Abstract
Background and methods
The role of the endogenous purine nucleoside, adenosine, in nociception is well established. Inhibition of the equilibrative nucleoside transporter (ENT1) prevents adenosine uptake into cells, and could therefore enhance the antinociceptive properties of adenosine. The effects of ENT1 inhibition were studied in two animal models of inflammatory pain. Analgesic activity was assessed in a complete Freund’s adjuvant (CFA)-induced and carrageenan-induced mechanical and thermal hyperalgesia model in the guinea pig.
Results
Draflazine, dipyridamole, dilazep, lidoflazine, soluflazine, and KF24345 showed efficacy in the CFA thermal hyperalgesia model. Draflazine, the most potent compound in this test, was further characterized in the CFA model of mechanical hyperalgesia and the carrageenan inflammation model of thermal and mechanical hyperalgesia, where it completely reversed the hypersensitivity. The antihyperalgesic effects of draflazine (10 mg/kg, administered subcutaneously) were attenuated by the A1 receptor antagonist, cyclopentyltheophylline (5–40 mg/kg, administered intraperitoneally), by the nonselective adenosine antagonist, caffeine (10–40 mg/kg intraperitoneally), and by the A2 antagonist, DMPX (10 mg/kg administered intraperitoneally).
Conclusion
ENT1 inhibition is an effective way of reversing mechanical and thermal inflammatory hyperalgesia in the guinea pig, and these effects are mediated by enhancement of endogenous adenosine levels. Both A1 and A2 adenosine receptor subtypes are likely to be involved.
Introduction
The role of adenosine, an endogenous purine nucleoside, in nociception has been well established over recent decades.Citation1–Citation8 Intravenous infusion was shown to be effective in neuropathic, experimental, and perioperative pain.Citation9 However, due to its short extracellular half-life, continuous infusion is necessary to maintain adequate tissue concentrations, which, in turn, is associated with a high incidence of side effects.Citation10–Citation14 Furthermore, analgesia might be achieved by the potent anti-inflammatory properties that have been attributed to adenosine by several investigators.Citation15–Citation21 To avoid the toxic effects of exogenously administered adenosine, a large volume of work has been done to study the increase in endogenously released adenosine. Adenosine is metabolized by adenosine kinase and adenosine deaminase, and inhibitors of both enzymes effectively increase adenosine levels, producing analgesia in animal models.Citation22–Citation25 Another way of increasing extracellular adenosine levels is by blocking the equilibrative nucleoside transporter, ENT1.Citation26 The equilibrative nucleoside transporters are widely distributed in a number of tissues and in the central nervous system,Citation27–Citation29 and are involved in glutamatergic neurotransmission by regulating local extracellular levels of adenosine.Citation30
The present study explored the hypothesis that inhibition of ENT1 increases extracellular adenosine levels in inflammatory conditions, and that such an increase leads to antinociception in guinea pigs.
Materials and methods
Animals
All experiments were conducted following the ethical guidelines of the International Association for the Study of PainCitation31 and were approved by the local animal care ethics committee. Male Dunkin Hartley guinea pigs (Charles River, Kissleg, Germany) weighing 300–400 g were used (n = 5–7 animals per group). Each animal was used only once. The animals were group-housed in standard rodent cages with sawdust bedding and food and water ad libitum, except during testing. The housing room was air-conditioned with a 12:12-hour day/night cycle. During the light period, a conventional radio was playing, providing background noise. During the experiments, comparable surrounding conditions were present in the laboratory. Habituation to laboratory conditions took place before testing, as explained below.
CFA-induced and carrageenan-induced inflammation
Inflammation of the left hind paw was induced by a 50 μL intraplantar injection of complete Freund’s adjuvant (CFA, 5 mg/mL) or lambda-carrageenan (2% solution). CFA consists of radiant-heat killed Mycobacterium butyricum suspended in paraffin oil. Subcutaneous injection of CFA produces a subacute, localized inflammatory reaction. Hyperalgesia and allodynia of the affected area usually develops within 24 hours after injection.Citation32 Carrageenan is a vegetable gelatin derived from Irish sea moss. The lambda fraction produces an acute inflammatory response in the injected tissue within a very short time frame, accompanied by typical clinical signs of inflammation (redness, warmth, pain behavior, and diminished function). Thermal and mechanical hyperalgesia develops relatively fast (within 4 hours) and allows for testing on the same day of injection, providing a more acute inflammatory condition.Citation33 Behavioral testing of animals started 24 hours after inoculation with CFA or 4 hours after subplantar injection with lambda-carrageenan.
Evaluation of thermal hyperalgesia
Thermal hyperalgesia was quantified with a plantar test analgesia meter (Model 336; IITC Life Sciences, Woodland Hills, CA) according to Hargreaves’ method.Citation33 As described earlier, the time course of thermal hyperalgesia after CFA injection in the rat and the guinea pig is very similar.Citation34 In short, the animals were placed in a plexiglass box on a thin glass plate, and after a habituation period of 60 minutes, radiant heat was applied to the plantar surface of the hind paw. Paw withdrawal latencies (seconds) on exposure to the thermal stimulus were recorded. Temperature settings of the radiant heat source were standardized and were such that a temperature of 33.0°C was measured on the glass surface at the cut-off time of 20 seconds. This cut-off time was chosen to prevent tissue damage and to allow for repeated measurements.
Evaluation of mechanical hyperalgesia
Mechanical hyperalgesia was measured using an Analgesy- Meter (model 7200; Ugo Basile, North America, Collegeville, PA) according to Randall and Selitto.Citation35 Increasing weight was applied to the paw using a wedge-shaped probe, and mechanical thresholds (in g) were determined. The mechanical threshold was reached on withdrawal of the paw or when the animal showed apparent signs of distress, like struggling or vocalization. Cut-off was set at 250 g to prevent any injury and to make repeated testing possible.
Temperature measurements
Body temperature measurements were obtained in a series of separate experiments. An esophageal temperature probe and thermocouple was used (Digitron Instruments Ltd, Thousand Palms, CA). Baseline body temperature was measured after a 1-hour habituation period in the Hargreaves setup. Animals were randomized to receive a subcutaneous injection of active compound or vehicle. Temperature was measured every 30 minutes for the first 2 hours and then every hour until 4 hours post injection. In antagonist experiments, the temperature was measured in all animals after the 2-hour testing period.
Experimental design
For all behavioral testing, the animals were habituated to laboratory conditions 24 hours prior to the experiment. They were habituated to the experimental setup and to the mode of stimulation. This was achieved by applying the stimulus to the left and to the right paw three times, with a 5–10-minute interval between each testing.
All dose-response experiments using nucleoside transport inhibitors were carried out in the CFA-induced thermal hyperalgesia model, because of the high reproducibility of development of thermal hyperalgesia in this model. For these experiments, the animals were trained 24 hours before the test as stated earlier, and were injected with CFA in the left hind paw after their training session. After the injection, the animals were returned to their cages.
On the day of the experiment, the animals were placed in the experimental setup 1 hour prior to measurement of baseline thermal thresholds of the noninflamed right paw and the inflamed left paw. Two measurements of each paw were performed at 5–10-minute intervals, and the average withdrawal latency (seconds) of each paw was calculated. The withdrawal latency of the inflamed hind paw needed to be at least 50% lower than the value for the noninflamed right paw for the animal to be included in the experiment. This inclusion criterion was implemented to ensure that all animals had a sufficient degree of inflammation. After establishment of baseline, the different treatments were randomized (nucleoside transport inhibitor in different doses or vehicles). Drugs were injected subcutaneously at T0, and thermal thresholds were measured every 15 minutes for a period of 2 hours.
This model was also chosen for the antagonism studies. In these experiments, the same protocols were adhered to, except that at T0 the antagonist was injected intraperitoneally immediately before subcutaneous injection of the nucleoside transport inhibitor at a fixed dose (draflazine 10 mg/kg) or the appropriate vehicle. In one antagonism experiment, the antagonist was injected intraperitoneally 45 minutes after dosing of the nucleoside transport inhibitor. One of the nucleoside transport inhibitors was chosen (on the basis of its activity profile) to be characterized further in the other inflammatory models.
In the carrageenan-induced thermal hyperalgesia model, the animals were trained in the experimental setup as described earlier. After this training session, the animals were returned to their normal housing. On the day of the experiment, the left hind paws were injected with carrageenan and the animals were allowed to recover in their cages. Three hours after the carrageenan injection, the animals were placed in the experimental setup, where they were allowed to acclimatize for 1 hour.
Four hours after carrageenan injection, thermal thresholds for the inflamed and the noninflamed paw were measured twice and the average of these two values was calculated. Again, the difference in withdrawal latency between the inflamed left paw and the noninflamed right paw needed to be at least 50% (left paw withdrawal latency 50% less than right paw withdrawal latency) for the animal to be included in the experiment. After establishment of baseline inflammation values, the treatments were randomized and the animals were injected subcutaneously with the drug at different doses or the drug vehicle. Withdrawal latencies in response to thermal stimulation were measured every 15 minutes for a period of 2 hours.
In the CFA-induced mechanical hyperalgesia model, the animals were trained in the experimental setup as stated before and were injected with CFA into the left hind paw at the end of the training session. On the day of the experiment, 24 hours after inoculation, mechanical thresholds were calculated for the inflamed left paw and the noninflamed right paw using the average of the mechanical threshold (in g) of two paw pressure measurements. Also in this model, the inflamed hind paw needed to be 50% more sensitive than the noninflamed paw before the animal was included in the experiment. Treatments were randomized, and the animals received a subcutaneous injection of a drug dose or the appropriate vehicle. The mechanical thresholds of the inflamed left paws were measured at T60 and T90 minutes. These time points were chosen according to the activity profile of the nucleoside transport inhibitor in the thermal hyperalgesia model. Because testing required handling of the animals, it was decided to perform two post drug measurements in order to minimize stress to the guinea pigs. Between measurements, the animals were left in their cages.
The carrageenan-induced mechanical hyperalgesia model was carried out in a similar way. The animals were trained on the day before the experiment as explained earlier. On the test day, carrageenan was injected into the left hind paw and baseline mechanical thresholds were established 4 hours after injection of carrageenan in the same way as for the CFA-induced mechanical hyperalgesia testing. Baseline inflammation values for inflamed left hind paws and noninflamed right hind paws were calculated, and the same inclusion criterion of a 50% difference between the inflamed left and noninflamed right paw was used. At this time point, the animals received a subcutaneous injection of a drug dose or vehicle and post drug thresholds were also measured at 60 and 90 minutes after dosing, at time points when activity in the thermal hyperalgesia model was maximal. In all experiments, the animals were observed for behavioral side effects, and for each animal, it was noted if there was ptosis or flattening, changes in body posture, or hyperactivity.
Drugs
Draflazine, lidoflazine, soluflazine, and dilazep were synthesized in our laboratories (Janssen Pharmaceutica, Beerse, Belgium). Dipyridamole was purchased from Ciba-Geigy (Dehli, India), KF24345 was purchased from Mercachem (Nijmegen, The Netherlands), caffeine was obtained from Acros Chimica (Geel, Belgium), chlordiazepoxide was purchased from Alpha Pharma (Mumbai, India), and cyclopentyltheophylline (CPT, A1 adenosine antagonist, 8-cyclopentyl-1, 3-dimethylxanthine), DMPX (A2 adenosine antagonist 3,7-dimethyl-1-propargylxanthine), and lambda-carrageenan were purchased from Sigma-Aldrich (St Louis, MO).
All ENT1 inhibitors were prepared as aqueous solutions. Draflazine and lidoflazine were dissolved in a 10% hydroxy- β-cyclodextrin solution and KF24345 was dissolved in a 20% hydroxy-β-cyclodextrin solution. Tartaric acid was added to aid dissolution for draflazine, dipyridamole, lidoflazine, and KF24345. Caffeine and DMPX were dissolved in H2O, and CPT was prepared as a suspension in Tween. Carrageenan-lambda was prepared as a 2% solution in normal saline. Drugs were injected subcutaneously or intraperitoneally. The volume of injection was 10 mL/kg bodyweight. Dose ranges were chosen from the dosing schedule 0.63, 2.5, 10, or 40 mg/kg, or according to the literature.
Data analysis
Randomization principles were applied in all experiments. In each treatment group, 5–7 animals were tested. All data are presented as the mean and the standard error of the mean, unless otherwise stated. Statistical significance between experimental conditions was calculated using the Wilcoxon-Mann- Whitney test with StatXact 4.0.1 software. Corrections for repeated measures were applied where appropriate. P < 0.05 (two-tailed) was considered to be statistically significant.
Results
Effect of various ENT1 inhibitors on CFA-induced thermal hyperalgesia
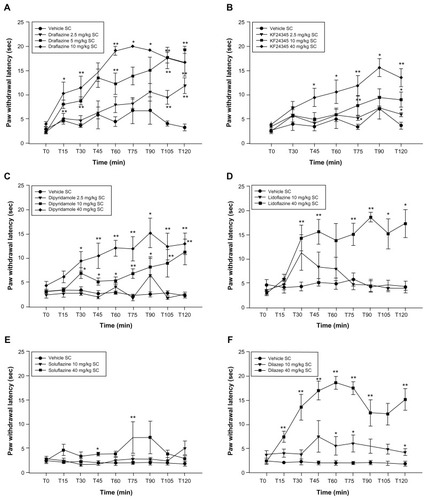
Mean paw withdrawal latency for the CFA-inflamed paw was 3.16 ± 0.13 seconds and 12.85 ± 0.28 seconds for the noninflamed paw in these experiments. Nearly all the ENT1 inhibitors tested increased paw withdrawal latencies in response to the thermal stimulus within 30 minutes after injection when compared with vehicle-treated animals (). KF24345 and soluflazine became active at later time points. This effect lasted throughout the entire 2-hour testing period for all compounds tested. The maximal effect was seen at 60 and 90 minutes after dosing; after this time point, paw withdrawal latencies began to decrease. The lowest active doses were 40 mg/kg for lidoflazine, 10 mg/kg for dipyridamole and dilazep, and 2.5 mg/kg for draflazine and KF24345. Soluflazine administration produced minimal yet statistically significant effects at the highest dose tested (40 mg/kg).
Figure 1 Effect of various subcutaneously administered nucleoside transport inhibitors on complete Freund’s adjuvant-induced thermal hyperalgesia.
Draflazine in acute and subacute inflammatory models
Draflazine, the most active ENT1 inhibitor in the CFA-induced thermal hyperalgesia assay, was chosen for further characterization in other inflammatory models. First, it was tested if draflazine was able to reverse the mechanical hyperalgesia caused by CFA injection in the hind paw. The mechanical threshold in these experiments was on average 50 ± 4 g for left CFA-inflamed paws, whereas for the right noninflamed paws, it was 127 ± 9 g. Subcutaneous administration of draflazine attenuated mechanical hyperalgesia after 60 and 90 minutes, the two time points tested, as compared with vehicle-treated animals. The lowest active dose of draflazine in this model was 5 mg/kg, where activity was seen at 60 minutes after drug treatment. A dose of 10 mg/kg completely reversed mechanical hyperalgesia at both 60 and 90 minutes after injection ().
Figure 2 (A) Effect of draflazine in various doses (2.5–10 mg/kg) and vehicle on complete Freund’s adjuvant-induced mechanical hyperalgesia. Shown are the mean and standard error of the mean values for mechanical withdrawal threshold in grams for the inflamed left paw; 5–7 animals per dose group were tested. Dose groups were compared with the vehicle group using the Wilcoxon-Mann-Whitney test, with correction for repeated measures. *P < 0.05; **P < 0.01. (B) Effect of draflazine at various doses (2.5–10 mg/kg) and vehicle on carrageenan-induced thermal hyperalgesia. Shown are the mean and standard error of the mean values for thermal paw withdrawal latency in seconds for the inflamed left paw; 5–7 animals per dose group were tested. Dose groups were compared with the vehicle group using the Wilcoxon-Mann-Whitney test, with correction for repeated measures. *P < 0.05; **P < 0.01. (C) Effect of draflazine at various doses (2.5–10 mg/kg) and vehicle on carrageenan-induced mechanical hyperalgesia. Shown are the mean and standard error of the mean values for mechanical withdrawal threshold in grams for the inflamed left paw; 5–7 animals per dose group were tested. Dose groups were compared with the vehicle group using the Wilcoxon-Mann-Whitney test, with correction for repeated measures. *P < 0.05; **P < 0.01.
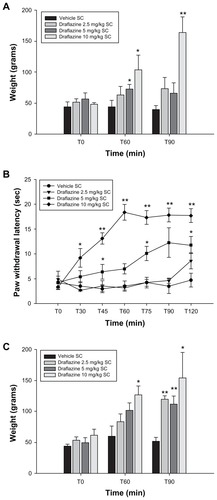
The effects of draflazine were then further investigated in two additional acute inflammatory models of carrageenan-induced hyperalgesia. Carrageenan inflammation significantly decreased withdrawal latencies of the affected paw in response to heat stimulation. An animal with a carrageenan-inflamed paw withdrew the paw after a mean 4.19 ± 0.57 seconds of heat stimulation, where as for the noninflamed right paw, the mean withdrawal latency was 16.0 ± 0.92 seconds. Administration of draflazine successfully attenuated carrageenan-induced thermal hyperalgesia within 30 minutes after injection. The antihyperalgesic effect was maximal after 60 minutes for 10 mg/kg (the high dose) and after 90 minutes for 5 mg/kg (the intermediate dose). At these doses, complete reversal of thermal hyperalgesia was observed (). Carrageenan inflammation also caused a decrease in mechanical threshold from 110 ± 6 g for the noninflamed paws to 51 ± 4 g in the carrageenan-inflamed paws. Draflazine dosed at 10 mg/kg subcutaneously partially normalized the mechanical thresholds within 60 minutes after injection as compared with vehicle. At 90 minutes after administration, draflazine 10 mg/kg completely reversed mechanical hyperalgesia. Lower doses up to 2.5 mg/kg had some effects ().
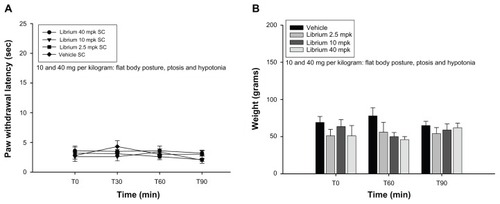
In all these experiments, it was noted that animals treated with draflazine showed signs of sedation (flat body posture, ptosis). This effect was present at 10 mg/kg and became less prominent in the lower dose ranges, where there was a variable degree of ptosis only at the later time points. It was investigated whether sedation could influence nociceptive testing, and high doses of chlordiazepoxide, a sedative benzodiazepine, were tested. Doses of 40 mg/kg produced an obvious flat body posture and ptosis, but did not have any influence on paw withdrawal latencies in the CFA-induced thermal and mechanical hyperalgesia assay ().
Figure 3 (A) Effect of chlordiazepoxide at various doses (2.5–40 mg/kg) and vehicle on complete Freund’s adjuvant-induced thermal hyperalgesia. Shown are the mean and standard error of the mean values for thermal withdrawal latency in seconds for the inflamed left paw; 5–7 animals per dose group were tested. Dose groups were compared with the vehicle group using the Wilcoxon-Mann-Whitney test, and no statistically significant differences were found. (B) Effect of chlordiazepoxide at various doses (2.5–40 mg/kg) and vehicle on complete Freund’s adjuvant-induced mechanical hyperalgesia. Shown are the mean and standard error of the mean values for the mechanical withdrawal threshold in grams for the inflamed left paw; 5–7 animals per dose group were tested. Dose groups were compared with the vehicle group using the Wilcoxon- Mann-Whitney test, and no statistically significant differences were found.
It was also noted that animals treated with a nucleoside transport inhibitor had a decrease in body temperature. Decreases in temperature in draflazine-treated and KF24345- treated animals were dose-dependent. Half an hour after administration, there was a slight decrease in temperature (1°C for the highest doses tested). One hour after administration of the drug, temperature was above 36.5°C for all doses tested.
Involvement of adenosine pathway: antagonism studies
In order to evaluate the adenosine receptors involved in the nucleoside transport inhibition effect, a series of experiments with adenosine receptor antagonists was performed. To do so, a 10 mg/kg dose of draflazine was selected because this dose significantly attenuated the thermal hyperalgesia induced by CFA injection into the left hind paw in these experiments. Intraperitoneal administration of vehicle had no effect on draflazine-induced antihyperalgesic effects in this model. Administration of caffeine 2.5–40 mg/kg, CPT 5–40 mg/kg, and DMPX 0.1–1 mg/kg had no effects on paw withdrawal latency when administered alone (data not shown). Further, CPT 10 mg/kg and 40 mg/kg induced motor activation when it was administered alone. When caffeine, a selective adenosine antagonist, was coadministered with draflazine, a significant decrease in the draflazine effect was noted. At 40 mg/kg, caffeine was able to reverse the antihyperalgesic effect of draflazine fully. At 10 mg/kg, caffeine still partially reversed the effects of draflazine ().
Figure 4 (A) Effect of caffeine at various doses (2.5–40 mg/kg) on the antihyperalgesic effect of draflazine (10 mg/kg) in the CFA-induced thermal hyperalgesia model. Caffeine was injected intraperitoneally immediately after subcutaneous injection of draflazine at time point T0. Shown are the mean and standard error of the mean values for thermal paw withdrawal latency in seconds for the inflamed left paws; 5–7 animals per dose group were tested. First, the draflazine + vehicle group was compared with the vehicle + vehicle group using the Wilcoxon-Mann-Whitney test. $P < 0.05; $$P < 0.01. Comparisons were then made between the draflazine + caffeine dose groups and the draflazine + vehicle group using the Wilcoxon-Mann-Whitney test, with correction for repeated measures. *P < 0.05; **P < 0.01. (B) Effect of adenosine A1 antagonist, CPT, at various doses (5–40 mg/kg) on the antihyperalgesic effect of draflazine (10 mg/kg) in the CFA-induced thermal hyperalgesia model. CPT was injected intraperitoneally immediately after subcutaneous injection of draflazine at time point T0. Shown are the mean and standard error of the mean values for thermal paw withdrawal latency in seconds for the inflamed left paws; 5–7 animals per dose group were tested. First, the draflazine + vehicle group was compared with the vehicle + vehicle group using the Wilcoxon-Mann-Whitney test. $P < 0.05; $$P < 0.01. Comparisons were then made between the draflazine + CPT dose groups and the draflazine + vehicle group using the Wilcoxon-Mann-Whitney test, with correction for repeated measures. *P < 0.05; **P < 0.01. (C) Effect of A1 adenosine antagonist, CPT, in various doses (5–40 mg/kg) on the antihyperalgesic effect of draflazine (10 mg/kg) in the CFA-induced thermal hyperalgesia model. CPT was injected intraperitoneally 45 minutes after subcutaneous injection of draflazine at T0. Shown are the mean and standard error of the mean values for thermal paw withdrawal latency in seconds for the inflamed left paws; 5–7 animals per dose group were tested. First, the draflazine + vehicle group was compared with the vehicle + vehicle group using the Wilcoxon-Mann-Whitney test. $P < 0.05; $$P < 0.01. Comparisons were then made between the draflazine + CPT dose group and the draflazine + vehicle group using the Wilcoxon-Mann-Whitney test, with correction for repeated measures.*P < 0.05; **P < 0.01. (D) Effect of A2 antagonist, DMPX, at various doses (0.1–10 mg/kg) on the antihyperalgesic effect of draflazine (10 mg/kg) in the CFA-induced thermal hyperalgesia model. CPT was injected intraperitoneally immediately after subcutaneous injection of draflazine at time point T0. Shown are the mean and standard error of the mean values for thermal paw withdrawal latencies in seconds for the inflamed left paws; 5–7 animals per dose group were tested. First, the draflazine + vehicle group was compared with the vehicle + vehicle group using the Wilcoxon-Mann-Whitney test. $P < 0.05; $$P < 0.01. Comparisons were then made between the draflazine + DMPX dose group and the draflazine + vehicle group using the Wilcoxon-Mann-Whitney test, with correction for repeated measures.*P < 0.05; **P < 0.01.
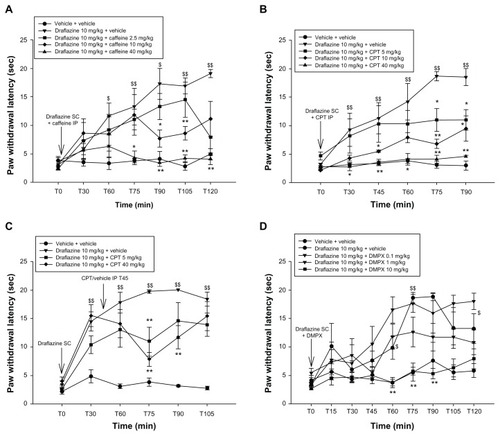
Intraperitoneal injection of CPT, a specific A1 adenosine antagonist, administered immediately after subcutaneous injection of draflazine, completely blocked the antihyperalgesic effects of draflazine. This complete block was noted at a 40 mg/kg dose of CPT; doses of 5 and 10 mg/kg only partly antagonized draflazine (). When CPT 5–40 mg/kg was injected intraperitoneally 45 minutes after administration of draflazine, the antagonist significantly decreased the antihyperalgesic effect of draflazine at all doses tested, with a peak effect 30 minutes after administration (); however, no complete reversal was achieved.
The selective A2A antagonist, DMPX (10 mg/kg), injected intraperitoneally immediately after draflazine, completely reversed the effect of draflazine. The lower doses (0.1–1 mg/kg) had no statistically significant effect on the antihyperalgesic effect of the nucleoside transport inhibitor, although there was a tendency towards lower latencies in animals receiving the 1 mg/kg dose of DMPX, that almost reached statistical significance 90 minutes after administration (P = 0.0556, ).
The adenosine antagonists also attenuated the sedative effects of draflazine, with no obvious sedation noted in animals treated with CPT 40 mg/kg, DMPX 10 mg/kg, or caffeine 10 and 40 mg/kg (observational data, not shown). Furthermore, the adenosine antagonists prevented the decrease in body temperature caused by administration of draflazine. Caffeine 40 mg/kg and DMPX 10 mg/kg when coadministered with draflazine 10 mg/kg completely blocked the temperature decrease in the animals (data not shown). CPT was not tested in this regard.
Discussion
The present study demonstrates that systemically administered nucleoside transport inhibitors can reverse inflammation-induced thermal and mechanical hyperalgesia in the guinea pig in a consistent and dose-dependent fashion. Further, the antinociceptive activity of ENT1 inhibition was antagonized by adenosine antagonists, suggesting an important role in the enhancement of endogenous adenosine levels acting at adenosine receptors. This is direct evidence that systemically administered nucleoside transport inhibitors have antinociceptive properties in vivo. Nucleoside transport inhibitors enhance extracellular levels of adenosine by blocking adenosine reuptake into cells, which is reported to be one of the key mechanisms by which adenosine is cleared from the extracellular space.Citation36 The high local turnover of adenosine during states of inflammationCitation37 makes this condition ideally suited for testing the effects of nucleoside transport inhibition.
In addition to having a strong antinociceptive effect, side effects related to activation of adenosine receptors (sedation, hypothermia) were noted. Previous authors have reported that increased levels of adenosine are associated with sedationCitation38 and that nucleoside transport inhibitors can enhance the sedative effect of exogenously administered adenosine agonists.Citation39 Our experiments confirmed these observations because adenosine antagonists reversed the sedative effects of draflazine. However, the effect on paw withdrawal latency seems limited, given that comparable sedation with chlordiazepoxide did not influence paw withdrawal latency in the CFA-induced thermal hyperalgesia assay or mechanical thresholds in CFA-induced mechanical hyperalgesia testing. This observation provides indirect evidence that clinically relevant sedation does not influence thermal and mechanical hyperalgesia. However, these are observational data. Quantitative characterization of the degree of sedation was not performed, but might have resulted in additional value for this experiment.
Treatment with draflazine and KF24345 was associated with hypothermia, an effect that was counteracted by adenosine antagonists. This confirms previous reports that hypothermia is related to adenosine receptor stimulation.Citation40 Other analgesics, like opioids and cannabinoids, have been reported to produce hypothermia, an effect distinct from their analgesic properties.Citation41 We have strong indications that hypothermia did not influence the antinociceptive effects. First, low doses of draflazine and KF24345 (2.5 mg/kg) induced significant antihyperalgesic effects, with only minimal effects on body temperature. Second, the hypothermic response to nucleoside transport inhibitors is temporally distinct from analgesic activity, ie, antihyperalgesia was observed at early time points with a marginal decrease in body temperature, ie, <1°C 30 minutes after drug administration. The antihyperalgesic effect subsided before the hypothermia resolved.
All adenosine antagonists, whether nonselective (caffeine), A1-selective (CPT), or A2-selective (DMPX), were capable of reversing the antihyperalgesic effects of draflazine. However, the occurrence of motor activation after high doses of CPT suggests an adjuvant A2-inhibiting effect. This has been described previously.Citation42 Likewise, only high doses of DMPX were able to block the antihyperalgesic effects of draflazine, presumably due to limited selectivity. The analgesic effect seems to be both peripherally and centrally mediated. Adenosine has been recognized to have potent anti-inflammatory properties, working mainly through the A2A receptor, located on the cell surface of endothelial and inflammatory cells. ENTs are widely distributed in various tissues and in the central nervous system.Citation27–Citation29
There is high expression of ENT1 receptors across the superficial dorsal horn laminae, and ENT1 is especially abundant in the substantia gelatinosa. It is also present in the dorsal root ganglia.Citation43 At these important loci for pain processing, the ENT 1 is colocalized with the A1 adenosine receptor, which is highly implicated in modulation of pain transmission at the spinal level.Citation5,Citation44,Citation45 The antinociceptive effects of nucleoside transport inhibitors are adenosine-mediated and both A1 receptors and A2 receptors are likely to be involved in this process.
Some analgesic activity of nucleoside transport inhibitors and, more specifically the effects of elevated adenosine levels, has already been demonstrated previously in the mouse tail flick assay after intrathecal administration of dilazep.Citation46 Further, the antihyperalgesic activity of nucleoside transport inhibition is supported by experiments with propentofylline, an atypical methylxanthine with weak adenosine reuptake inhibitor activity, in nerve-injured rats.Citation47 Amitriptyline, a tricyclic antidepressant commonly used to treat neuropathic pain syndromes, produces antinociception together with an increase in endogenous adenosine levels when injected into the rat hind paw. These effects could be reversed by adenosine antagonists and were partly attributed to the nucleoside transport blocking activity of the drug.Citation48
Although the number of clinical trials investigating the analgesic effects of ENT inhibition is limited, there are some reports indicating a favorable effect in the clinical setting. Merskey and Hamilton reported beneficial effects of the ENT inhibitor, dipyridamole, in chronic pain in patients,Citation49 and the combination of dipyridamole and a low dose of prednisolone improved pain and functionality in patients with osteoarthritis.Citation50
In conclusion, we have demonstrated that nucleoside transport inhibitors show antinociceptive activity in a guinea pig model of inflammatory hyperalgesia and that both adenosine A1 and A2 receptors seem to be involved. The results of our experiments show that nucleoside transport inhibition could be a promising target in inflammatory pain states. More studies are obviously needed to investigate their therapeutic potential.
Disclosure
The authors report no conflicts of interest in this work.
References
- BurnstockGWoodJNPurinergic receptors: their role in nociception and primary afferent neurotransmissionCurr Opin Neurobiol199665265328794102
- DunwiddieTVMasinoSAThe role and regulation of adenosine in the central nervous systemAnn Rev Neurosci200124315511283304
- DickensonAHSuzukiRReeveAJAdenosine as a potential analgesic target in inflammatory and neuropathic painsCNS Drugs2000137785
- SawynokJSweeneyMIThe role of purines in nociceptionNeuroscience1989325575692689906
- SawynokJAdenosine receptor activation and nociceptionEur J Pharmacol19983471119650842
- SawynokJLiuXJAdenosine in the spinal cord and periphery: release and regulation of painProg Neurobiol20036931334012787573
- SolleviAAdenosine for pain controlActa Anaesthesiol Scand Suppl19971101351369248564
- WuWPHaoJXHalldnerLIncreased nociceptive response in mice lacking the adenosine A1 receptorPain200511339540415661449
- HayashidaMFukudaKFukunagaAClinical application of adenosine and ATP for pain controlJ Anesth20051922523516032451
- JacobsonKAvan GalenPJWilliamsMAdenosine receptors: pharmacology, structure-activity relationships, and therapeutic potentialJ Med Chem1992354074221738138
- DawickiDDAgarwalKCParksREJrAdenosine metabolism in human whole blood. Effects of nucleoside transport inhibitors and phosphate concentrationBiochem Pharmacol1988376216263342099
- PlagemannPGWohlhueterRMKrauppMAdenosine uptake, transport, and metabolism in human erythrocytesJ Cell Physiol19851253303363877060
- YeungPKMosherSJMacraeDAKlassenGAEffect of diltiazem and its metabolites on the uptake of adenosine in blood: an in-vitro investigationJ Pharm Pharmacol1991436856891682442
- YanLBurbielJCMaassAMullerCEAdenosine receptor agonists: from basic medicinal chemistry to clinical developmentExpert Opin Emerg Drugs2003853757614662005
- CronsteinBNRosensteinEDKramerSBWeissmannGHirschhornRAdenosine: a physiologic modulator of superoxide anion generation by human neutrophils. Adenosine acts via an A2 receptor on human neutrophilsJ Immunol1985135136613712989364
- CronsteinBNLevinRIPhilipsMHirschhornRAbramsonSBWeissmannGNeutrophil adherence to endothelium is enhanced via adenosine A1 receptors and inhibited via adenosine A2 receptorsJ Immunol1992148220122061347551
- Le VrauxChenYLMassonIInhibition of human monocyte TNF production by adenosine receptor agonistsLife Sci199352191719248505858
- ParmelyMJZhouWWEdwardsCKBorcherdingDRSilversteinRMorrisonDCAdenosine and a related carbocyclic nucleoside analogue selectively inhibit tumor necrosis factor-alpha production and protect mice against endotoxin challengeJ Immunol19931513893968326132
- FiresteinGSBulloughDAErionMDInhibition of neutrophil adhesion by adenosine and an adenosine kinase inhibitor. The role of selectinesJ Immunol19951543263347527814
- RosengrenSWongGWFiresteinGSAnti-inflammatory effects of an adenosine kinase inhibitor. Decreased neutrophil accumulation and vascular leakageJ Immunol1995154544454517730646
- BlackburnMRVanceCOMorschlEWilsonCNAdenosine receptors and inflammationHandb Exp Pharmacol200919321526919639284
- McGaraughtySCowartMJarvisMFBermanRFAnticonvulsant and antinociceptive actions of novel adenosine kinase inhibitorsCurr Top Med Chem20055435815638777
- KowalukEAMikusaJWismerCTABT-702 (4-amino-5-(3- bromophenyl)-7-(6-morpholino-pyridin-3-yl)pyrido[2,3-d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti-inflammatory properties. II. In vivo characterization in the ratJ Pharmacol Exp Ther20002951165117411082454
- PoonASawynokJAntinociceptive and anti-inflammatory properties of an adenosine kinase inhibitor and an adenosine deaminase inhibitorEur J Pharmacol19991912313810611432
- WangBTangJWhitePFThe effect of GP683, an adenosine kinase inhibitor, on the desflurane anesthetic requirement in dogsAnesth Analg1997856756809296430
- Van BelleHNucleoside transport inhibition: a therapeutic approach to cardioprotection via adenosine?Cardiovasc Res19932768768458034
- BaldwinSABealPRYaoSYKingAECassCEYoungJDThe equilibrative nucleoside transporter family, SLC29Pflugers Arch200444773574312838422
- WilliamsTCJarvisSMMultiple sodium-dependent nucleoside transport systems in bovine renal brush-border membrane vesiclesBiochem J1991274Pt 127332001243
- AndersonCMXiongWYoungJDCassCEParkinsonFEDemonstration of the existence of mRNAs encoding N1/cif and N2/ cit sodium/nucleoside cotransporters in rat brainBrain Res Mol Brain Res1996423583619013795
- AckleyMAGovernoRJMCassCEYoungJDBaldwinSAKingAEControl of glutamatergic neurotransmission in the rat spinal dorsal horn by the nucleoside transporter ENT1J Physiol200354850751712611914
- ZimmermannMEthical guidelines for investigation of experimental pain in conscious animalsPain1983161091106877845
- SteinCMillanMJHerzAUnilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholdsPharmacol Biochem Behav1988314454513244721
- HargreavesKDubnerRBrownFFloresCJorisJA new and sensitive method for measuring thermal nociception in cutaneous hyperalgesiaPain19883277883340425
- DjouhriLDawbarnDRobertsonANewtonRLawsonSNTime course and nerve growth factor dependence of inflammation-induced alterations in electrophysiological membrane properties in nociceptive primary afferent neuronsJ Neurosci2001218722873311698584
- RandallLOSelittoJJA method for measurement of analgesic activity on inflamed tissueArch Int Pharmacodyn Ther195711140941913471093
- NojiTTakayamaMMizutaniMKF24345, an adenosine uptake inhibitor, suppresses lipopolysaccharide-induced tumor necrosis factor-alpha production and leukopenia via endogenous adenosine in miceJ Pharmacol Exp Ther200230020020511752117
- CronsteinBNAdenosine, an endogenous anti-inflammatory agentJ Appl Physiol1994765138175547
- Porkka-HeiskanenTStreckerREThakkarMBjorkumAAGreeneRWMcCarleyRWAdenosine: a mediator of the sleep-inducing effects of prolonged wakefulnessScience1997276126512689157887
- CrawleyJNPatelJMarangosPJAdenosine uptake inhibitors potentiate the sedative effects of adenosineNeurosci Lett1983361691746306520
- YarbroughGGMcGuff in-ClineschmidtJCIn vivo behavioral assessment of central nervous system purinergic receptorsEur J Pharmacol1981761371446895875
- MagumaHTaylorDAThe effect of chronic opioid vs cannabinoid exposure on the expression of tolerance to morphine- or WIN-55, 212-2-induced analgesia and hypothermia in the guinea pigEur J Pharmacol201166033434021514292
- JacobsonKANikodijevicOPadgettWLGallo-RodriguezCMaillardMDalyJW8-(3-chlorostyryl)caffeine (CSC) is a selective A2-adenosine antagonist in vitro and in vivoFEBS Lett19933231411448495727
- GovernoRJDeucharsJBaldwinSAKingAELocalization of the NBMPR-sensitive equilibrative nucleoside transporter, ENT1, in the rat dorsal root ganglion and lumbar spinal cordBrain Res2005105912913816226730
- ChocaJIProudfitHKGreenRDIdentification of A1 and A2 adenosine receptors in the rat spinal cordJ Pharmacol Exp Ther19872429059103656118
- ChocaJIGreenRDProudfitHKAdenosine A1 and A2 receptors of the substantia gelatinosa are located predominantly on intrinsic neurons: an autoradiography studyJ Pharmacol Exp Ther19882477577643183969
- KeilGJDeLanderGETime-dependent antinociceptive interactions between opioids and nucleoside transport inhibitorsJ Pharmacol Exp Ther1995274138713927562512
- RaghavendraVTangaFRutkowskiMDDeLeoJAAnti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokinesPain200310465566412927638
- SawynokJReidARLiuXJParkinsonFEAmitriptyline enhances extracellular tissue levels of adenosine in the rat hindpaw and inhibits adenosine uptakeEur J Pharmacol200551811612216156010
- MerskeyHHamiltonJTAn open label trial of the possible analgesic effects of dipyridamoleJ Pain Symptom Manage1989434372703736
- KvienTKSlatkowsky-ChristensenBFjeldEEfficacy and safety of a novel syncretic drug candidate-CRX-102 in hand osteoarthritisAnn Rheum Dis20086794294817962237