. Considering the cell volume and current amplitude of TTX-R sodium channels, the TTX-R sodium current density may also increase in these DRG neurons following chronic visceral inflammation and morphine treatment. In each experiment, the DRG neuronal status, membrane potentials, and basic properties of the neurons were tested using current-clamp mode, prior to adding TTX in the extracellular solution. We also found that in both experiment groups, the threshold potential of the activation of the current was decreased, which suggests that their excitability was also increased ().
Figure 3 Representative activating and steady-state inactivating curves of TTX-R sodium current from individual DRG neurons. (A) The activation and inactivation of the TTX-R Na+ current from a small control neuron. (B) The activation and inactivation of the TTX-R Na+ current from a small neuron of the visceral pain group. (C) The activation and inactivation of the TTX-R Na+ current from a small neuron of the morphine tolerance group.
Abbreviations: TTX-R, tetrodotoxin-resistant; DRG, dorsal root ganglion.
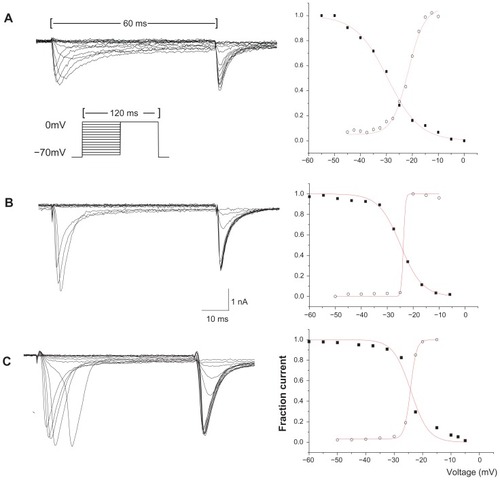
Table 2 The difference in average diameter of DRG neurons among control, visceral inflammatory pain and morphine tolerance groups
Table 3 Effect of visceral inflammatory pain or morphine tolerance on threshold potential (TP) of activation of TTX-R Na+ current in DRG neurons
Morphine treatment and visceral inflammation alter the kinetic properties of TTX-R Na+ current in DRG neurons
A change in the voltage dependence of Na+ currents may alter the excitability of DRG neurons, and these gating-control parameters are not affected by the cell volume and current amplitude; therefore, we measured the voltage-dependent characters of activation and steady-state inactivation for TTX-R Na+ channels from different groups (data are shown in , and ).
Figure 4 Effect of visceral inflammatory pain or morphine tolerance on voltage dependence of activation and steady-state inactivation of TTX-R Na+ current in DRG neurons.
Abbreviations: TTX-R, tetrodotoxin-resistant; DRG, dorsal root ganglion.
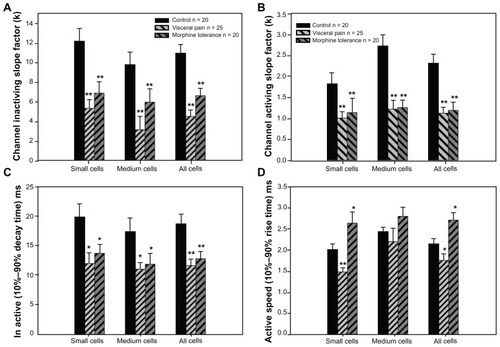
In terms of the activation properties of TTX-R sodium currents, the average V1/2max (half-maximal activation potential) of the TTX-R sodium currents in the DRG neurons from the two experiment groups exhibited negative shifts. In addition, the DRG neurons from the experimental groups exhibited significant differences from the control group when comparing the small cells of the visceral pain group and all of the detected cells in the morphine tolerance group (); no differences were detected between the medium cells in the visceral pain group. Meanwhile, the half-maximal inactivation V1/2max did not exhibit significant increases in the either morphine tolerance or visceral inflammation groups (). Also, we found that the slope factors (k) of both the activation () and inactivation () curves in the experimental groups were significantly decreased when compared with controls. Taken together, these results indicated that these curves became steep, and the activating and inactivating rates increased quickly in the visceral inflammatory pain or morphine tolerance groups.
Then the effects of visceral inflammatory pain or morphine tolerance on the activating and inactivating speed of the maximal TTX-R Na+ current in the DRG neurons were investigated. A 10%–90% rise or decay time was obtained by determining of maximal spikes of the Na+ current. The activating and inactivating speed of the maximal TTX-R Na+ current in the DRG neurons increased after visceral inflammatory pain; furthermore, the inactivating speed increased and the activating speed decreased after morphine tolerance ().
Discussion
In the present report, we described experiments in which two experimental manipulations – chronic visceral pain and morphine tolerance – both led to some plastic alteration in TTX-resistant sodium currents in small and medium DRG neurons. These changes were manifested in both the amplitude of the sodium current as well as in some of their kinetic properties.
The alterations in the channel properties or densities of the TTX-R Na+ may be responsible for some of the changes in the excitability of DRG neurons given that the Na+ channel is the primary inward charge carrier for action potentials in most DRG neurons.Citation21 TTX-R Na+ channels are especially expressed in nociceptive neurons, therefore, they are of considerable interest in pain research and therapeutics. In damaged DRG neurons, various channels are recruited allowing the membrane to achieve a hyperexcitable state, which is associated with hyperalgesia and other chronic pain states.Citation10,Citation19,Citation27,Citation38,Citation39 Our results also showed that there was a significant amplitude increase in the TTX-R current after these pathological changes occurred, which is similar to previous findings in other models.Citation3,Citation17,Citation35,Citation41,Citation42 Therefore, the density of the currents may have increased in small or medium-sized neurons following chronic visceral inflammation, and following morphine tolerance in the medium-sized neurons, likely due to the fact that there were no differences in the size of the cells in the respective groups according to density calculation formula assessing cell volume and current amplitude. This increase in density could be primarily due to the increase in this channel across a number of small neurons expressing this current. Therefore, we have reason to suppose that the increase appears to be primarily a result of the increase in protein or gene expression of TTX-R (SNS or PN3 type) sodium channels in small and medium DRG neurons; however, this conclusion should be confirmed through further molecular biological testing.
Our results differ in some ways from those of other investigators studying TTX-R Na+ currents in C-type DRG neurons after chronic constriction nerve injuries or peripheral axotomy; however, it should be noted that these researchers used the somatic injury models, which were different from our visceral inflammation and morphine tolerance models.Citation21 Our results are more consistent with the results found in investigations using a skin inflammatory injury model (ie, the changes in the carageenan-challenged small and medium-sized neurons).Citation19 These results also demonstrated that the amplitude (or density) of the TTX-R sodium channels, as well as the mRNA expression of the sodium channels SNS or PN3 (associated with TTX-R sodium channel) in small DRG neurons projecting to the inflamed limb were significantly higher than those on the contralateral side 4 days postinjection.Citation19 The results illustrated that there was more coincidence between the test results of somatic and visceral inflammation than there was in the case of neuropathologic pain.Citation44 Based on our results, we might infer that an increase in the density of the TTX-R Na+ current would accompany a left-shift of its voltage dependence on activation. The persistent changes in the channels occurring after visceral inflammation may be the result of long-term exposure to inflammatory autacoids.
Recent studies show that many animal models of inflammatory pain are characterized by the release of neurotrophic factors,Citation28,Citation38,Citation40,Citation42 and that nerve growth factor may function as a key molecular switch that alters dopamine receptor, DOPr, function under sustained opioid stimulation.Citation42,Citation45 Therefore, we also think that the mechanisms in both our models are all involved in the changes among the neurotrophic factors.
It should be noted that our results from these two models of visceral pain and morphine tolerance also have some differences in addition to the similarities. For instance, when comparing both models, the hyperalgesia happened early on (around the 1st day) in visceral inflammation, whereas hyperalgesia from morphine started on the 3rd day of treatment as shown in . That means the hyperalgesia developed from morphine tolerance or addiction is a chronic process.
There are also some differences in the electrophysiological changes in voltage-gated sodium channels after examining the two different models. The current amplitude of TTX-R in small and medium cells in the visceral pain group was significantly increased; in contrast, this effect only occurred in medium cells and did not occur in small neurons in the morphine tolerance group. For the visceral pain model, the inflammatory factor (bee venom) was injected into the colon submucosa, which led to a direct noxious stimulus on afferent C fibers and Aδ fibers, which was related to nociceptive imports. In the development of the morphine tolerance model, there was no direct injury stimulation on enteric organs, but visceral hyperalgesia was only formed in the late phase. From the literature, only Aδ and C fibers carry nociceptive signals and are associated with small to medium diameter neurons; comparatively, non-nociceptive Aβ fibers belong to larger diameter neurons.Citation19,Citation24 The researchers indicated that both C fibers and Aδ fibers are involved in central nervous system sensitization. This is also the reason why we analyzed changes in both medium and small cells.
In the morphine tolerance group, there was increased excitability of small (C-type) and medium (controlled by Aδ fiber) DRG neurons, which was different from the results found among the visceral inflammation group. The average V1/2max values exhibited negative shifts and there were significant differences only in the small cells of the visceral pain group; however, this was also noted in the medium cells in morphine tolerance group. It may be reasonable to assume that more noxious stimulating C fibers were activated in the pain transduction rather than the visceral inflammation situation, but this could be different from the morphine treatment status. In the meantime, the inactivating speed was only increased during visceral inflammation, and the activating speed decreased after morphine tolerance. The latter reaction could be explained by the decreased neuronal reaction in the rats with morphine tolerance. The alteration in the TTX-R Na+ channel property might also be responsible for some of the changes in the excitability of the DRG neurons. The channel may support prolonged firing during chronic depolarization and increase the repertoire of cell burst firing.Citation29
In our results, most changes were concentrated in the small and medium neurons relative to hyperalgesia. The two pathological models produced some similar changes in these cells. The results showed that the average activating V1/2max underwent a negative shift and both the slope factors of the activating and inactivating curves were significantly decreased; moreover, the activating and inactivating rate increased in both experiment groups. The threshold potentials of the two pathological groups were decreased, but the activating and inactivating speed of activation of the TTX-R Na+ current was increased. In addition, the activating V1/2max exhibited a negative shift, but half-maximal inactivation Vmax had a tendency to increase (right shift). This phenomenon may result in an increase in the duration of action potentials and increase the release of Ca2+-related transmitters, thus reinforcing the afferent neuron response to noxious information. This may be another explanation behind the induction of visceral hyperalgesia, but it still needs further evidence.
The net result of this investigation was the neuroplastic changes in both activation and steady-state inactivation properties of the current, which means that both the chronic administration of morphine and colonic inflammation could lead to an increase in neuronal excitability. The channels may be easily activated, and this was evidenced when the neurons became easily excited after receiving a subthreshold stimulus. The frequency of the action potential in the unit may be increased, which could be the major mechanism underlying visceral hyperalgesia. This change in the activation properties is similar to those produced by some hyperalgesic autacoids, and may contribute to the increase in primary afferent excitability and hyperalgesia that occur after these pathological changes. Our results were also consistent with an increase in excitability in small and some medium-sized neurons after chronic constrictive injuries. The results indicate that the current is involved in the development of hyperexcitability associated with visceral inflammation and morphine tolerance. It also suggests that although hyperalgesia has very different causes such as chronic pain and morphine tolerance, each of these causes may actually share at least some mechanistic similarities.Citation30
In one of our recent studies where we used the dextran sulfate sodium model to induce a similar colonic inflammation, our in vivo studies have proven that dextran sulfate sodium-induced colitis sensitized spinal afferent DRG neurons, which led to visceral hyperalgesia. This was evidenced by an increase in the spontaneous activity of the neurons, and was associated with an increase in the high threshold of the afferent discharge response to CRD (data not shown here). The results from this study are very similar to our previous study in which we used a bee venom induced visceral inflammatory model. We are performing more mechanism studies investigating the mRNA or protein expression of the voltage-gated sodium channels, SNS or PN3 (associated with the TTX-R sodium channel) in small and medium DRG neurons after visceral inflammation. Several studies have shown that colonic inflammation, elicited by different chemical or biological agents, induces a state of hypersensitivity that can be observed as increased pain responses during CRD. The results of these future investigations may further confirm and support the findings we noted in the present study about visceral hypersensitivity.
Significance
As discussed above, following the development of morphine tolerance and visceral inflammation, the TTX-R channels in the DRG neurons underwent similar changes. These neuroplastic changes may be involved, at least partially, in the molecular pathologic mechanisms of hyperalgesia induced by chronic morphine treatment or visceral inflammation. That means that there are some correlated mechanisms at the channel and molecular levels that act during both morphine tolerance and visceral hyperalgesia, which provides a promising common target for pharmaceutical intervention. Repeated opioid intake could simulate ongoing nociceptive input.Citation2 Therefore, the demonstration of the interrelationships between the neural mechanisms underlying hyperalgesia and morphine tolerance may lead to a better understanding of these two pathological phenomena. Our further understanding of the common mechanisms underlying hyperalgesia, opioid tolerance, or addiction, and their interactions, would advance the clinical management of intractable visceral pain syndromes and provide a scientific basis for improved pain management with opiate analgesics.
Taken together, pain is a complex process involving considerably more processes than simply the modulation of a single class of ion channels. Nevertheless, this study focused on the TTX-R sodium channel, which has increased our understanding of the neurobiology of pain. Furthermore, as a result of the greater understanding that the nociceptor channels and their plastic alterations are a result of selective targeting of the channel (Nav 1.8 and Nav 1.9), neuroscience may yield a new strategy for the development of analgesics to treat chronic pain with a better safety profile. Our results also suggest that pharmacological therapies designed to specifically block TTX-R INa might effectively treat pain syndromes characterized by visceral hyperalgesia.
Acknowledgment
Part of the project was supported by the US Army Research Office under Contract/Grant No W911NF-08-1-0511 and by the Texas Norman Hackerman Advanced Research Program under Grant No 003599-0001-2009.
Disclosure
The authors report no conflicts of interest in this work.
References
- BuenoLFioramontiJDelvauxMFrexinosJMediators and pharmacology of visceral sensitivity: from basic to clinical investigationsGastroenterology19971125171417439136853
- MayerDJMaoJHoltJPriceDDCellular mechanisms of neuropathic pain, morphine tolerance, and their interactionsProc Natl Acad Sci U S A199996147731773610393889
- LaulinJPLarcherACelerierELe MoalMSimonnetGLong-lasting increased pain sensitivity in rat following exposure to heroin for the first timeEur J Neurosci19981027827859749743
- ChenJHLiuYGongZHQinBYRelated neural mechanisms between visceral hyperalgesia and morphine toleranceChinese Journal of Pharmacology and Toxicology2002165321327
- AkopainANSivilottiLWoodJNA tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neuronsNature199637965622572628538791
- AkopianANSouslovaVEnglandSThe tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathwaysNat Neurosci19992654154810448219
- EglenRMHunterJCDrayAIons in the fire: recent ion-channel research and approaches to pain therapyTrends Pharmacol Sci199920833734210431213
- GoldMSReichlingDBShusterMJLevineJDHyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptorsProc Natl Acad Sci U S A1996933110811128577723
- GoldMSTetrodotoxin-resistant Na+ currents and inflammatory hyperalgesiaProc Natl Acad Sci U S A199996147645764910393874
- GoldMSWeinreichDKimCSRedistribution of Na(V)1.8 in uninjured axons enables neuropathic painJ Neurosci200323115816612514212
- KhasarSGGoldMSLevineJDA tetrodotoxin-resistant sodium current mediates inflammatory pain in the ratNeurosci Lett1998256117209832206
- GoldMSZhangLWrigleyDLTraubRJProstaglandin E(2) modulates TTX-R I(Na) in rat colonic sensory neuronsJ Neurophysiol20028831512152212205171
- LaiJGoldMSKimCSInhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8Pain2002951–214315211790477
- Silos-SantiagoIThe role of tetrodotoxin-resistant sodium channels in pain states: are they the next target for analgesic drugs?Curr Opin Investig Drugs2008918389
- JarvisMFHonorePShiehCCA-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the ratProc Natl Acad Sci U S A2007104208520852517483457
- EkbergJJayamanneAVaughanCWmuO-conotoxin MrVIB selectively blocks Nav1.8 sensory neuron specific sodium channels and chronic pain behavior without motor deficitsProc Natl Acad Sci U S A200610345170301703517077153
- NordinMNystromBWallinUHagbarthKEEctopic sensory discharges and paresthesiae in patients with disorders of peripheral nerves, dorsal roots and dorsal columnsPain19842032312456096790
- WaxmanSGDib-HajjSCumminsTRBlackJASodium channels and painProc Natl Acad Sci U S A199996147635763910393872
- WaxmanSGCumminsTRDib-HajjSFjellJBlackJASodium channels, excitability of primary sensory neurons, and the molecular basis of painMuscle Nerve19992291177118710454712
- StudyREKralMGSpontaneous action potential activity in isolated dorsal root ganglion neurons from rats with a painful neuropathyPain1996652–32352428826512
- KralMGXiongZStudyREAlteration of Na+ currents in dorsal root ganglion neurons from rats with a painful neuropathyPain1999811–2152410353489
- ZhangJMDonnellyDFSongXJLamotteRHAxotomy increases the excitability of dorsal root ganglion cells with unmyelinated axonsJ Neurophysiol1997785279027949356426
- FangXMcMullanSLawsonSNDjouhriLElectrophysiological differences between nociceptive and non-nociceptive dorsal root ganglion neurones in the rat in vivoJ Physiol2005565Pt 392794315831536
- HarperAALawsonSNConduction velocity is related to morphological cell type in rat dorsal root ganglion neuronesJ Physiol198535931463999040
- MiampambaMChery-CrozeSGorryFBergerFChayvialleJAInflammation of the colonic wall induced by formalin as a model of acute visceral painPain19945733273347524010
- QiaoZJA new integrable equation with cuspons and W/M-shape-peaks solitonsJ Math Phys20064711112701112709
- AlvaresDFitzgeraldMBuilding blocks of pain: the regulation of key molecules in spinal sensory neurons during development and following peripheral axotomyPain1999Suppl 6S71S8510491975
- XanthosDNKumarNTheodorssonECoderreTJThe roles of nerve growth factor and cholecystokinin in the enhancement of morphine analgesia in a rodent model of central nervous system inflammationNeuropharmacology200956368469119103210
- RushAMBrauMEElliottAAElliottJRElectrophysiological properties of sodium current subtypes in small cells from adult rat dorsal root gangliaJ Physiol1998511Pt 37717899714859
- GoldMSLevineJDCorreaAMModulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitroJ Neurosci1998182410345103559852572
- BrockJAMcLachlanEMBemlonteCTetrodotoxin-resistant impulses in single nociceptor nerve terminals in guinea-pig corneaJ Physiol1998512Pt 12112179729630
- ChenJHLiuYGongZHColitis pain model induced by colonic submucosal injection of bee venom in ratsChinese Journal of Pharmacology and Toxicology20062017175
- Dib-HajjSBlackJAFeltsPWaxmanSGDown-regulation of transcripts for Na channel alpha-SNS in spinal sensory neurons following axotomyProc Natl Acad Sci U S A1996932514950149548962162
- EnglandSBevanSDochertyRJPGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurons via the cyclic AMP-protein kinase A cascadeJ Physiol1996495Pt 24294408887754
- Omana-ZapataIKhabbazMAHunterJGBleyKRQX-314 inhibits ectopic nerve activity associated with neuropathic painBrain Res199777122282379401743
- PorrecaFLaiJBianDA comparison of the potential of the tetrodotoxin-insensitive sodium channels PN3/SNS and NaN/SNS2, in rat models of chronic painProc Natl Acad Sci U S A199996147640764410393873
- RizzoMASuccessful treatment of painful traumatic mononeuropathy with carbamazepine: insights into a possible molecular pain mechanismJ Neurol Sci199715211031069395132
- RosenLBGreenbergMEStimulation of growth factor receptor signal transduction by activation of voltage-sensitive calcium channelsProc Natl Acad Sci U S A1996933111311188577724
- UdaRHoriguchiSItoSHyodoMHyaishiONociceptive effects induced by intrathecal administration of prostaglandin D2, E2, or F2 alpha to conscious miceBrain Res1990510126322322844
- TanakaMCumminsTRIshikawaKDib-HajjSDBlackJAWaxmanSGSNS Na+ channel expression increases in dorsal root ganglion neurons in the carageenan inflammatory pain modelNeuroreport1998969679729601651
- WaxmanSGThe molecular pathophysiology of pain: abnormal expression of sodium channel genes and its contributions to hyperexcitability of primary sensory neuronsPain1999Suppl 6S133S14010491982
- WoolfCJSafieh-GarabedianBMaQPCrillyPWinterJNerve growth factor contributes to the generation of inflammatory sensory hypersensitivityNeuroscience19946223273317530342
- YakshTLSpinal systems and pain processing: development of novel analgesic drugs with mechanistically defined modelsTrends Pharmacol Sci199920832933710431212
- YoshimuraNSekiSNovakovicSDThe involvement of the tetrodotoxin-resistant sodium channel Na(v)1.8 (PN3/SNS) in a rat model of visceral painJ Neurosci200121218690869611606657
- BieBZhangZCaiYQNerve growth factor-regulated emergence of functional delta-opioid receptorsJ Neurosci201030165617562820410114